Introduction
Cancer remains the leading cause of death worldwide.
Pain resulting from the abnormal growth of malignant cells is the
most common and most feared symptom in cancer patients. Beyond
that, the metastatic carcinomas and typically anti-neoplastic
approaches involving chemotherapy, radiotherapy also cause
substantial pain that is required to be treated with analgesic
drugs (1). For these reasons,
analgesics and chemotherapeutic drugs are often used concurrently
in antitumor therapy. Thus, there is an urgent need to explore the
potential influence of analgesic agents on the antitumor efficacy
of antineoplastic drugs and the mechanism of any such effects.
Gap junctions are formed when two hemichannels, in
neighboring cells, align and then dock with each other to form a
full gap junction channel that spans the space between the two
closely apposed cells. Each hemichannel is composed of 6 connexin
(Cx) subunits. The six connexin subunits can be homogeneous or
heterogeneous. The formation of stable gap junctions allows for
direct communication between the cytoplasmic compartments of the
two neighbouring cells, as well as providing a conduit for
electrical signaling. The channels are large enough to allow for
small hydrophilic metabolites and messenger molecules to pass
between cells. In humans, 21 members of the Cx family have been
identified and are named by their molecular weights. Among them,
Cx43 is the most ubiquitously expressed and is critical for
regulating biological and pathological processes in a variety of
tissues (2,3).
Gap junction intercellular communication (GJIC)
sensitizes cancer cells to chemotherapeutic agents, radiation and
other antitumor therapies. Some metabolites or death signal
triggered in target cancer cells by those antineoplastic treatments
may transfer through gap junctions, thereby substantially
increasing their antitumor efficacy (4–6).
Such toxicity amplifying phenomenon mediated by GJIC is termed as
bystander effect. Specifically, gap junction composed of Cx43 has
been shown to increase the cytotoxicity induced by etoposide and
cisplatin in several cell lines (7,8).
Moreover, Cx43 participate in various essential cellular processes
including tumor cell proliferation, migration and chemotherapeutic
sensitivity independent of gap junction formation (9,10).
Gap junction dependent or independent effects thereby strongly
suggest that regulation of Cx43 or gap junctions by pharmacological
or biologic approaches would modulate the efficacy of antitumor
strategies.
Our research has previously proved that analgesics
tramadol and flurbiprofen inhibited gap junction channels in HeLa
cells expressing Cx32, and in this case, decreased cisplatin
cytotoxicity (11). Although Cx43
is ubiquitously expressed in majority of tissues, there is no
evidence of the effect of these analgesic drugs on Cx43 and its
constituent gap junctions. Whether Cx43 or Cx43-composed gap
junction channels will be affected by analgesics thereby altering
chemotherapeutic efficiency has not been investigated.
Temozolomide (TMZ) is used as a first-line
antineoplastic drug against Cx43 expressing glioblastoma (GBM)
(12). We investigated the
interplay between the analgesic tramadol, Cx43 and Cx43-composed
gap junction, and the cytotoxicity of TMZ in U87 glioblastoma
cells. In the present study, we report that long-term treatment of
tramadol attenuated TMZ cytotoxicity through the inhibition of
Cx43-composed gap junction.
Materials and methods
Materials
TMZ was purchased from Shanghai Selleck Chemicals
Co., Ltd. (Shanghai, China). Tramadol, 18-α-glycyrrhetinic acid
(18-α-GA) and retinoic acid (RA) were from Sigma-Aldrich (St.
Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), penicillin/streptomycin, calcein-acetoxymethyl
ester and CM-DiI were obtained from Invitrogen (Carlsbad, CA, USA).
All the antibodies and other reagents were from Sigma-Aldrich
unless otherwise indicated.
Cell lines and cell culture
U87 cells were obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA). Cells were maintained
in DMEM containing 10% FBS and 1% penicillin/streptomycin at 37°C
in an atmosphere of 5% CO2 in air.
Drug treatment and gap junction
inhibition
TMZ was reconstituted in dimethyl sulfoxide (DMSO)
at the concentration of 50 mM, and then aliquots were stored at
−20°C. Exposures to TMZ were performed for indicated times in the
dark with or without tramadol at various concentrations dissolved
in phosphate-buffered saline (PBS). Cells were incubated with the
gap junction inhibitor 18-α-GA (50 µM in DMSO) or
potentiator RA (20 µM in DMSO) for 4 h before exposure to
TMZ and during TMZ treatment.
Transfection
We used the stable cell lines expressing endogenous
shRNA to downregulate Cx43 expression as described (12,13).
The PRP.EX3d-MCS1>shCx43>PGK/puro plasmid expressing green
fluorescence protein (GFP), shRNA targeting Cx43 (target sequence:
5'-CAATTCTTCTTGCC GCAATTACTCGAGTAATTGCGGCAAGAAGAATTG-3') and the
PRP.EX3d-MCS1>negative control>PGK/puro plasmid were
purchased from Cyagen Biosciences, Inc. (Santa Clara, CA, USA).
Plasmids were separately transfected into U87 cells, and
transfected cells were selected on 0.3 mg/ml G418 for 30 days.
Before the application of stable transfection cell lines, western
blot analysis was used to examine the expression of Cx43 and a
'parachute' dye-coupling assay was performed to examine the dye
spread through GJIC.
