Introduction
Primary effusion lymphoma (PEL) is a rare subtype of
large B-cell lymphoma and strongly linked with Kaposi's
sarcoma-associated herpesvirus (KSHV) infection (1,2). PEL
is also termed body cavity-based lymphoma, and is classified as a
type of non-Hodgkin's B-cell lymphoma affecting with
immunocompromised patients, such as AIDS patients or those who have
undergone organ transplantation (1,2). PEL
is usually present as a lymphomatous effusion in body cavities and
is treated with CHOP therapy either alone or in combination with
rituximab. However, PEL is resistant to these chemotherapy regimens
(3), and is associated with a very
poor prognosis (4–6). Currently, there is no effective
treatment for PEL. KSHV, also known as human herpesvirus 8 (HHV8),
is an oncogenic DNA virus and is classified in the γ-herpesvirus
subfamily. As with other human herpesviruses, KSHV exists in either
a latent or lytic infection state, which can persist long-term in
the host after primary infection (7). During latency, the KSHV genome
circularizes and forms a double-stranded episomal DNA molecule in
infected cells such as PEL cells. KSHV expresses only a limited set
of genes, including latency-associated nuclear antigen (LANA),
v-FLIP, v-Cyclin and microRNAs, all of which strongly contribute to
establishing latent infection (7,8).
These viral molecules constitutively and/or transiently activate
signaling pathways, including nuclear factor-κB (NF-κB), Akt, Wnt
and extracellular signal-regulated kinase (ERK), which are
essential for the malignant phenotype and cell survival of PEL
(8–10). After re-activating the lytic
infection state, most viral genes are expressed, and these viral
gene products facilitate viral replication and the production of
mature virions (11). We
previously demonstrated that diallyl trisulfide, pyrrolidinium
C60 fullerene, or sangivamycin induce the apoptosis of
PEL cells via the inhibition of NF-κB (12), Akt (13), or ERK signaling (14), respectively. In addition, the
unfolded protein response (UPR) pathway is downregulated in PEL
cells; namely, IRE1α and PERK are downregulated under normal
culture conditions (15).
Therefore, endoplasmic reticulum (ER)-stress inducers, including
methylseleninic acid, sodium selenite and thapsigargin cause severe
ER stress and subsequent apoptosis through pro-apoptotic UPR
activation in PEL cells (15,16).
Arctigenin, a representative dibenzylbutyrolactone
lignin, is an extract from the burdock plant, Arctium lappa,
whose seed has traditionally been used in Japanese Kampo medicine
for detoxification and inflammation, including in mastitis.
Arctigenin has been reported to exhibit many biological functions,
such as anticancer, anti-inflammatory (17), antiviral (18,19),
immunomodulatory, antioxidant (20), neuroprotective (20) and antidiabetic (21) activities, and has been shown to
modulate cell signaling pathways, such as signal transducer and
activator of transcription 3 (STAT3) (22), Akt (23,24),
NF-κB (24), p38 mitogen-activated
protein kinase (p38 MAPK) (25,26)
and ERK (24,27). In addition to affecting cell
signaling, arctigenin influences ER stress and inhibits or
activates the UPR, resulting in apoptosis or protection against ER
stress (28–30). Arctigenin has been shown to exhibit
anticancer activity in numerous types of cancer, including
hepatocellular carcinoma (23),
colon cancer (25), gastric cancer
(31), pancreatic cancer (32), gallbladder cancer (27), breast cancer (24,26),
ovarian cancer (33) and
pancreatic cancer (34,35). One of the interesting anticancer
properties of arctigenin is that it can preferentially induce the
apoptosis of cancer cells under conditions of glucose starvation
(34,36). Furthermore, arctigenin (GBS-01) has
presented a high safety profile and good therapeutic effects in a
phase I clinical trial in patients with gemcitabine-refractory
pancreatic cancer due to its anticancer and anti-austerity activity
(35). However, the effects of
arctigenin on aggressive PEL phenotypes remain unclear. In the
present study, the effects of arctigenin on PEL cells and the
underlying molecular mechanisms were investigated.
