Introduction
According to the World Health Organization,
colorectal cancer (CRC) is the third leading cause of
cancer-related death in the world after lung and liver cancers. Of
the 8.8 million deaths reported in 2015, 774,000 cases were
attributed to CRC (1). There are
at least four types of human colorectal carcinogenesis, namely
adenoma-carcinoma, hereditary non-polyposis colorectal cancer
(HNPCC), de novo cancer, and colitis cancer (2). Many cases of CRC are related to
environmental or dietary factors rather than heritable genetic
changes. These factors include the environmental and food-borne
mutagens, specific intestinal commensals, pathogens, and chronic
intestinal inflammation, which subsequently induce tumor
development. The progression from adenoma to cancer and metastatic
stage involves the reciprocal failure of protective mechanisms such
as adenomatous polyposis coli (APC), p53, and transforming growth
factor β (TGF-β) as well as the induction of oncogenic pathways
such as K-RAS and β-catenin (3–6).
For the past decade, the development of CRC is
seldom being linked to infectious diseases. However, recent studies
showed that the proteins immediate early 1 (IE1) and pp65 of human
cytomegalovirus (HCMV) were detected in colorectal polyps and
adenocarcinomas but not the adjacent non-neoplastic colon biopsy
samples (7). The presence of HCMV
proteins, mRNA of early genes, and DNA was demonstrated through
immunochemical staining, in situ hybridization, and polymerase
chain reaction (PCR), respectively (7,8). In
addition, our previous study reported the presence of HCMV nucleic
acids in the tumorous epithelium of CRC. Furthermore, the existence
of HCMV in CRC was correlated with the poor outcome in elderly
group but better outcome in the younger group (8,9).
Dimberg et al showed that the HCMV-DNA-positive rate was
significantly higher in cancerous tissue as compared with the
paired normal tissue (10).
Growing evidence demonstrates that HCMV infection occurs in tumor
tissues and its gene products may promote important oncogenic
pathways in CRC (11).
Human cytomegalovirus belongs to the subfamily of
β-herpesviruses. Upon infection, it gets adapted and remains
lifelong in the host. The viral replication cycle is reactivated
whenever the host immunity is impaired, resulting in disease
relapse (12). HCMV comprises a
genome of ~235 kb with >200 open reading frames (ORFs) that
encode >180 proteins. Among these proteins, some are essential
for its replication and a vast majority may interfere with the
cellular and immunological functions to enable the virus to coexist
with its host (13). Several
studies provide evidence that HCMV proteins and nucleic acids are
frequently detected in tissue specimens from patients with cancers
of different origin, including cancer of colon (7,8–11),
breast (14), prostate (15), and mucoepidermoid salivary gland
(16) as well as glioblastoma
(17–19) and neuroblastoma (20). In addition, HCMV proteins are
believed to function as 'oncomodulators' in cancer. There have been
a number of studies suggesting HCMV proteins such as IE, US28,
pp65, non-coding RNA β 2.7kb (β 2.7 kb) and other transcripts
enable the virus to provide mechanisms for oncomodulation, thus
enable the virus to evade from host immune and aid in the oncogenic
transformation (21–23). Some of the HCMV gene products and
proteins are known to accelerate cancer progression via certain
pathways. Some of these pathways are involved in the suppression of
the local immune response against tumors, while others are involved
in the promotion of cell proliferation, apoptosis, angiogenesis and
metastasis.
Increasing evidence revealed HCMV infection in
glioblastoma multiforme (GBM) and glioma stem cell (GSC), which are
believed to cause the recurrence of GBM after the surgery or
therapy (24–27). However, the impact of HCMV
infection in CRC and developing tumors is questionable, especially
in colon cancer stem cell (CSC). To date, there is no well
establish cell model to study the interaction of HCMV and CRC. In
this direction, we studied the influence and effect of HCMV in
CRC-derived cell lines.
Materials and methods
Virus infection
The laboratory-adapted strain of HCMV AD169 obtained
from American Type Culture Collection (ATCC, USA) was propagated in
confluent monolayers of MRC-5 cells (ATCC) in minimal essential
medium (MEM) (Gibco, Life Technologies, CA, USA) supplemented with
10% fetal bovine serum (FBS) (Hyclone, USA). Supernatants were
harvested from MRC-5 cells displaying 90–100% cytopathic effects
(CPE) and the aliquots were store at −80°C. Infectious titers of
all virus stocks were determined by performing the plaque assay on
MRC-5 cells. Virus propagation was carried out by low multiplicity
of infection (MOI).
Cell culture
HT29 and SW480 cells were provided by Professor
Hsei-Wei Wang of National Yang-Ming University, Taiwan. HT29 cells
were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco,
Life Technologies) and SW480 cells were cultured in Leibovitz's
L-15 medium (Gibco, Life Technologies) supplemented with 10% FBS
and 1% penicillin/streptomycin (Gibco, Life Technologies). MRC-5
cells (ATCC) were cultured in MEM supplemented with 10% FBS. All
cells were cultured at 37°C with 5% CO2.
Sphere formation assays
For stem-like culture, HT29 and SW480 parental cells
were resuspended in serum-free DMEM/F12 medium supplemented with 1X
N-2 supplement (Gibco, Life Technologies), 10 ng/ml recombinant
human epidermal growth factor (EGF) (Sigma-Aldrich, USA), 10 ng/ml
basic fibroblast growth factor (bFGF) (Sigma-Aldrich), and 1%
penicillin/streptomycin. Cells were plated at a density of
102, 103 or 104 cells/well, as per
the experimental requirement, and monitored for 2–3 weeks until
spheroids were formed.
Flow cytometry analysis
Flow cytometry assay was used to analyze the
expression profile of the cancer stem cell marker CD44. Briefly,
~106 cells were washed with phosphate-buffered saline
(PBS; Amresco, USA) and labeled with FITC-conjugated anti-CD44
(Miltenyi Biotec, Auburn, CA, USA) in the dark for 30 min at room
temperature. Following incubation, cells were washed twice with PBS
and analyzed using a Cytomics FC 500 Series flow cytometry system
(Beckman Coulter, Indianapolis, IN, USA).
Immunofluorescence assays and
determination of the infectivity rate of HCMV in CRC derived
cells
To determine the HCMV infection in cells,
immunofluorescence assays were carried out by labeling the
non-infected and infected cells with cytomegalovirus immediate
early (IE) antibody (GeneTex, USA) and anti-cytomegalovirus pp65
antibody (Abcam, USA). To determine viral infectivity,
103 parental and stem-like HT29 cells were infected with
HCMV AD169 at MOI of 5 on coverslips. After 24, 48 and 72 h of
infection, infected and non-infected cells were fixed with ice-cold
methanol for 10 min at room temperature and washed thrice with PBS.