'Parachute' dye-coupling assay
To evaluate gap junction function, 'parachute'
dye-coupling assay was performed as described by Goldberg et
al (14) and Koreen et
al (15). Donor and receiver
cells were grown to confluence. The donor cells were double-labeled
with 5 µM CM-DiI, a membrane-permeable dye that does not
spread to coupled cells, and with 5 µM calcein-AM, which is
converted intracellularly into the gap junction permeable dye
calcein. The donor cells were washed and unincorporated dye was
removed. Then, cells were trypsinized and seeded onto the receiver
cells at a 1:150 donor/receiver ratio. The donor cells attached to
the mono-layer of receiver cells and form gap junctions for 4 h at
37°C. Then examined with a fluorescence microscope. For each
experimental condition, the average number of receiver cells
containing dye per donor cell was determined and normalized to that
of control cultures. To detect functional gap junctions in
GFP-expressing cells, we reduced the exposure time to a suitable
value to exclude the interference of GFP. The donor and receiver
cells were exposed to tramadol during indicated periods in which
the donor cells were plated onto the receive cell monolayer.
Western blotting
Whole-cell lysates for western blotting were
prepared by cell incubation in lysis buffer with
protease/phosphatase inhibitors at 4°C. Then, cell lysates (10
µg) were separated by SDS/PAGE in 13% Tris-glycine mini-gels
and transferred to a PVDF membrane. Membranes were blocked with 5%
skim milk-TBST for 30 min. Individual membranes were probed with
specifical primary antibody against Cx43 diluted to 1:4,000, p-Cx43
and caspase-3 diluted to 1:2,000, β-tubulin diluted to 1:10,000,
ERK, JNK and p38 diluted to 1:1,000 (Cell Signaling Technology,
Danvers, MA, USA). After washing with TBST, membranes were
incubated with horseradish peroxidase (HRP)-conjugated secondary
antibodies dissoved in 5% milk-TBST for 30 min at 37°C. The
immunoreactive bands were visualized by ECL plus western blotting
detection systerm (GE Healthcare, Piscataway, NJ, USA). All western
blot exposures were in the linear range of detection, and the
intensities of the resulting bands were quantified by Quantity One
software on GS-800 densitometer (Bio-Rad Laboratories, Hercules,
CA, USA).
CCK-8 assay
To test the cytotoxicity of TMZ, we used the Cell
Counting kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto,
Japan) which utilizes a highly water-soluble tetrazolium salt for
the cell proliferation assay. U87 cells were seeded at a density of
3×103/well in 96-well plates. After treatment with TMZ
with or without tramadol for indicated time periods, CCK-8 solution
and DMEM at a ratio of 1:9 was added to each well to a total volume
of 100 µl. After 2-h cell incubation, an ultraviolet
spectrophotometer was used to determine the optical density at 450
nm (IX51; Beckman Coulter, Brea, CA, USA). Samples for each group
were analyzed in triplicate. Cell survival rates with various
treatments were calculated using the following formula: Survival
rate = Optical density drug/Optical density control.
Standard colony-forming assay
Cell survival was assayed by a standard
colony-forming assay, adapted for use at high and low cell density,
corresponding to conditions in which junctional channel formation
was permitted or not, respectively (5). In the high density condition, cells
were seeded at 30,000 cells/cm2 so that cultures were
70%–100% confluent during TMZ exposure. Cells were treated with TMZ
for 1 week, then washed with PBS, harvested by trypsinization,
counted, diluted and seeded into 6-well dishes (500
cells/cm2). Colony formation was assessed 5–7 days later
by fixation and staining with crystal violet. Colonies containing
50 or more cells were scored. In the low density condition, cells
were directly seeded at the density of 500 cells/cm2
into 6-well plates and treated with TMZ after attachment. They were
rinsed and assessed for colony formation as above. Colony formation
was normalized to the colony forming efficiency of non-TMZ treated
cells. There was no significant difference in plating efficiency
between the low- and high-density cultures in the untreated samples
(data not shown). Tramadol was added to the culture medium during
TMZ treatment.
Annexin V/PI double-staining assay
Apoptosis was analyzed using flow cytometry with
Annexin V/PI double-staining to detect membrane events. U87 cells
(3.0×104/ml) were seeded into 6-well plates and treated
with TMZ with or without 30 µM tramadol for 1 week. Both
floated and attached cells were collected, washed with ice-cold PBS
twice, and then incubated at room temperature in the presence of
media-binding buffer containing Annexin V (2.5 µg/ml) and PI
(2 ng/ml) for 15 min in the dark. Apoptosis was quantified by flow
cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA) immediately
and analyzed by the FlowJo software. The cytogram of the four
quadrants in the figures was used to distinguish normal (Annexin
V−/PI−), early apoptotic (Annexin
V+/PI−), late apoptotic (Annexin
V+/PI+) and necrotic (Annexin
V−/PI+) cells. Early apoptotic cells appeared
in the lower right (Annexin V+/PI−)
quadrant.
Statistical analysis
Statistical package for social sciences (SPSS)
version 13.0 was used and differences with P<0.05 are considered
statistically significant. Two-group comparisons were analyzed
using an unpaired Student's t-test. A one-way ANOVA with Tukey's
post hoc test was used to analyze the multiple group comparisons.
All analyses were plotted using SigmaPlot (Jandel Scientific, Inc.,
San Rafael, CA, USA).