Materials and methods
Cell lines and reagents
KSHV-positive PEL cell lines (BC3, BCBL1, BC2 and
HBL6) are derived from patients with PEL. PEL and KSHV-negative
B-lymphoma cell lines (BJAB, Ramos and DG75) were kindly provided
by Dr S.D. Hayward (Johns Hopkins University School of Medicine,
Baltimore, MD, USA) and were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum. 2-Deoxyglucose, was
dissolved in distilled sterile water, and arctigenin (both from
Wako, Osaka, Japan), SB203580 (p38 MAPK inhibitor; Nacalai Tesque,
Kyoto, Japan), SB202190 (p38 MAPK inhibitor; Nacalai Tesque) and
U0126 (ERK inhibitor; Cell Signaling Technology, Beverly, MA, USA)
were dissolved in dimethyl sulfoxide (DMSO).
Cell viability assay
The PEL cells and KSHV-negative cells were
maintained in RPMI-1640 medium supplemented with 10% FCS. The cells
were seeded in 96-well plates at 1×104 cells/well in 100
µl of RPMI-1640 (or glucose-free RPMI-1640) with or without
the compound at various concentrations and incubated for 24 h. The
number of viable cells was estimated using Cell Count Reagent SF
(Nacalai Tesque), as previously described (12). The optical density at 450 nm of
each sample was measured using a microplate spectrophotometer
(Tecan M200; Tecan, Kanagawa, Japan) and expressed as a percentage
of the value obtained from untreated cells (defined as 100%).
Cell viability assay in the presence of
2-deoxyglucose
The cells (1×104 cells/well) were seeded
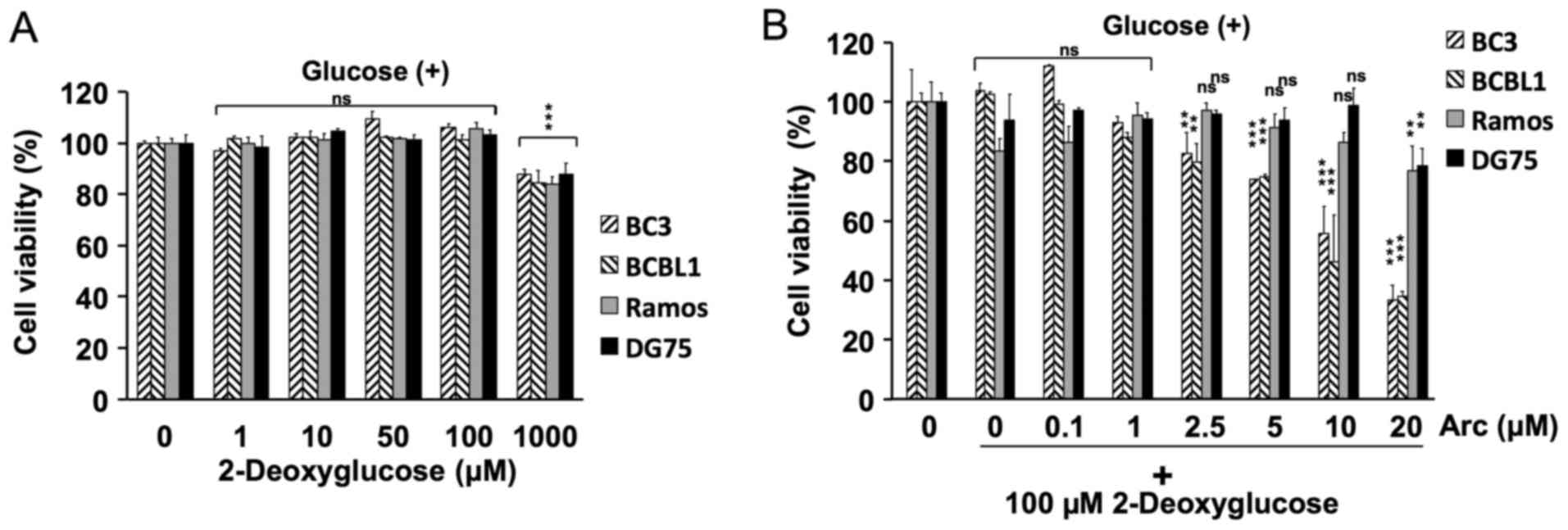
onto 96-well plates in 100 µl of RPMI-1640 medium. The cells
were incubated in medium with 0, 1, 10, 50, 100 or 1,000 µM
2-deoxyglucose for 24 h (Fig. 2A),
or with 0, 0.1, 1, 2.5, 5, 10 or 20 µM arctigenin in
combination with 100 µM 2-deoxyglucose for 24 h (Fig. 2B). The number of viable cells was
evaluated using Cell Count Reagent SF (Nacalai Tesque).