Cells were blocked with 1% bovine serum albumin (Sigma-Aldrich) for
1 h at room temperature, followed by three washes of PBS. Both
infected and non-infected cells were stained with 1:20
cytomegalovirus IE antibody for 1 h in a humidified chamber at
37°C. Following incubation, cells were washed thrice with PBS and
probed with an anti-mouse IgG FITC (GeneTex) secondary antibody
(1:2,000 dilution) for 1 h in a humidified chamber at 37°C. Cells
were washed as described above and stained with
4′,6-diamidino-2-phenylindole (DAPI). The washing step was repeated
and a coverslip was placed on the slide with a mounting agent. The
slide was visualized using a fluorescence microscope fitted with a
camera for cell counting. Five low-magnification fields were
counted for each condition and the percentage of positive cells was
calculated by dividing the number of IE-positive cells with the
total number of nuclei, followed by multiplication with 100.
Cell viability and cell
proliferation
Cell proliferation was evaluated in triplicates by a
colorimetric WST-1 assay. The assay determines cellular viability
by measuring the metabolic conversion of a water-soluble
tetrazolium salt into a dark red formazan by mitochondrial
dehydrogenases. The amount of formazan produced is proportional to
the number of live cells, which is expressed as cellular viability.
Briefly, 102 non-infected and infected cells were seeded
in 96-well plates and incubated for 6, 12, 24, 48 and 72 h. The
assay was performed by adding WST-1 (Roche, Germany) directly to
culture wells, followed by incubation for 1 h at 37°C. Plates were
read using DS2® (Dynex, USA) by measuring the absorbance
of the dye at 450 nm wavelength, with 620 nm set as a reference
wavelength. Each experimental condition was performed in
triplicates.
To determine the growth effect of HCMV on infected
cells, the cell proliferation was evaluated by direct cell
counting. HCMV infected and non-infected cells were seeded at a
density of 104 cells/cm2 in 24-well plates
and cultured for 3, 6, 12, 24, 48 and 72 h. Following incubation,
cells were washed with PBS and harvested by trypsinization. The
cell number was determined following staining with 0.4% trypan blue
by Countess Automated Cell Counter (Invitrogen/Life Technologies).
The experiment was repeated thrice and each reaction condition was
performed in triplicates.
Migration assays
For Transwell migration assays, dissociated
stem-like or adherent parental HT29 and SW480 cells infected or
non-infected with AD169 were plated at 103
cells/cm2 on the top chambers containing non-coated
membrane with 8 µm pore size (Corning, NY, USA). Cells in
the top chamber were grown in 100 µl serum-free medium,
while the lower chamber was filled with 600 µl of 10%
FBS-supplemented DMEM/F12. After 6, 12, 24, 48 and 72 h of
infection, cells on the upper side were removed and those under the
surface were fixed and stained with crystal violet. The cell number
was counted using a microscope at five magnification fields. All
assays were performed in triplicates.
Reverse transcription (RT)-PCR and
real-time PCR (quantitative PCR)
RNA was extracted using TRIzol (Sigma-Aldrich) and 2
µg of RNA from each sample was used for the synthesis of the
complementary DNA (cDNA). Reverse transcription was carried out in
a reaction containing 2 µl of 10X RT buffer, 2 µl of
10X RT random primers, 0.8 µl of 100 mM dNTP mix, 1
µl of MultiScribe™ reverse transcriptase, 1 µl of
RNase inhibitor (ABI, USA), and diethyl pyrocarbonate
(DEPC)-treated water (total volume of 10 µl). Following
reaction, 2 µg of total RNA in a total volume of 10
µl was added to the master mix. The reverse transcription
condition was as follows: 25°C for 10 min and 37°C for 120 min,
heat inactivation at 85°C for 5 min, and cooling on ice. The cDNA
synthesized was stored at −20°C until its use for PCR. Real-time
PCR reaction was performed by mixing 12.5 µl 2X SYBR master
mix (ABI), 2 µl cDNA, 0.25 µl primer pair mix (0.1
µM/µl each primer), and 23 µl water with the
PCR reaction. The PCR cycle was as follows: 1 cycle at 50°C for 2
min, 1 cycle at 95°C for 10 min, 40 cycles of 95°C for 15 sec, 60°C
for 30 sec, 72°C for 30 sec, and a final extension at 72°C for 10
min. Real-time PCR was performed in CFX Connect™ Real-Time
Detection System (Bio-Rad, USA) and the results were analyzed with
the CFX Manager™ Software (Bio-Rad). Gene expression level was
normalized with that of glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) and the fold change was calculated as 2−ΔΔCt,
which is the normalized gene expression 2−ΔCt in the
infected sample divided by the normalized gene expression
2−ΔCt in the non-infected sample.
For RT-PCR, the PCR mixture was prepared in a total
volume of 25 µl and included 2X Taq PLUS PCR Smart mix 1
(SolGent™, Korea) and the forward and reverse primers at a
concentration of 0.3 µM each. The volume was adjusted with
DEPC-treated water. PCR was performed with an initial denaturation
at 94°C for 2 min, followed by 35 cycles of 94°C for 30 sec, 55°C
for 30 sec, 72°C for 30 sec, and a final elongation step for 4 min
at 72°C. The primers used in this study are summarized in Table I.
 | Table IThe primers used for RT-PCR and
real-time PCR. |
Table I
The primers used for RT-PCR and
real-time PCR.