Results
The effect of Cx43 and Cx43-composed gap
junctions on TMZ-induced cytotoxicity in U87 cells
To evaluate TMZ cytotoxicity, U87 cells were treated
with TMZ for indicated time periods and then the cell viability was
assessed using CCK-8 assays (Fig.
1). The cell optical density after TMZ treatment relative to
the optical density of control is shown as cell survival rates.
Fig. 1A illustrated that TMZ
treatment (from 0 to 1,000 µM) for 48, 72 h and 1 week
significantly reduced U87 cell survival in a
concentration-dependent manner. The survival rates of U87 cells
after 48 h TMZ treatment was similar with that of 72 h. Though cell
surival of U87 cells after 0–800 µM TMZ exposure for 1 week
was much lower than that in both 48- and 72-h treatment groups,
similar inhibition of the cell survival was observed after 1,000
µM TMZ treatment for all indicated times.
To investigate the effect of intercellular
communication by gap junction on cytotoxicity, cells exposed to TMZ
were concurrently treated with the gap junction inhibitor 18-α-GA
or a gap junction enhancer RA. Western blot analysis showed that
neither 20 µM RA nor 50 µM 18-α-GA affected Cx43
expression (Fig. 2A). Data from a
parachute assay is shown in Fig.
2B: 18-α-GA inhibited dye transfer between U87 cells while RA
enhanced dye transfer. In this case, dye transfer is used as a
surrogate of cell coupling via gap junctions. Additionally, stable
cell lines expressing endogenous Cx43-shRNA were used to evaluate
the effect of Cx43 on TMZ cytotoxicity. As shown in Fig. 2C and D, both Cx43 expression and
gap junction communication were drastically downregulated in
Cx43-shRNA transfected U87 (U87GFP-Cx43shRNA) cells.
As shown in Fig.
1B, neither 18-a-GA nor RA had any effect by themselves on U87
cell survival. In contrast, TMZ by itself significantly reduced
cell survival in U87 cells (800 µM TMZ for 72 h). RA (20
µM) significantly enhanced TMZ-induced cytotoxicity,
resulting in a lower survival of U87 cells when the drugs were
applied concurrently. Conversely, TMZ-induced cytotoxicity was
significantly suppressed by 18-α-GA (50 µM), resulting in
higher survival when U87 cells were treated with both drugs. These
results suggest an important role for gap junctions in determining
U87 cell survival following exposure to TMZ. In Fig. 1C, U87GFP-Cx43shRNA cells
were much more sensitive to TMZ, yielding a reduced survival
fraction when treated with 400 or 800 µM TMZ as compared
with U87GFP-NC cells. This indicated Cx43 may act as a
single protein to decrease the sensitivity of U87 cells to TMZ.
Tramadol attenuates TMZ-induced
cytotoxicity in U87 cells in the presence of Cx43, but not in its
absence
Fig. 3 indicates
the effect of tramadol on cell response to TMZ using CCK-8 assay.
Treatment with different doses of tramadol alone for as long as 1
week did not alter cell survival in U87 cells (Fig. 3A). Tramadol alleviated TMZ
cytotoxicity in a dose-dependent manner, resulting in substantially
higher U87 cell survival rates when U87 cells were treated with
both drugs for 48, 72 h and 1 week (Fig. 3B). However, tramadol treatment for
72 h had no effect on TMZ cytotoxicity in
U87GFP-Cx43shRNA cells. No alteration in TMZ
cytotoxicity was observed even with prolonged tramadol treatment
for 1 week (Fig. 3C). Our results
here strongly suggest that the reduction of TMZ cytotoxicity by
tramadol was dependent on Cx43.
Tramadol attenuates TMZ-induced apoptosis
in U87 cells in the presence of Cx43, but not in its absence
Flow cytometry with Annexin V and PI double labeling
was used to assess the effect of tramadol on TMZ-induced early
stage apoptosis in U87 cells. TMZ treatment for 1 week induced
early apoptosis in U87 cells. In the absence of any drug, the
apoptosis rate in U87 cells was ~5% and was not changed following
exposure to tramadol. In contrast, tramadol significantly reduced
TMZ-induced apoptosis by almost 50%. The apoptosis rate was reduced
from 23% when cells were treated with TMZ alone to 12% when
simultaneously treated with tramadol and TMZ (Fig. 4A and B).
Caspase cascade systems always play an important
role in the induction, transduction and amplification of
intracellular apoptotic signals. There are two classic caspase
pathways including the participation of mitochondria or the
interaction of death receptors with its ligands. Both of them may
finally lead to caspase-3 activation, which is considered to be the
most important of the executioner caspases. Therefore, cleaved
caspase-3 expression (active caspase-3) was determinated after
treatment with TMZ with or without tramadol. In
U87GFP-NC and U87GFP-Cx43shRNA cells,
tramadol was unable to induce cleaved caspase-3 expression. TMZ
induced apparent expression of cleaved caspase-3. In
U87GFP-NC cells, tramadol significantly reduced
caspase-3 expression induced by TMZ. On the contrary, tramadol
showed no effect on TMZ-induced caspase-3 expression in
U87GFP-Cx43shRNA cells (Fig. 4C and D). These findings indicated
tramadol suppressed TMZ-induced apoptosis in the presence of
Cx43.