Western blot analysis and antibodies
The cells (1×106 cells) were lysed in 200
µl of sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) sample buffer containing 0.5 mM
phenylmethylsulfonyl fluoride, 1 µg/ml pepstatin, 5
µg/ml aprotinin and 1% 2-mercaptoethanol, boiled for 5 min,
and sonicated for 30 sec with an immersible tip-type sonicater in
order to shear the chromosomal DNA. The resulting lysate was
subjected to SDS-PAGE on 8 or 12% polyacrylamide gel followed by
western blot analysis. The proteins were transferred onto a
ClearTrans nitrocellulose membrane (Wako) and, a membrane was
incubated with 3% non-fat dry milk in phosphate-buffered saline
(PBS) containing 0.1% Tween-20 (PBS-T) for 1 h at room temperature.
The membrane was then incubated with the primary antibody
(1,000-fold dilution) and subsequently with the secondary antibody
(horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG
antibody) (3,000-fold dilution) in Can Get Signal Immunoreaction
Enhancer Solution (Toyobo, Osaka, Japan). The antibody-reactive
bands were visualized by ECL Western Blotting Detection Reagents
(GE Healthcare Life Sciences, Chicago, IL, USA), and the bands were
visualized with X-ray film (Fujifilm Corp., Tokyo, Japan). The
primary antibodies used in these experiments were as follows:
β-actin (sc-69879), ribosomal protein S6 kinase A1/RSK-1
(sc-393147) and activating transcription factor (ATF)6α (H-280)
(sc-22799) (all from Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA), cleaved caspase-3 (#9661), cleaved caspase-9 (#9501),
cleaved PARP (#9541), Thr202/Tyr204-phospho-ERK1/2 (#9101),
Ser380-phospho-p90RSK (#11989) (all from Cell Signaling Technology,
Danvers, MA, USA), Thr180/Tyr182-phospho-p38 MAPK (612288), p38
MAPK (612168), Thr183/Tyr185-phospho-SAPK/ JNK (612540), SAPK/JNK
(610627), Thr202/Tyr204-phospho-ERK1/2 (20A) (612359), ERK1/2
(610103) and pan ERK (610123) (all from BD Biosciences, Franklin
Lakes, NJ, USA).
JC-1 staining for assessment of
mitochondrial membrane potential
The inner mitochondrial membrane potential was
evaluated by cell staining with JC-1 (PromoCell GmbH, Heidelberg,
Germany), a cationic fluorescent dye that exhibits a fluorescence
emission shift upon aggregation from 530 nm (green 'JC-1 monomer')
to 590 nm (red 'JC-1 aggregates'). In healthy cells with a high
mitochondrial membrane potential, JC-1 enters the mitochondrial
matrix in a potential-dependent manner and forms JC-1-aggregates,
resulting in a red fluorescence signal, whereas mitochondrial
damage induces mitochondrial depolarization and monomeric JC-1,
resulting in a green fluorescence signal. The cells in a 96-well
plate were cultured with 2 mg/ml JC-1 for 30 min and washed with
PBS. The cells were then incubated with or without arctigenin for
30 min, and red fluorescence (535 nm excitation and 590 nm
emission) and green fluorescence (485 nm excitation and 530 nm
emission) were measured using a 96-well plate fluorometer (Tecan
M200). The polarization of mitochondrial membrane potential, i.e.,
normality, are shown as the ratio of red/green fluorescence.