| Gene | Forward
(5′–3′) | Reverse
(5′–3′) |
|---|
| RT-PCR | | |
| IE1 |
CGACGTTCCTGCAGACTATG |
TCCTCGGTCACTTGTTCAAA |
| US28 |
GTGAACCGCTCATATAGACC |
GAAACAGGCAGTGAGTAACG |
| β 2.7 kb |
AAGATGTTGCGATGCGGTTG |
CGGTCAGCAGCCAAACAATC |
| GAPDH |
ACCACAGTCCATGCCATCAC |
TCCACCACCCTGTTGCTGTA |
| qPCR | | |
| IE1 |
AAGCGGCCTCTGATAACCAAG |
GAGCAGACTCTCAGAGGATCG |
| US28 |
GTACCACAGCATGAGCTTTTC |
GTATAATTTGTGAGACGCGACA |
| β 2.7 kb |
AAGATGTTGCGATGCGGTTG |
CGGTCAGCAGCCAAACAATC |
| Wnt 11 |
GACACAAGACAGGCAGTG |
GTCCTTGAGCAGAGTCCT |
| FZD7 |
AAGACTTGCAGGACGATGCT |
TGTATCTCCCACTCGCCTTC |
| CTNNB |
GTGCTATCTGTCTGCTCT |
CATCCCTTCCTGTTTAGTTG |
| GSK3β |
AAGTTAGCAGAGACAAGGA |
CGCAATCGGACTATGTTAC |
| GAPDH |
CTGCCCCCTCTGCTGATG |
TCCACGATACCAAAGTTGTCATG |
Analysis of epithelial to mesenchymal
transition (EMT) pathways
HCMV AD169 was used to infect 106 HT29
stem-like cells at MOI of 5. Cells were harvested and total RNA
extracted with TRIzol. The extracted total RNA was sent to
Genomics, Taiwan, for human epithelial to mesenchymal transition
(EMT) RT™ Profiler™ PCR Array (SABiosciences/Qiagen, Germany)
analysis. Briefly, the total RNA was reverse-transcribed and the
resulting cDNA analyzed with a 96-well plate quantitative PCR
array. The array includes 84 key genes of EMT signal pathways. Gene
expression levels were quantified and analyzed with the vendor's
web-based software module. Data were collected and normalized based
on the mean Ct value from five housekeeping genes in the arrays
(ACTB, B2M, GAPDH, HPRT1 and RPLP0) and further normalized to the
untreated control sample. For fold-change comparisons in cells,
non-infected cells were used as the control sample. The fold-change
of gene expression was calculated as 2−ΔΔCt, which is
the normalized gene expression 2−ΔCt in the infected
sample divided by the normalized gene expression 2−ΔCt
in the non-infected sample.
Statistical analyses
Data shown represent results of two independent
experiments, with each reaction performed in triplicates.
Statistical analyses were carried out by the two-way analysis of
variance (ANOVA) and Tukey's multiple comparisons test using
GraphPad Prism software to compare between groups. We used the
Student's t-test to evaluate two independent experimental groups. A
value of p<0.05 was considered as statistically significant.
Results
Increased HCMV infectivity in CRC derived
stem-like cells
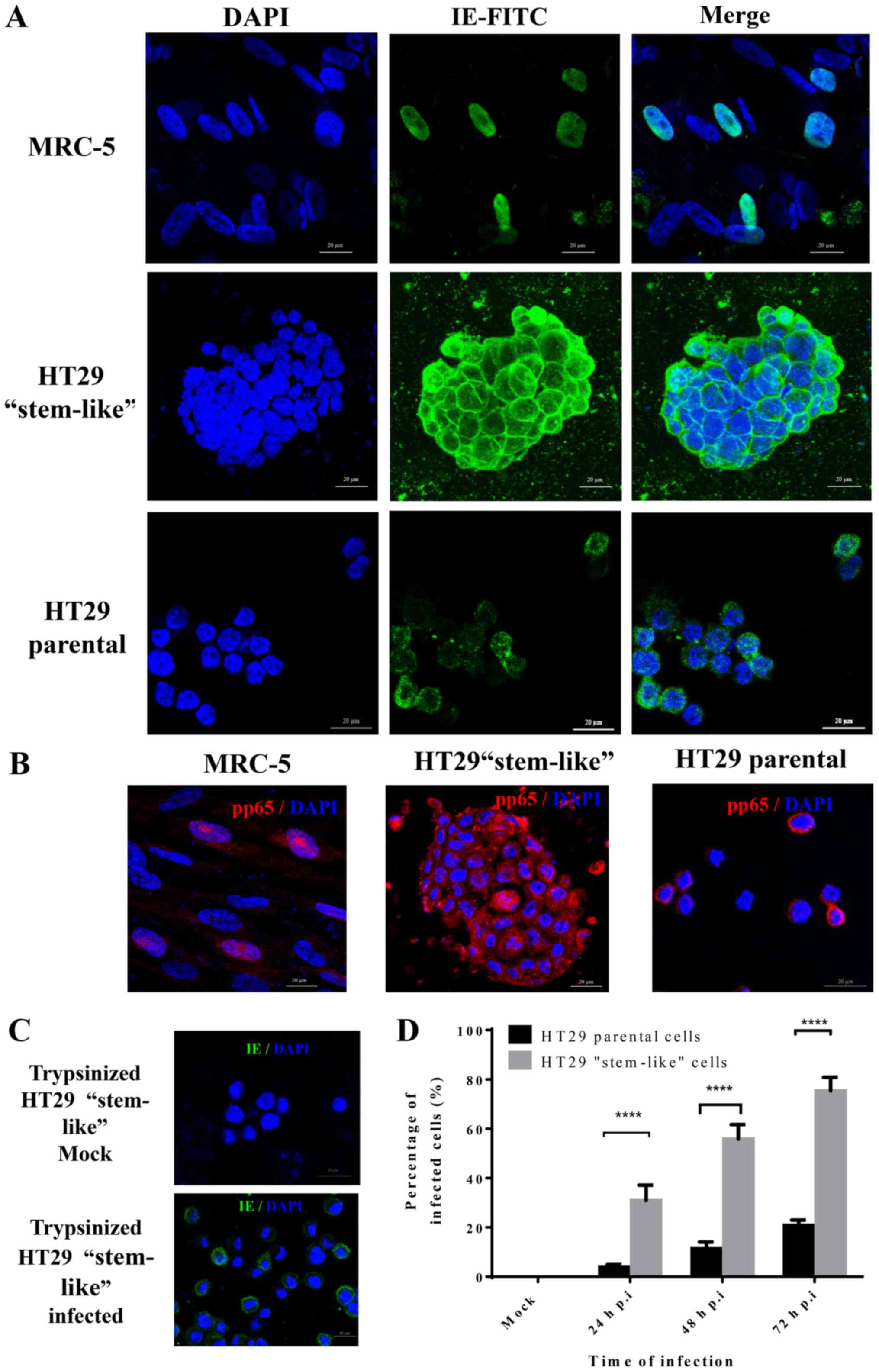
We assessed HCMV infection in CRC using parental and
stem-like HT29 cells as models (Fig.