Effect of tramadol on TMZ-induced
cytotoxicity is dependent on gap junction composed of Cx43
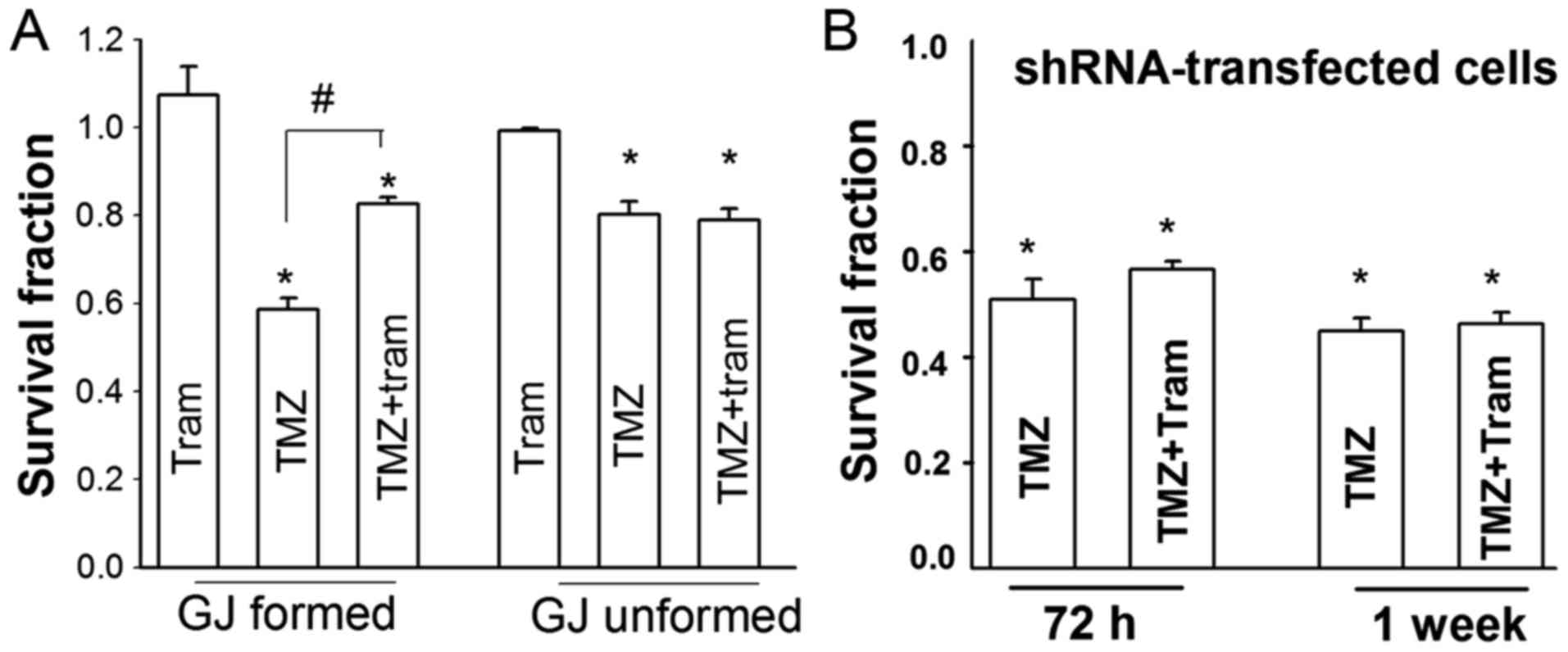
To investigate whether Cx43-dependence influences
tramadol on TMZ toxicity is associated with gap junctions or Cx43
protein itself, cell proliferation after TMZ treatment with or
without tramadol were assessed by standard colony formation assay
under conditions where gap junction formation was not possible (low
density; 500 cells/cm2; cells not in direct contact with
each other) or possible (at high density 30,000
cells/cm2) (Fig. 5A). A
total of 800 µM TMZ treatment for 1 week reduced the
clonogenic survival of U87 cells at both gap junction-formed and
unformed cells. However, the clonogenic survival of gap
junction-formed cells exposed to TMZ was substantially less than
gap junction unformed cells. Tramadol reduced the suppression of
clonogenic survival induced by TMZ in gap junction-formed cells,
while tramadol could not affect TMZ-induced suvival suppression in
gap junction unformed cells. Additionally, neither tramadol
treatment for 72 h nor 1 week exert effect on TMZ-induced
proliferation inhibition in shRNA-transfected cells (Fig. 5B). Thus, our results here suggest
the inhibition of tramadol on TMZ cytotoxicity is dependent on gap
junctions composed of Cx43.
Tramadol inhibited dye coupling through
gap junction composed of Cx43 in U87 cells
We then assessed the effect of tramadol on gap
junction function by parachute assay. In Fig. 6, tramadol inhibited the gap
junction mediated dye spread at 4 h and continued to 72 h. Fig. 6A shows micrographs to illustrate
the spreading of dye between cells. Fig. 6B plots the inhibition of dye
coupling as a function of tramadol concentration. The inhibition of
dye coupling was gradually augmented with increasing concentration
of tramadol from 7.5 to 30 µM for 4 h and maintained to be
the highest until 90 µM. The dye spread after 30 µM
tramadol treatment for 48 and 72 h were decreased compared with
that for 4 h, and it was maintained at 62 and 60% of control,
respectively (Fig. 6C).