Determination of cellular ATP
concentration
The cells were seeded in 96-well plates at
5×104 cells/well in 0.1 ml of medium and incubated with
or without arctigenin for 3 h. The cellular ATP concentration was
measured using the ATP assay reagent (Wako) according to the
manufacturer's instructions. Luminescence was measured using a
luminescence microplate leader (Tecan M200). The ATP concentration
of the untreated cells was defined as 100%.
Cell viability assay using the kinase
inhibitor
The cells (1×104 cells/well) were
cultured in 100 µl of glucose-free RPMI-1640 with or without
the kinase inhibitors (U0126, SB202190 or SB203580) at various
concentrations for 24 h. The number of viable cells was estimated
using Cell Count reagent SF (Nacalai Tesque).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was purified and extracted from
1×106 cells using RNAiso Plus (Takara Bio Inc., Shiga,
Japan), as previously described (12). First Strand cDNA was synthesized
from 20 ng of total RNA using the ReverTra Ace qPCR RT kit
(Toyobo). To quantify cDNA, polymerase chain reaction (PCR) was
performed using GoTaq Flexi DNA Polymerase (Promega, Madison, WI,
USA). The PCR products were analyzed by electrophoresis on 2%
agarose gels and staining with ethidium bromide. The nucleotide
sequences of oligonucleotides used for RT-PCR primers are shown in
Table I.
 | Table IPrimers for used for RT-PCR. |
Table I
Primers for used for RT-PCR.
| Molecule | Forward | Reverse |
|---|
| EDEM2 |
5′-TGCCTTTCCCTTCGATGAGC-3′ |
5′-ACATCTTCGAACACCGGGTC-3′ |
| EDEM1 |
5′-TGGACTGCAGGTGCTGATAG-3′ |
5′-GACTTTCCCCCTTGGTCCTG-3′ |
| GADD34 |
5′-CGAGGAAGAGGGAAGTTGCTG-3′ |
5′-CTCCATCCTTCTCAGCTGCC-3′ |
| GRP94 |
5′-GCCAGTTTGGTGTCGGTTTC-3′ |
5′-GTGATGAATACACGGCGCAC-3′ |
| CHOP |
5′-GGTACCTATGTTTCACCTCCTG-3′ |
5′-GAGCCGTTCATTCTCTTCAGC-3′ |
| GRP78 |
5′-AGGAAAGACAATAGAGCTGTGC-3′ |
5′-TGTCTTTTGTCAGGGGTCTTTC-3′ |
| ATF6α |
5′-CTTTACTAGGCCACCCCGTCTCG-3′ |
5′-CAATCCAACTCCTCAGGAAC-3′ |
| GAPDH |
5′-TGACCACAGTCCATGCCATC-3′ |
5′-GGGGAGATTCAGTGTGGTGG-3′ |
Luciferase reporter assay
HeLa cells were obtained from RIKEN BioResource
Center (Ibaragi, Japan) and used for transient transfection. HeLa
cells (1×105) were transfected with 2 µg of a
luciferase reporter plasmid containing either the promoter of the
glucose-regulated protein 78 (GRP78)/BiP gene (pGL3-BiP) or the
promoter of the ATF6 gene plasmid (pGL3-ATF6), together with 1
µg of a pSV-β-Gal plasmid (Promega) by the Chen and Okayama
calcium-phosphate method (37).
pGL3-BiP and pGL3-ATF6 which bear the promoter region of the
GRP78/BiP gene and ATF6α gene upstream of the luciferase gene,
respectively, were kind gifts from Takayanagi et al
(38). pSV-β-Gal was used as an
internal control for the determination of the transfection
efficiency. The transfected cells were incubated in glucose-free
medium with arctigenin (or kinase inhibitor) for 4 h. The cells
were resuspended in 0.1 ml of lysis buffer for luciferase and β-Gal
assay. Luciferase activity was measured using GloMax 20/20
luminometer (Promega). The luciferase activity divided by
β-galactosidase activity in untreated cells cultured in
glucose-free medium was defined as 100%.