1A). The enriched HT29 stem-like cells were analyzed using the
CD44 marker. Flow cytometry data showed that CD44+ cell
population was significantly higher (97.31%) in the HT29 stem-like
cells as compared with the parental HT29 cells (5.42%) (Fig. 1B). Both cell types were infected
with the laboratory strain AD169 and the infection was confirmed by
immunofluorescence staining with CMV IE and pp65 (Fig. 2A and B). Infection efficiency was
defined as the number of IE-positive cells divided by the total
number of nucleated cells. We observed a significantly large number
of HT29 stem-like cells infected with AD169 as compared with the
HT29 parental cells. To facilitate the HT29 stem-like cells count,
the infected spheroid cells were dispersed by trypsinization
(Fig. 2C). After 24-h infection,
IE-positive cells were ~3% in HT29 parental cells as compared to
29% in HT29 stem-like cells. After 72-h infection, ~70% of HT29
stem-like cells were IE-positive as compared to only 20%
IE-positive HT29 bulk cells (Fig.
2D).
HCMV gene expression
To evaluate the mechanism of HCMV infection in HT29
stem-like cells, we infected these cells with AD169 at MOI of 5 and
determined the expression pattern of HCMV genes at different time
post-infection using RT-PCR and qPCR. RT-PCR data showed different
HCMV gene expression (Fig. 3A).
Following AD169 infection, the HCMV IE1 transcript was detected at
24 h, but it decreased at 72 h and day 7. A small increase in the
expression of US28 late gene and β 2.7 kb was reported at 24 h, 72
h, and day 7 after infection.
We used SYBR Green real-time PCR for accurate
quantification of the mRNA expression of HCMV genes. Following
infection, IE1 expression surged at day 1 but decreased at day 3
and 7 (Fig. 3B), while the IE1
mRNA level decreased at all time-points. On the other hand, no
significant differences in US28 and β 2.7 kb mRNA expression levels
were observed after day 1, 3 or 7 of HCMV infection. In HT29
stem-like cells with 4 weeks prolonged AD169 infection, we found
that the IE proteins were still expressed (Fig. 3C).
Cell proliferation and viability
We used the colorimetric WST-1 assay to evaluate the
viability of HT29 and SW480 cells after AD169 infection. HT29 and
SW480 parental and stem-like cells infected with AD169 at different
time-points along with the non-infected control were prepared in
triplicates. We observed increased cell viability in HT29 and SW480
stem-like cells infected with AD169 (Fig. 4A and B). In comparison to the
non-infected cells, HT29 stem-like cells infected with AD169 showed
a significant increase in cell proliferation after 12 (0.96±0.05,
p=0.011), 48 (3.10±0.04, p<0.001), and 72 h (3.31±0.03,
p<0.0001) of infection. Besides, we also observed a significant
increase in SW480 stem-like cells infected with AD169 after 24
(1.49±0.02, p<0.0001), 48 (2.79±0.06, p<0.0001), and 72 h
(3.10±0.04, p<0.0001) compared to the non-infected cells. No
significant growth difference was observed between the HT29 and
SW480 parental cells infected with AD169 and non-infected
cells.
We evaluated the proliferation of AD169-infected
cells by direct cell counting. HT29 and SW480 parental and
stem-like cells infected with AD169 at different time-points were
harvested, stained with trypan blue, and counted using the Countess
Automated Cell Counter. The results revealed that AD169 infection
promoted cell growth. Both HT29 and SW480 parental and stem-like
cells showed a significant increase in growth following infection
(p<0.001) (Fig. 4C and D).
However, the growth rate was higher for the infected HT29 or SW480
stem-like cells as compared to the infected parental cells at all
time-points. In comparison to the infected parental cells, infected
stem-like cells showed a 1.3, 2.1, 1.4, and 1.6-fold increase in
the growth rate after 12, 24, 48 and 72 h of infection,
respectively. The infected HT29 stem-like proliferated at a
significantly faster rate compared either with the non-infected
HT29 stem-like or HT29 infected and non-infected parental cells. We
observed that SW480 infected stem-like cells also proliferated
faster than the parental infected cells. The growth rates after 12,
24, 48 and 72 h of infection were 1.5, 1.4, 1.7 and 1.8-fold higher
than those of the parental infected cells. These data suggest that
HCMV infection may promote cell proliferation.
HCMV infection increased cell migration
ability
We used Transwell migration assays to study the
migration ability of HT29 and SW480 cells with or without AD169
infection. In comparison to the non-infected cells, the infected
HT29 and SW480 parental and stem-like cells showed significantly
higher migration ability (p<0.001). However, the number of
migrated cells was more in the stem-like cells as compared with the
infected parental cells (Fig. 5).
In addition, the number of migrated SW480 infected stem-like cells
was higher than that of HT29 infected stem-like cells. Thus, HCMV
infection increased the migration ability of colorectal-derived
cells.
EMT RT2 PCR analyses
To investigate the involvement of EMT in the
increased migration ability of these cells, we evaluated the
expression of EMT-associated genes in infected and non-infected
HT29 stem-like cells using the RT2 Profiler™ PCR array.
The heat map (Fig. 6A) showed that
most of the EMT-related genes were upregulated after HCMV
infection. After 24-h infection, 62 genes were upregulated and 27
genes, downregulated. On the other hand, 35 and 57 EMT-related
genes were upregulated and 49 and 27 genes were downregulated after
72 h and 7 days of infection, respectively.
The array data indicated an increase in the
expression of mesenchymal markers such as N-cadherin and
fibronectin at all time-points. On the other hand, E-cadherin was
downregulated across the infection time. In addition, the
expression of EMT drivers such as SNAIL1, SNAIL2/SLUG, ZEB1, and
TWIST1 was upregulated following infection (Fig. 6B). We observed an increase in the
level of WNT11, frizzled-7 (FZD7), glycogen synthase kinase 3β
(GSK3β), and β-catenin (CTNNB1) during HCMV infection (Fig. 6C). In the subsequent analysis, we
designed a panel of primers against WNT signaling (Table I) and evaluated the expression of
WNT11, FZD7, GSK3β, and β-catenin by real-time PCR. As shown in
Fig. 6D, we observed that the
results were compatible with the results of the array.
In comparison to the non-infected cells, those
infected showed a significant increase (6-fold) in the expression
of WNT11 at day 7 following infection (p<0.001). The expression
of FZD7 in the infected cells was 2.2±0.94-fold higher than that in
the control cells. No significant different in GSK3β expression was
noted. Although there was an increase in the expression of
β-catenin during infection, the difference was not statistically
significant.