Effects of tramadol on Cx43 and p-Cx43
expression in U87 cells
Not only the expression of Cxs affect gap junction
formation and its function, but Cx43 phosphorylation also plays an
important role in regulation of gap junction functions through
direct action or by indirectly modulating Cx43 disassembly and
internalization from the cell surface. We then determined whether
the inhibitory effect of tramadol on gap junctional communication
was the result of regulation on Cx43 or by phosphorylation
(Fig. 7). Western blotting
demonstrated that 30 µM tramadol treatment slightly reduced
total Cx43 expression for 4 h, but not for 72 h. Phosphorylated
Cx43 expression was altered by tramadol exposure for only 72 h.
Moreover, both phosphorylated JNK and ERK were significantly
reduced after tramadol exposure for 72 h but not 4 h.
Phosphorylated p38 expression was unchanged by tramadol treatment
for 4 and 72 h. These results indicated that decreased gap
junctional intercellular communication between U87 cells by
long-term tramadol treatment may likely relate to the reduction of
Cx43 phosphorylation via ERK and JNK.
Discussion
The data outlined in the present study suggest that
TMZ toxicity is strongly dependent on the presence of functional
gap junctions in U87 cells. In addition, Cx43 by itself, in a gap
junction independent manner, appears to also modify TMZ toxicity.
Moreover, we observed that long-term treatment with tramadol, at an
appropriate analgesic concentration, reduced TMZ toxicity through
depressing Cx43-mediated gap junctional intercellular communication
that was probably associated with downregulation of Cx43
phosphorylation via MAPKs. The present study highlights the
importance of making a rational choice for the analgesic that would
be routinely provided concurrently with antineoplastic agents.
In the central nervous system, GBM is the most
common malignancy and accounts for more than 45% of all malignant
brain tumors. The typical therapies for GBM involves surgery,
chemotherapy, radiotherapy or combination therapy. TMZ is a
front-line chemo-alkylating agent used to treat glioblastoma
(16). TMZ causes DNA damage
through the DNA alkylation, which induces mismatches in the DNA
repair pathway and finally leads to cell death. The concentrations
of TMZ we used are similar to those employed in other clinically
relevant studies (17). Using this
concentration paradigm make the GJIC-mechanism observed in this
study. Nevertheless, a much higher concentration than that may
result in extensive cell death and no GJIC-mediated effects will be
identified. Of course, at higher concentration TMZ will also have
significantly larger effects on non-cancerous cells and will
thereby produce even greater side-effects.
The present study establishes the role for
functional gap junctions in TMZ toxicity in U87 cells. Other
studies have previously established roles for gap junctions
composed of Cx43 on the cytotoxicity induced by radiotherapy and
different antineoplastic drugs including cisplatin, etoposide, 5-FU
and others (8,18–20).
Glioblastoma is a heterogeneous disease in which Cx43 expression is
still present. It has been reported that thymidine
kinase/ganciclovir suicide gene therapy targeting glioblastoma was
strongly inhibited by gap junction inhibitors (12). The bystander effect of gap
junctions composed of Cx43 was shown to enhance the antitumor
effect of miR-124-3p in glioblastoma cell lines and xenograft
models (13). A different study
showed an enhancing effect of gap junctions on cisplatin
cytotoxicity in U87 cells (21).
Our results showing the involvement of functional Cx43-containing
gap junctions in TMZ cytotoxicity, provide key insight into the
importance of the bystander effect in glioblastoma cells treated
with TMZ. Further research will be needed to determine whether this
bystander effect is observed ubiquitously for antineoplastic
therapies in glioblastoma or not.
We also found that Cx43 expression was inversely
correlated to TMZ sensitivity in a gap junction-dependent as well
as gap junction independent manner. If the entire Cx43 effect on
TMZ toxicity was mediated by gap junctions, then inhibiting gap
junctions with a drug, 18-α-GA, should have produced a change in
chemotherapeutic sensitivity equivalent to disrupting gap junctions
by downregulating Cx43 with a shRNA construct. It did not, which
suggests that there is an important component of TMZ toxicity that
relates to a completely different function of Cx43. This finding is
consistent with the results of others (22,23).
The carboxyl tail of Cx43 can be post-translationally modified and
contains the largest part number of potential binding sites for
other proteins. These sites are thought to be the basis for Cx43
forming signaling complexes through direct or indirect interaction
with other proteins known to participate in chemotherapeutic
resistance, potentially leading to the modulation of the
sensitivity of cells to antineoplastic agents. Alternatively, and
more related to our study, 'toxic' or 'death' signals triggered by
antitumor agents are supposed to pass through gap junctions and
enhance cell death and apoptosis in adjacent cells (4,5,13).
Enhanced gap junction function would therefore increase the
cytotoxicity induced by chemotherapeutic. Our results show that the
gap junction mediated effect is quite different from the gap
junction independent effect.
Cx43 appears to have the ability to bi-directionally
alter TMZ sensitivity. Alteration in gap junction function without
affecting protein expression decreases TMZ toxicity, while
decreasing all Cx43 expression increases toxicity. Such Cx-mediated
effects similarly exist in other cell settings and in different
stages of tumorigenesis (9,24,25).
However, on confluent U87 cells where Cx43 was expressed normally
and functional gap junctions between neighboring cells were formed,
neither 18-α-GA and RA nor prolonged treatment with tramadol
induced any change in Cx43 expression, which excludes changes in
Cx43 protein from producing gap-junction independent alterations in
TMZ sensitivity. However, more work needs to be done to better
elucidate the interplay between the two pathways.