Real-time PCR for the measurement of
viral production
The measurement of lytic virus production was
performed as previously described (12). Briefly, the BC3 cells were treated
with 3 mM sodium butyrate (NaB) together with arctigenin for 48 h,
and the culture media (300 µl) were harvested. To obtain
only enveloped and encapsulated viral genomes, media were incubated
with 20 units of DNase I (Wako) for 30 min, and viral DNA was
purified using a QIAamp DNA blood mini kit (Qiagen, Hilden,
Germany). Viral DNA was quantified by real-time PCR using an ORF50
(RTA)-specific primer set (forward, 5′-ATA ATC CGA ATG CAC ACA TCT
TCC ACC AC-3′ and reverse, 5′-TTC GTC GGC CTC TCG GAC GAA CTG
A-3′).
Statistical analysis
All data are presented as the means ± standard
deviation (SD) from at least 3 independent experiments. Statistical
significance was determined by one-way ANOVA followed by Tukey's
test for multiple comparisons using GraphPad PRISM 7 (GraphPad
Software, La Jolla, CA, USA).
Results
Arctigenin exhibits preferential
cytotoxicity against PEL cells under conditions of glucose
deprivation
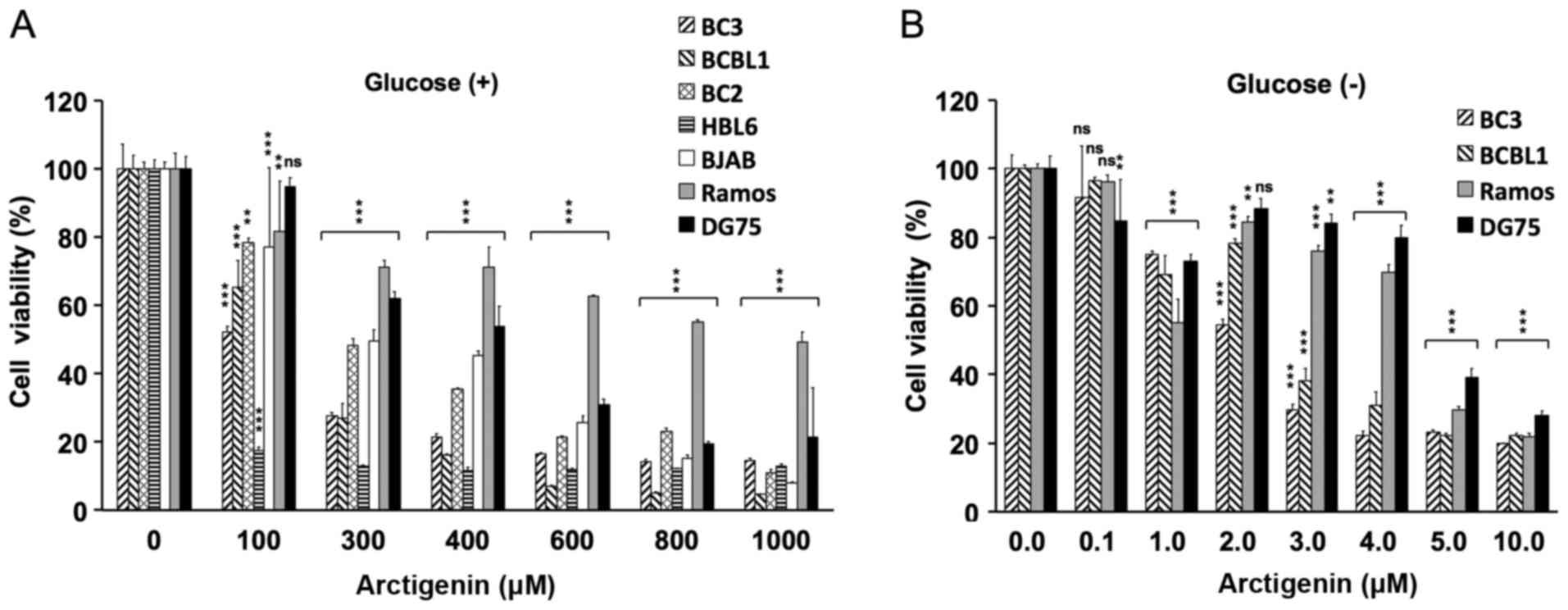
First, the proliferation of PEL cells and
KSHV-uninfected B-cell lymphoma cells was measured in normal medium
containing arctigenin, and the cytotoxicity was evaluated by
analyzing the viability of the treated vs. the untreated cells
(Fig. 1A). Arctigenin
preferentially inhibited the proliferation of the PEL cell lines
(BC3, BC2, BCBL1 and HBL6) compared with the KSHV-uninfected
B-lymphoma cell lines (BJAB, DG75 and Ramos). We then evaluated the
cytotoxic effects of arctigenin on PEL cells cultured in
glucose-free medium. As a result, at low concentrations (2–4
µM), arctigenin exerted preferential growth inhibitory
effects on PEL cells (Fig. 1B). In
particular, the viability of the BC3 and BCBL1 cells was decreased
by 70% following treatment with 3 µM arctigenin in
glucose-deprived medium (Fig. 1B).
By contrast, a 100-fold greater concentration of arctigenin (300
µM) was needed to decrease the viability of these cells by
70% in glucose-containing media (Fig.
1A). The cytotoxic effects (CC50) of arctigenin on
B-lymphoma cells under the glucose-supplied and glucose-deprived
conditions are summarized in Table