Discussion
In this study, we demonstrated that the HCMV strain
AD169 infected HT29 stem-like cells with higher efficiency than the
HT29 parental cells. The infection rate increased in a
time-dependent manner. This result was consistent with a previous
study, wherein the efficiency of HCMV infection was higher in 387
and 3832 GSCs as compared with the standard glioma cell line U87
and T98G (26). Fornara et
al found that glioblastoma cells infected with HCMV exhibited
the ability to grasp those GSC from differentiated condition,
thereby enhancing the stem cell phenotype. Consequently, there was
an increase in the number of GSCs (28). CSCs have been implicated in the
colon carcinogenesis, however their existence had not been
experimentally demonstrated until recently. Due to the complexity
of their biology and technical problems, the definite
identification and isolation is still under debate (29,30).
The cell adhesion molecule CD44 was one of the proposed CSC
markers. CD44-positive cells seem to exhibit CSC properties, such
as a single cell could form a sphere and a xenograft tumor that
resembled the original lesion (31). Therefore, in this study we used
CD44 as the CSC marker to verify the enriched tumor sphere HT29
cells. Du et al (31)
showed that the CD44 was expressed at the bottom of the crypt in
colon tissues where the stem cells are distributed as reported
(32). Previously, we showed that
HCMV viral nucleic acids were mostly found localized at the basal
layer of crypt (8) and in this
study, we demonstrated that the infection rate was higher in the
HT29 stem-like cells. This suggests that HCMV may favor cancer stem
cell-like cells for infection.
In the present study, we observed that the patterns
of HCMV IE and pp65 localization in HT29 stem-like cells were
different from the permissive cell MRC-5. In MRC-5 infected cells,
IE and pp65 were expressed in the nucleus while in HT29 stem-like
cells they were detected in both nucleus and cytoplasm, but mostly
in the cytoplasm. In previous study, the β2.7 kb RNA was detected
in both intranuclear and cytoplasmic human fibroblasts, but in
non-permissive cells, the early transcript is only present within
the nucleus (33). In this case,
the HT29 stem-like cells might behave as fibroblasts, permissive
cells. As showed in this study, HT29 stem-like cells infected with
HCMV upregulated TWIST and SNAIL expression and enhanced EMT by
downregulating E-cadherin and upregulating the N-cadherin,
fibronectin and vimentin (34,35).
As anticipated, the resulting cells may acquire fibroblast-like
properties. Further study should be carried out to clarify this
topographic pattern. We observed a different trend in gene
expression at all time-points (Fig. 3A
and B). The expression of IE1 gene decreased over time, while
expression of US28 and β 2.7 kb transcripts showed fluctuations,
which may be attributed to the different stages in the virus life
cycle. HCMV has an organized genome expression. Its replication
starts with the immediate early gene expression, followed by the
expression of early and late genes. IE1, expressed in the initial
phase, regulated the expression of other viral genes such as US28
and β 2.7 kb. The fluctuation in gene expression reported at
certain time-points may be a signaling change in the virus
replication (36–38).
In this study, we used two CRC-derived cell lines,
HT29 and SW480 cells to verify the proliferation after AD169
infection. We found that either HT29 or SW480 infected stem-like
cells proliferated more than the non-infected cells or the infected
parental and non-infected parental cells. In line with our finding,
Fiallos et al, showed that a long-term infection of HCMV in
GSC also promoted cell proliferation (26). At the same time, Fornara et
al, claimed that the HCMV IE expression induced GBM cells to
display stem-like phenotypes and promoted the growth of glioma
cancer stem cells (GCSCs) (28).
Significant proliferation was shown in the stem-like cells. This is
consistent with previous clinical finding in our group where the
presence of HCMV in CRC patients with stage II, III and IV had a
poor outcome (9,39). The tumoral presence of HCMV was
associated with a decreased disease-free survival and this might
due to the recurrence of CRC. The dysregulation of cell growth
signaling in cancerous cells sustains chronic proliferation.
Phosphatidylinositol-3-kinase/protein kinase (PI3K/AKT) and
mitogen-activated protein kinase (MAPK) activation as well as
phosphatase and tensin homologue (PTEN) mutation is known to
promote tumorigenesis, as these signaling events stimulate growth,
proliferation, and survival of cancer cells (40–43).
It had been reported that HCMV gB induced activation of
platelet-derived growth factor receptor α (PDGFRα) and PI3K/AKT,
which increased growth and promoted survival and motility in cancer
cells (44). HCMV IE proteins have
been shown to induce expression of nuclear factor κB (NF-κB)
subsequently activating the cell survival pathways in tumor cells
(45). HCMV IE1 and IE2 were shown
to interact with p53 suppressor to prevent the infected cells from
undergoing cell growth arrest and apoptosis (46). In addition, HCMV encoded the
protein pUL38, which mimics the mammalian target of rapamycin
complex-1 by blocking the function of tuberous sclerosis protein 2
(TSC2). Inhibition of TSC2 by pUL38 dysregulates the mTOR pathway
and induces survival signal in infected cells (47). Furthermore, Reeves et al,
showed that β 2.7 kb interacts with the mitochondrial respiratory
chain complex I in neuronal U373 cells and prevents cell death
(23).
Accumulated evidence indicates that the HCMV protein
US28 is one of the potential proteins that play an important role
in tumor progression. US28 was found to induce an invasive and
angiogenic phenotype in GBM. US28 has the ability to promote cell
growth and induce progression of cell cycle and expression of
vascular endothelial growth factor, a proangiogenic factor in
NIH3T3. In intestinal cells, US28 was shown to activate β-catenin
by inhibiting GSK3β. At the same time, it dysregulated the WNT
signaling target genes such as cyclin D, survivin and
c-myc, which are important for controlling cell
proliferation (48–53). Furthermore, US28 promotes cell
migration through the chemokines RANTES and monocyte
chemoattractant protein 1 (MCP-1) (48). Another study showed that the
interaction of integrin ανβ3 and PDGFRα with glioma cells resulted
in an increase in the cell migratory ability (54). This explains the phenomenon
observed in our study, wherein the infected colorectal cells showed
greater migration ability.