Because of its limited opioid induced side-effects
and because it blocks serotonin and norepinephrine re-uptake in a
manner similar to antidepressant medications, tramadol is widely
used in treatment of moderate to moderately severe pain including
post-operative pain, chronic neuropathic pain and cancer pain
(26). We showed prolonged
tramadol treatment by itself produced no toxicity, but when
co-administered with TMZ attenuated apoptosis and toxicity only in
the presence of Cx43. More importantly, tramadol attenuates TMZ
cyto-toxicity only under high density condition where gap junction
formation is possible. Given that tramadol treatment reduced gap
junction function, our results strongly suggest that the underlying
mechanism associated with the inhibitory effect on TMZ cytotoxicity
is from the suppression of gap junctions. The concentration of
tramadol we used is higher than the reported plasma concentration
(2467±540 ng/ml) in patients given a single dose of tramadol
(27). Nonetheless, the dosage
used in this study are similar and even lower relative to those
given in other in vitro and in vivo experiments
(28–30). Much higher doses of tramadol have
been administered clinically over months in cancer patients with an
expected linear increase of the serum concentration (31,32).
Furthermore, a wide range of actual serum concentrations of
tramadol is expected due to the large differences in liver
metabolic enzyme activity.
Tramadol which may rapidly penetrate the blood-brain
barrier (30), exerts
antinociceptive action as a weak agonist of the μ opioid receptor
and inhibits synaptic norepinephrine and serotonin re-uptake. There
is no evidence that noradrenaline and 5-HT transporters exist in
glioblastoma cells and the relationship between the pathway of
noradrenaline and 5-HT and gap junction function have not been
documented. Even though μ opioid receptor was expressed in some
glioblastoma cells (33), tramodol
is recognized to exhibit relatively weak activity on opioid
receptor. No direct evidence has verified its existance in U87
cells. Phosphorylation participates in the regulation of gap
junction function by direct action on channel activity or by
triggering Cx disassembly, degradation and internalization. Cx43 is
easily phosphorylated by a multiplicity of phosphorylation protein
kinases such as MAPKs (34). In
the present study, tramadol treatment for 4 h decreased Cx43
expression without changing phosphorylated Cx43, indicating the
suppression effect of gap junction function by short-term treatment
of tramadol was due to the reduced Cx43 level. Tramadol treatment
for 72 h showed no effect on Cx43 expression, but reduced Cx43
phosphorylation accompanied with the downregulation of p-JNK and
p-ERK expression. Similar effect of tramadol on pERK expression was
also shown in breast cancer cells (29). Since other studies have shown that
the phosphorylation of Cx43 may increase gap junctional
intercellular communication and the suppression of Cx43
phosphorylation induced the reduction of gap junction functions in
some cell lines (35,36). We suppose that the deceasing GJIC
induced by prolonged tramadol exposure is likely associated with
the downregulation of Cx43 phosphorylation through the ERK and JNK
molecular pathway.
Gap junctions, formed by Cxs, have important roles
in maintenance of tissue homeostasis and regulation of cell growth
and differentiation (3,37). Reduced expression and disruption of
gap junction function is usually correlated with tumorigenic
phenotypes in various cancer cells. However, studies have shown
that increased levels of Cxs including Cx43 and constituent gap
junctions are involved in metastasis, invasiveness and
extravasation (38,39). Primary tumors that initially
exhibit impaired intercellular communication via gap junction
exhibit functional gap junctions at the metastatic stage (40). In addition to glioblastoma cells,
suppression of gap junction function by tramadol here is a clue to
its possible therapeutic effect on advanced tumors expressing Cx43.
Since increased Cx43 and GJIC in spinal cord astrocytes are closely
associated with neuropathic pain (41), our finding also suggests a probable
Cx43-related mechanism for the anti-nociception actions of tramadol
in the spinal cord.
The present study reveals that a previously unknown
consequence of long-term treatment with tramadol is to reduce Cx43
containing gap junction function thereby counteracting TMZ-induced
toxicity in glioblastoma cells. Our results indicate a profitable
strategy whereby augmenting gap junction function could increase
the sensitivity of TMZ in glioblastoma and perhaps other
TMZ-resistant cancers. Moreover, this study has clinical
implication in optimizing the selection of analgesic for
glioblastoma patients concurrently receiving chemotherapy agents.
That would be for example, where possible, analgesics with no
effect on gap junctions other than tramadol might be preferable to
use to control cancer pain in glioblastoma patients. Otherwise, if
tramadol should be used, it may be advisable to increase the dose
of antitumor drugs to preserve their effectiveness. Finally, it is
important to investigate the possible interplay between analgesics
with different mechanism of anti-nociceptive action, various gap
junctions and chemotherapeutic agents in specific cancers to
develop an effective combination strategy in antitumor
treatment.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81401017), the Pearl River
S&T Nova Program of Guangzhou (no. 201610010060) and the
Foundation of Guangdong Traditional Chinese Medicine Bureau
(2015KT1741). Many thanks to Dr Zheng Xie and Dr Aaron Fox in the
Department of Anesthesia and Critical Care in University of Chicago
for their help on language.
Glossary
Abbreviations
Abbreviations:
|
GJIC
|
gap junctional intercellular
communication
|
|
TMZ
|
temozolomide
|
|
18-α-GA
|
18-α-glycyrrhetinic acid
|
|
RA
|
retinoic acid
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
GFP
|
green fluorescence protein
|
References
|
1
|
Leppert W, Zajaczkowska R, Wordliczek J,
Dobrogowski J, Woron J and Krzakowski M: Pathophysiology and
clinical characteristics of pain in most common locations in cancer
patients. J Physiol Pharmacol. 67:787–799. 2016.
|
|
2
|
Grek CL, Rhett JM, Bruce JS, Ghatnekar GS
and Yeh ES: Connexin 43, breast cancer tumor suppressor: Missed
connections? Cancer Lett. 374:117–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Willebrords J, Crespo Yanguas S, Maes M,
Decrock E, Wang N, Leybaert L, da Silva TC, Veloso Alves Pereira I,
Jaeschke H, Cogliati B, et al: Structure, regulation and function
of gap junctions in liver. Cell Commun Adhes. 22:29–37. 2015.
View Article : Google Scholar
|
|
4
|
Hong X, Wang Q, Yang Y, Zheng S, Tong X,
Zhang S, Tao L and Harris AL: Gap junctions propagate opposite
effects in normal and tumor testicular cells in response to
cisplatin. Cancer Lett. 317:165–171. 2012. View Article : Google Scholar
|
|
5
|
Jensen R and Glazer PM:
Cell-interdependent cisplatin killing by Ku/DNA-dependent protein
kinase signaling transduced through gap junctions. Proc Natl Acad
Sci USA. 101:6134–6139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao J, Zhang G, Li B, Wu Y, Liu X, Tan Y
and Du B: Dioscin augments HSV-tk-mediated suicide gene therapy for
melanoma by promoting connexin-based intercellular communication.
Oncotarget. 8:798–807. 2017.
|
|
7
|
Kim YJ, Kim J, Tian C, Lim HJ, Kim YS,
Chung JH and Choung YH: Prevention of cisplatin-induced ototoxicity
by the inhibition of gap junctional intercellular communication in
auditory cells. Cell Mol Life Sci. 71:3859–3871. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang L, Fu Y, Peng J, Wu D, Yu M, Xu C,
Wang Q and Tao L: Simvastatin-induced up-regulation of gap
junctions composed of connexin 43 sensitize Leydig tumor cells to
etoposide: An involvement of PKC pathway. Toxicology. 312:149–157.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sin WC, Aftab Q, Bechberger JF, Leung JH,
Chen H and Naus CC: Astrocytes promote glioma invasion via the gap
junction protein connexin43. Oncogene. 35:1504–1516. 2016.
View Article : Google Scholar
|
|
10
|
Plante I, Stewart MK, Barr K, Allan AL and
Laird DW: Cx43 suppresses mammary tumor metastasis to the lung in a
Cx43 mutant mouse model of human disease. Oncogene. 30:1681–1692.
2011. View Article : Google Scholar
|
|
11
|
He B, Tong X, Wang L, Wang Q, Ye H, Liu B,
Hong X, Tao L and Harris AL: Tramadol and flurbiprofen depress the
cytotoxicity of cisplatin via their effects on gap junctions. Clin
Cancer Res. 15:5803–5810. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suzhi Z, Liang T, Yuexia P, Lucy L,
Xiaoting H, Yuan Z and Qin W: Gap junctions enhance the
antiproliferative effect of MicroRNA-124-3p in glioblastoma cells.
J Cell Physiol. 230:2476–2488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cottin S, Gould PV, Cantin L and Caruso M:
Gap junctions in human glioblastomas: Implications for suicide gene
therapy. Cancer Gene Ther. 18:674–681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goldberg GS, Bechberger JF and Naus CC: A
pre-loading method of evaluating gap junctional communication by
fluorescent dye transfer. Biotechniques. 18:490–497.
1995.PubMed/NCBI
|
|
15
|
Koreen IV, Elsayed WA, Liu YJ and Harris
AL: Tetracycline-regulated expression enables purification and
functional analysis of recombinant connexin channels from mammalian
cells. Biochem J. 383:111–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Su J, Cai M, Li W, Hou B, He H, Ling C,
Huang T, Liu H and Guo Y: Molecularly targeted drugs plus
radiotherapy and temozolomide treatment for newly diagnosed
glioblastoma: A meta-analysis and systematic review. Oncol Res.
24:117–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng L, Ng L, Ozawa T and Stella N:
Quantitative analyses of synergistic responses between cannabidiol
and DNA-damaging agents on the proliferation and viability of
glioblastoma and neural progenitor cells in culture. J Pharmacol
Exp Ther. 360:215–224. 2017. View Article : Google Scholar
|
|
18
|
Autsavapromporn N, Suzuki M, Funayama T,
Usami N, Plante I, Yokota Y, Mutou Y, Ikeda H, Kobayashi K,
Kobayashi Y, et al: Gap junction communication and the propagation
of bystander effects induced by microbeam irradiation in human
fibroblast cultures: The impact of radiation quality. Radiat Res.
180:367–375. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lawrence TS, Rehemtulla A, Ng EY, Wilson
M, Trosko JE and Stetson PL: Preferential cytotoxicity of cells
transduced with cytosine deaminase compared to bystander cells
after treatment with 5-flucytosine. Cancer Res. 58:2588–2593.