Studies have proposed that the EMT pathway drives
the progression of cancer to an aggressive metastatic stage. During
the metastatic stage, tumor cells lose their adhesiveness with the
adjacent cells, become more invasive, and develop cancer stemness
properties. Several signaling pathways such as WNT/β-catenin,
Notch, TGF-β, hedgehog, and EGFR are involved in the EMT process
(55–60). In our study, we found that the
expression of mesenchymal markers in EMT such as N-cadherin and
fibronectin were increased upon infection with HCMV. On the other
hand, E-cadherin was suppressed during HCMV infection. E-cadherin
functions as a mediator during cell-cell adhesion and the loss of
E-cadherin is known to induce EMT. β-catenin is thought to be
secluded in E-cadherin adherent junction. APC malfunction in CRC
may result in GSK3β inhibition by the WNT signaling pathway,
leading to the accumulation of β-catenin in the cell cytoplasm.
This will induce the expression of target genes such as
c-myc and cyclin D by the TCF/LEF-1 family
transcription factors. Activation of these genes usually stimulates
tumor progression (61–64).
The array data show that the expression of WNT11 was
high during the infection time course. Therefore, we repeated the
experiment by evaluating the expression of related genes such as
WNT11, FZD7, GSK3β, and β-catenin. It was confirmed that the
expression of these genes was higher in the infected cells as
compared with the non-infected cells. Previous studies (65,66)
revealed that either the canonical or non-canonical WNT signaling
was implicated during the dynamic and reversible EMT and
mesenchymal-epithelial transition during CRC progression. The high
expression of WNT11 and its ligand FZD7 is reported to enhance
proliferation and migration/invasion activities in the colon cancer
cells (67,68). These findings explain the
phenotypic changes observed in our study, wherein HCMV infection
enhanced the proliferation and migratory ability of cells. This
study successfully established a CRC culture model with high HCMV
infectivity to facilitate investigation of the underlying
mechanisms. Further studies are needed to identify the HCMV genes
involved in these phenotypic changes.
Acknowledgments
This study was supported by the grants from the
Ministry of Science and Technology (MOST 105-2314-B-075-055-MY2-1
to Y-J.C. and MOST 104-2320-B-010-017 to J.C.H.). We also thank Dr
Hui-Yu Chuang for her experimental assistance.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
HNPCC
|
hereditary non-polyposis colorectal
cancer
|
|
APC
|
adenomatous polyposis coli
|
|
TGF-β
|
transforming growth factor β
|
|
HCMV
|
human cytomegalovirus
|
|
IE-1
|
immediate early 1
|
|
PCR
|
polymerase chain reaction
|
|
ORF
|
open reading frame
|
|
GBM
|
glioblastoma multiforme
|
|
GSC
|
glioma stem cell
|
|
ATCC
|
American Type Culture Collection
|
|
MEM
|
minimal essential medium
|
|
FBS
|
fetal bovine serum
|
|
CPE
|
cytopathic effect
|
|
MOI
|
multiplicity of infection
|
|
EGF
|
epidermal growth factor
|
|
bFGF
|
basic fibroblast growth factor
|
|
IE
|
immediate early
|
|
PBS
|
phosphate-buffered saline
|
|
EMT
|
epithelial to mesenchymal
transition
|
|
RT-PCR
|
reverse transcription polymerase chain
reaction
|
|
cDNA
|
complementary DNA
|
|
DEPC
|
diethyl pyrocarbonate
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
ANOVA
|
analysis of variance
|
|
FZD7
|
frizzled-7
|
|
GSK3β
|
glycogen synthase kinase 3β
|
|
PI3K/AKT
|
phosphatidylinositol-3-kinase/protein
kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
PTEN
|
phosphatase and tensin homologue
|
|
PDGFRα
|
platelet-derived growth factor
receptor α
|
|
TSC2
|
tuberous sclerosis protein 2
|
References
|
1
|
World Health Organization fact sheet
Cancer. February. 2017, http://www.who.int/mediacentre/factsheets/fs297/en/.
|
|
2
|
Tanaka T: Colorectal carcinogenesis:
Review of human and experimental animal studies. J Carcinog.
8:52009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moore HG, Baxter NN and Guillem JG:
Colorectal cancer: Epidemiology, etiology, and molecular basis. The
ASCRS Textbook of Colon and Rectal Surgery. 2nd edition. Beck D:
Springer Science, Business Media; pp. 669–690. 2011, View Article : Google Scholar
|
|
4
|
Colussi D, Brandi G, Bazzoli F and
Ricciardiello L: Molecular pathways involved in colorectal cancer:
Implications for disease behavior and prevention. Int J Mol Sci.
14:16365–16385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harkins L, Volk AL, Samanta M, Mikolaenko
I, Britt WJ, Bland KI and Cobbs CS: Specific localisation of human
cytomegalovirus nucleic acids and proteins in human colorectal
cancer. Lancet. 360:1557–1563. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen HP, Jiang JK, Chen CY, Chou TY, Chen
YC, Chang YT, Lin SF, Chan CH, Yang CY, Lin CH, et al: Human
cytomegalovirus preferentially infects the neoplastic epithelium of
colorectal cancer: A quantitative and histological analysis. J Clin
Virol. 54:240–244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen HP, Jiang JK, Lai PY, Chen CY, Chou
TY, Chen YC, Chan CH, Lin SF, Yang CY, Chen CY, et al: Tumoral
presence of human cytomegalovirus is associated with shorter
disease-free survival in elderly patients with colorectal cancer
and higher levels of intratumoral interleukin-17. Clin Microbiol
Infect. 20:664–671. 2014. View Article : Google Scholar
|
|
10
|
Dimberg J, Hong TT, Skarstedt M, Löfgren
S, Zar N and Matussek A: Detection of cytomegalovirus DNA in
colorectal tissue from Swedish and Vietnamese patients with
colorectal cancer. Anticancer Res. 33:4947–4950. 2013.PubMed/NCBI
|
|
11
|
Bai B, Wang X, Chen E and Zhu H: Human
cytomegalovirus infection and colorectal cancer risk: A
meta-analysis. Oncotarget. 7:76735–76742. 2016.PubMed/NCBI
|
|
12
|
Crough T and Khanna R: Immunobiology of
human cytomegalovirus: From bench to bedside. Clin Microbiol Rev.
22:76–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murphy E, Yu D, Grimwood J, Schmutz J,
Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM and Shenk TE:
Coding potential of laboratory and clinical strains of human
cytomegalovirus. Proc Natl Acad Sci USA. 100:14976–14981. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harkins LE, Matlaf LA, Soroceanu L, Klemm
K, Britt WJ, Wang W, Bland KI and Cobbs CS: Detection of human
cytomegalovirus in normal and neoplastic breast epithelium.
Herpesviridae. 1:82010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Samanta M, Harkins L, Klemm K, Britt WJ
and Cobbs CS: High prevalence of human cytomegalovirus in prostatic
intraepithelial neoplasia and prostatic carcinoma. J Urol.