1998.PubMed/NCBI
|
|
20
|
Zhang Y, Tao L, Fan L, Peng Y, Yang K,
Zhao Y, Song Q and Wang Q: Different gap junction-propagated
effects on cisplatin transfer result in opposite responses to
cisplatin in normal cells versus tumor cells. Sci Rep. 5:125632015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Wang X, Wang Q, Ge H and Tao L:
Propofol depresses cisplatin cytotoxicity via the inhibition of gap
junctions. Mol Med Rep. 13:4715–4720. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murphy SF, Varghese RT, Lamouille S, Guo
S, Pridham KJ, Kanabur P, Osimani AM, Sharma S, Jourdan J, Rodgers
CM, et al: Connexin 43 inhibition sensitizes chemoresistant
glioblastoma cells to temozolomide. Cancer Res. 76:139–149. 2016.
View Article : Google Scholar :
|
|
23
|
Gielen PR, Aftab Q, Ma N, Chen VC, Hong X,
Lozinsky S, Naus CC and Sin WC: Connexin43 confers Temozolomide
resistance in human glioma cells by modulating the mitochondrial
apoptosis pathway. Neuropharmacology. 75:539–548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naiki-Ito A, Asamoto M, Naiki T, Ogawa K,
Takahashi S, Sato S and Shirai T: Gap junction dysfunction reduces
acetaminophen hepatotoxicity with impact on apoptotic signaling and
connexin 43 protein induction in rat. Toxicol Pathol. 38:280–286.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kameritsch P, Pogoda K and Pohl U:
Channel-independent influence of connexin 43 on cell migration.
Biochim Biophys Acta. 1818:1993–2001. 2012. View Article : Google Scholar
|
|
26
|
Grond S and Sablotzki A: Clinical
pharmacology of tramadol. Clin Pharmacokinet. 43:879–923. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Edmondson MA, Duran SH, Boothe DM, Stewart
AJ and Ravis WR: Pharmacokinetics of tramadol and its major
metabolites in alpacas following intravenous and oral
administration. J Vet Pharmacol Ther. 35:389–396. 2012. View Article : Google Scholar
|
|
28
|
Li C, Chen SQ, Chen BX, Huang WQ and Liu
KX: The antinociceptive effect of intrathecal tramadol in rats: The
role of alpha 2-adrenoceptors in the spinal cord. J Anesth.
26:230–235. 2012. View Article : Google Scholar
|
|
29
|
Xia M, Tong JH, Zhou ZQ, Duan ML, Xu JG,
Zeng HJ and Wang SH: Tramadol inhibits proliferation, migration and
invasion via α2-adrenoceptor signaling in breast cancer cells. Eur
Rev Med Pharmacol Sci. 20:157–165. 2016.
|
|
30
|
Sheikholeslami B, Gholami M, Lavasani H
and Rouini M: Evaluation of the route dependency of the
pharmacokinetics and neuro-pharmacokinetics of tramadol and its
main metabolites in rats. Eur J Pharm Sci. 92:55–63. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pergolizzi JV Jr, Taylor R Jr and Raffa
RB: Extended-release formulations of tramadol in the treatment of
chronic pain. Expert Opin Pharmacother. 12:1757–1768. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leppert W and Mikolajczak P: Analgesic
effects and assays of controlled-release tramadol and
o-desmethyltramadol in cancer patients with pain. Curr Pharm
Biotechnol. 12:306–312. 2011. View Article : Google Scholar
|
|
33
|
Pan EC, Bohn LM, Belcheva MM, Thomas GE,
Manepalli AN, Mamone JY, Johnson FE and Coscia CJ: Kappa-opioid
receptor binding varies inversely with tumor grade in human
gliomas. Cancer. 83:2561–2566. 1998. View Article : Google Scholar
|
|
34
|
Solan JL and Lampe PD: Connexin43
phosphorylation: Structural changes and biological effects. Biochem
J. 419:261–272. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao Y, Yu L, Xu S, Qiu F, Fan Y and Fu G:
Down-regulation of connexin43 gap junction by serum deprivation in
human endothelial cells was improved by (-)-epigallocatechin
gallate via ERK MAP kinase pathway. Biochem Biophys Res Commun.
404:217–222. 2011. View Article : Google Scholar
|
|
36
|
TenBroek EM, Lampe PD, Solan JL, Reynhout
JK and Johnson RG: Ser364 of connexin43 and the upregulation of gap
junction assembly by cAMP. J Cell Biol. 155:1307–1318. 2001.
View Article : Google Scholar
|
|
37
|
Mathews J and Levin M: Gap junctional
signaling in pattern regulation: Physiological network connectivity
instructs growth and form. Dev Neurobiol. 77:643–673. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brockmeyer P, Jung K, Perske C,
Schliephake H and Hemmerlein B: Membrane connexin 43 acts as an
independent prognostic marker in oral squamous cell carcinoma. Int
J Oncol. 45:273–281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang A, Hitomi M, Bar-Shain N, Dalimov Z,
Ellis L, Velpula KK, Fraizer GC, Gourdie RG and Lathia JD: Connexin
43 expression is associated with increased malignancy in prostate
cancer cell lines and functions to promote migration. Oncotarget.
6:11640–11651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Czyz J: The stage-specific function of gap
junctions during tumourigenesis. Cell Mol Biol Lett. 13:92–102.
2008. View Article : Google Scholar
|
|
41
|
Chen MJ, Kress B, Han X, Moll K, Peng W,
Ji RR and Nedergaard M: Astrocytic CX43 hemichannels and gap
junctions play a crucial role in development of chronic neuropathic
pain following spinal cord injury. Glia. 60:1660–1670. 2012.
View Article : Google Scholar : PubMed/NCBI
|