170:998–1002. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Melnick M, Sedghizadeh PP, Allen CM and
Jaskoll T: Human cytomegalovirus and mucoepidermoid carcinoma of
salivary glands: Cell-specific localization of active viral and
oncogenic signaling proteins is confirmatory of a causal
relationship. Exp Mol Pathol. 92:118–125. 2012. View Article : Google Scholar
|
|
17
|
Cobbs CS, Harkins L, Samanta M, Gillespie
GY, Bharara S, King PH, Nabors LB, Cobbs CG and Britt WJ: Human
cytomegalovirus infection and expression in human malignant glioma.
Cancer Res. 62:3347–3350. 2002.PubMed/NCBI
|
|
18
|
Rahbar A, Orrego A, Peredo I, Dzabic M,
Wolmer-Solberg N, Strååt K, Stragliotto G and Söderberg-Nauclér C:
Human cytomegalovirus infection levels in glioblastoma multiforme
are of prognostic value for survival. J Clin Virol. 57:36–42. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stragliotto G, Rahbar A, Solberg NW, Lilja
A, Taher C, Orrego A, Bjurman B, Tammik C, Skarman P, Peredo I, et
al: Effects of valganciclovir as an add-on therapy in patients with
cytomegalovirus-positive glioblastoma: A randomized, double-blind,
hypothesis-generating study. Int J Cancer. 133:1204–1213. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wolmer-Solberg N, Baryawno N, Rahbar A,
Fuchs D, Odeberg J, Taher C, Wilhelmi V, Milosevic J, Mohammad AA,
Martinsson T, et al: Frequent detection of human cytomegalovirus in
neuroblastoma: A novel therapeutic target? Int J Cancer.
133:2351–2361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Michaelis M, Doerr HW and Cinatl J Jr: The
story of human cytomegalovirus and cancer: Increasing evidence and
open questions. Neoplasia. 11:1–9. 2009. View Article : Google Scholar
|
|
22
|
Söderberg-Nauclér C and Nelson JY: Human
cytomegalovirus latency and reactivation - a delicate balance
between the virus and its host's immune system. Intervirology.
42:314–321. 1999. View Article : Google Scholar
|
|
23
|
Reeves MB, Davies AA, McSharry BP,
Wilkinson GW and Sinclair JH: Complex I binding by a virally
encoded RNA regulates mitochondria-induced cell death. Science.
316:1345–1348. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cobbs CS, Soroceanu L, Denham S, Zhang W,
Britt WJ, Pieper R and Kraus MH: Human cytomegalovirus induces
cellular tyrosine kinase signaling and promotes glioma cell
invasiveness. J Neurooncol. 85:271–280. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cobbs CS, Soroceanu L, Denham S, Zhang W
and Kraus MH: Modulation of oncogenic phenotype in human glioma
cells by cytomegalovirus IE1-mediated mitogenicity. Cancer Res.
68:724–730. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fiallos E, Judkins J, Matlaf L, Prichard
M, Dittmer D, Cobbs C and Soroceanu L: Human cytomegalovirus gene
expression in long-term infected glioma stem cells. PLoS One.
9:e1161782014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soroceanu L, Matlaf L, Khan S, Akhavan A,
Singer E, Bezrookove V, Decker S, Ghanny S, Hadaczek P, Bengtsson
H, et al: Cytomegalovirus immediate-early proteins promote stemness
properties in glioblastoma. Cancer Res. 75:3065–3076. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fornara O, Bartek J Jr, Rahbar A, Odeberg
J, Khan Z, Peredo I, Hamerlik P, Bartek J, Stragliotto G, Landázuri
N, et al: Cytomegalovirus infection induces a stem cell phenotype
in human primary glioblastoma cells: Prognostic significance and
biological impact. Cell Death Differ. 23:261–269. 2016. View Article : Google Scholar :
|
|
29
|
Abdul Khalek FJ, Gallicano GI and Mishra
L: Colon cancer stem cells. Gastrointest Cancer Res. (Suppl 1):
S16–S23. 2010.
|
|
30
|
Papailiou J, Bramis KJ, Gazouli M and
Theodoropoulos G: Stem cells in colon cancer. A new era in cancer
theory begins. Int J Colorectal Dis. 26:1–11. 2011. View Article : Google Scholar
|
|
31
|
Du L, Wang H, He L, Zhang J, Ni B, Wang X,
Jin H, Cahuzac N, Mehrpour M, Lu Y, et al: CD44 is of functional
importance for colorectal cancer stem cells. Clin Cancer Res.
14:6751–6760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gorham H, Sugino T, Woodman AC and Tarin
D: Cellular distribution of CD44 gene transcripts in colorectal
carcinomas and in normal colonic mucosa. J Clin Pathol. 49:482–488.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu TC, Lee WA, Pizzorno MC, Au WC, Chan
YJ, Hruban RH, Hutchins GM and Hayward GS: Localization of the
human cytomegalovirus 2.7-kb major early beta-gene transcripts by
RNA in situ hybridization in permissive and nonpermissive
infections. Am J Pathol. 141:1247–1254. 1992.PubMed/NCBI
|
|
34
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mocarski E and Shenk T: Cytomegaloviruses.
Fields Virology. 5th edition. Knipe D and Howley P: Lippincott
Williams and Wilkins; Philadelphia, PA: pp. 2701–2772. 2007
|
|
37
|
Fortunato EA and Spector DH: Regulation of
human cytomegalovirus gene expression. Adv Virus Res. 54:61–128.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Landolfo S, Gariglio M, Gribaudo G and
Lembo D: The human cytomegalovirus. Pharmacol Ther. 98:269–297.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen HP, Jiang JK, Chan CH, Teo WH, Yang
CY, Chen YC, Chou TY, Lin CH and Chan YJ: Genetic polymorphisms of
the human cytomegalovirus UL144 gene in colorectal cancer and its
association with clinical outcome. J Gen Virol. 96:3613–3623. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Suman S, Kurisetty V, Das TP, Vadodkar A,
Ramos G, Lakshmanaswamy R and Damodaran C: Activation of AKT
signaling promotes epithelial-mesenchymal transition and tumor
growth in colorectal cancer cells. Mol Carcinog. 53(Suppl 1):
E151–E160. 2014. View Article : Google Scholar
|
|
42
|
Porta C, Paglino C and Mosca A: Targeting
PI3K/Akt/mTOR signaling in cancer. Front Oncol. 4:642014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mayer IA and Arteaga CL: The PI3K/AKT
pathway as a target for cancer treatment. Annu Rev Med. 67:11–28.
2016. View Article : Google Scholar
|
|
44
|
Cobbs C, Khan S, Matlaf L, McAllister S,
Zider A, Yount G, Rahlin K, Harkins L, Bezrookove V, Singer E, et
al: HCMV glycoprotein B is expressed in primary glioblastomas and
enhances growth and invasiveness via PDGFR-alpha activation.
Oncotarget. 5:1091–1100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yurochko AD, Kowalik TF, Huong SM and
Huang ES: Human cytomegalovirus upregulates NF-kappa B activity by
transactivating the NF-kappa B p105/p50 and p65 promoters. J Virol.
69:5391–5400. 1995.PubMed/NCBI
|
|
46
|
Castillo JP, Yurochko AD and Kowalik TF:
Role of human cytomegalovirus immediate-early proteins in cell
growth control. J Virol. 74:8028–8037. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Moorman NJ, Cristea IM, Terhune SS, Rout
MP, Chait BT and Shenk T: Human cytomegalovirus protein UL38
inhibits host cell stress responses by antagonizing the tuberous
sclerosis protein complex. Cell Host Microbe. 3:253–262. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Streblow DN, Soderberg-Naucler C, Vieira
J, Smith P, Wakabayashi E, Ruchti F, Mattison K, Altschuler Y and
Nelson JA: The human cytomegalovirus chemokine receptor US28
mediates vascular smooth muscle cell migration. Cell. 99:511–520.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Maussang D, Verzijl D, van Walsum M, Leurs
R, Holl J, Pleskoff O, Michel D, van Dongen GA and Smit MJ: Human
cytomegalovirus-encoded chemokine receptor US28 promotes
tumorigenesis. Proc Natl Acad Sci USA. 103:13068–13073. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Maussang D, Langemeijer E, Fitzsimons CP,
Stigter-van Walsum M, Dijkman R, Borg MK, Slinger E, Schreiber A,
Michel D, Tensen CP, et al: The human cytomegalovirus-encoded
chemokine receptor US28 promotes angiogenesis and tumor formation
via cyclooxygenase-2. Cancer Res. 69:2861–2869. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Slinger E, Maussang D, Schreiber A,
Siderius M, Rahbar A, Fraile-Ramos A, Lira SA, Söderberg-Nauclér C
and Smit MJ: HCMV-encoded chemokine receptor US28 mediates
proliferative signaling through the IL-6-STAT3 axis. Sci Signal.
3:ra582010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bongers G, Maussang D, Muniz LR, Noriega
VM, Fraile-Ramos A, Barker N, Marchesi F, Thirunarayanan N, Vischer
HF, Qin L, et al: The cytomegalovirus-encoded chemokine receptor
US28 promotes intestinal neoplasia in transgenic mice. J Clin
Invest. 120:3969–3978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cai ZZ, Xu JG, Zhou YH, Zheng JH, Lin KZ,
Zheng SZ, Ye MS, He Y, Liu CB and Xue ZX: Human
cytomegalovirus-encoded US28 may act as a tumor promoter in
colorectal cancer. World J Gastroenterol. 22:2789–2798. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ding Q, Stewart J Jr, Olman MA, Klobe MR
and Gladson CL: The pattern of enhancement of Src kinase activity
on platelet-derived growth factor stimulation of glioblastoma cells
is affected by the integrin engaged. J Biol Chem. 278:39882–39891.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang J, Tian X-J and Xing J: Signal
transduction pathways of EMT induced by TGF-β, SHH, and WNT and
their crosstalks. J Clin Med. 5:412016. View Article : Google Scholar
|
|
56
|
Derynck R, Muthusamy BP and Saeteurn KY:
Signaling pathway cooperation in TGF-β-induced
epithelial-mesenchymal transition. Curr Opin Cell Biol. 31:56–66.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Takebe N, Harris PJ, Warren RQ and Ivy SP:
Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog
pathways. Nat Rev Clin Oncol. 8:97–106. 2011. View Article : Google Scholar
|
|
58
|
Takebe N, Miele L, Harris PJ, Jeong W,
Bando H, Kahn M, Yang SX and Ivy SP: Targeting Notch, Hedgehog, and
Wnt pathways in cancer stem cells: Clinical update. Nat Rev Clin
Oncol. 12:445–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jin D, Fang Y, Li Z, Chen Z and Xiang J:
Epithelial-mesenchymal transition-associated microRNAs in
colorectal cancer and drug-targeted therapies (Review). Oncol Rep.
33:515–525. 2015. View Article : Google Scholar
|
|
61
|
Bienz M and Clevers H: Linking colorectal
cancer to Wnt signaling. Cell. 103:311–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
63
|
Novellas de Munt L, Antas P and Li VS:
Targeting Wnt signaling in colorectal cancer. A review in the
theme: Cell signaling: proteins, pathways and mechanisms. Am J
Physiol Cell Physiol. 309:C511–C521. 2015. View Article : Google Scholar
|
|
64
|
Basu S, Haase G and X Ben-Ze'ev A: Wnt
signaling in cancer stem cells and colon cancer metastasis.
F1000Res 5: F1000 Faculty Rev. 699:2016.
|
|
65
|
Brabletz T, Jung A, Reu S, Porzner M,
Hlubek F, Kunz-Schughart LA, Knuechel R and Kirchner T: Variable
beta-catenin expression in colorectal cancers indicates tumor
progression driven by the tumor environment. Proc Natl Acad Sci
USA. 98:10356–10361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Howard S, Deroo T, Fujita Y and Itasaki N:
A positive role of cadherin in Wnt/β-catenin signalling during
epithelial-mesenchymal transition. PLoS One. 6:e238992011.
View Article : Google Scholar
|
|
67
|
Ueno K, Hazama S, Mitomori S, Nishioka M,
Suehiro Y, Hirata H, Oka M, Imai K, Dahiya R and Hinoda Y:
Down-regulation of frizzled-7 expression decreases survival,
invasion and metastatic capabilities of colon cancer cells. Br J
Cancer. 101:1374–1381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Nishioka M, Ueno K, Hazama S, Okada T,
Sakai K, Suehiro Y, Okayama N, Hirata H, Oka M, Imai K, et al:
Possible involvement of Wnt11 in colorectal cancer progression. Mol
Carcinog. 52:207–217. 2013. View Article : Google Scholar
|