II. Arctigenin was active against not only the PEL (BC3, BCBL1,
BC2 and HBL6), but also the KSHV-uninfected BJAB cells (Fig. 1A). These phenomena may be due to
differences in the genetic background or differentiation status of
the tested cells. Arctigenin exhibited potent anti-proliferative
activities in the BC3 and BCBL1 cells in a dose- and glucose
starvation-dependent manner, but did not have the same effects on
the DG75 and Ramos cells. Therefore, we focused and analyzed the
underlying molecular machinery in the BC3, BCBL1, Ramos and DG75
cells.
| Table IIThe CC50 value of
arctigenin in B-lymphoma cells under the glucose-supplied or
-deprived conditions. |
Table II
The CC50 value of
arctigenin in B-lymphoma cells under the glucose-supplied or
-deprived conditions.
| Cell line
|
|---|
| BC3 | BCBL1 | BC2 | HBL6 | BJAB | Ramos | DG75 |
|---|
Glucose
(+)
CC50 (µM) | 273.2 | 193.8 | 107.0 | 0.4 | 101.8 | 819.0 | 561.8 |
Glucose
(−)
CC50 (µM) | 2.8 | 2.3 | n.d. | n.d. | n.d. | 4.5 | 4.2 |
Combination of arctigenin and
2-deoxyglucose synergisti-cally suppresses the proliferation of PEL
cells
Arctigenin exhibited preferential cytotoxicity
against the PEL cells under conditions of glucose deprivation.
Therefore, we investigated whether treatment with a low
concentration of arctigenin in combination with 2-deoxyglucose, a
hexokinase inhibitor of glycolysis, inhibits the proliferation of
PEL cells under normal glucose conditions. First, we examined the
cytotoxic effects of 2-deoxyglucose on the PEL (BC3 and BCBL1) and
KSHV-uninfected (Ramos and DG75) cell lines cultured in
glucose-containing medium. Treatment with 2-deoxyglucose alone did
not inhibit the growth of these cell lines (Fig. 2A). We then combined arctigenin at
various concentrations (0–20 µM) with 100 µM of
2-deoxyglucose to examine the effects on PEL cells cultured in
glucose-containing medium. As shown in Fig. 2B, the combination of arctigenin and
2-deoxyglucose synergistically decreased the viability of the PEL
cells under normal glucose conditions, but did not decrease the
viability of the KSHV-uninfected cells. Hence, arctigenin inhibited
the proliferation of PEL cells under glucose-deprived conditions
more potently than under normal glucose conditions.
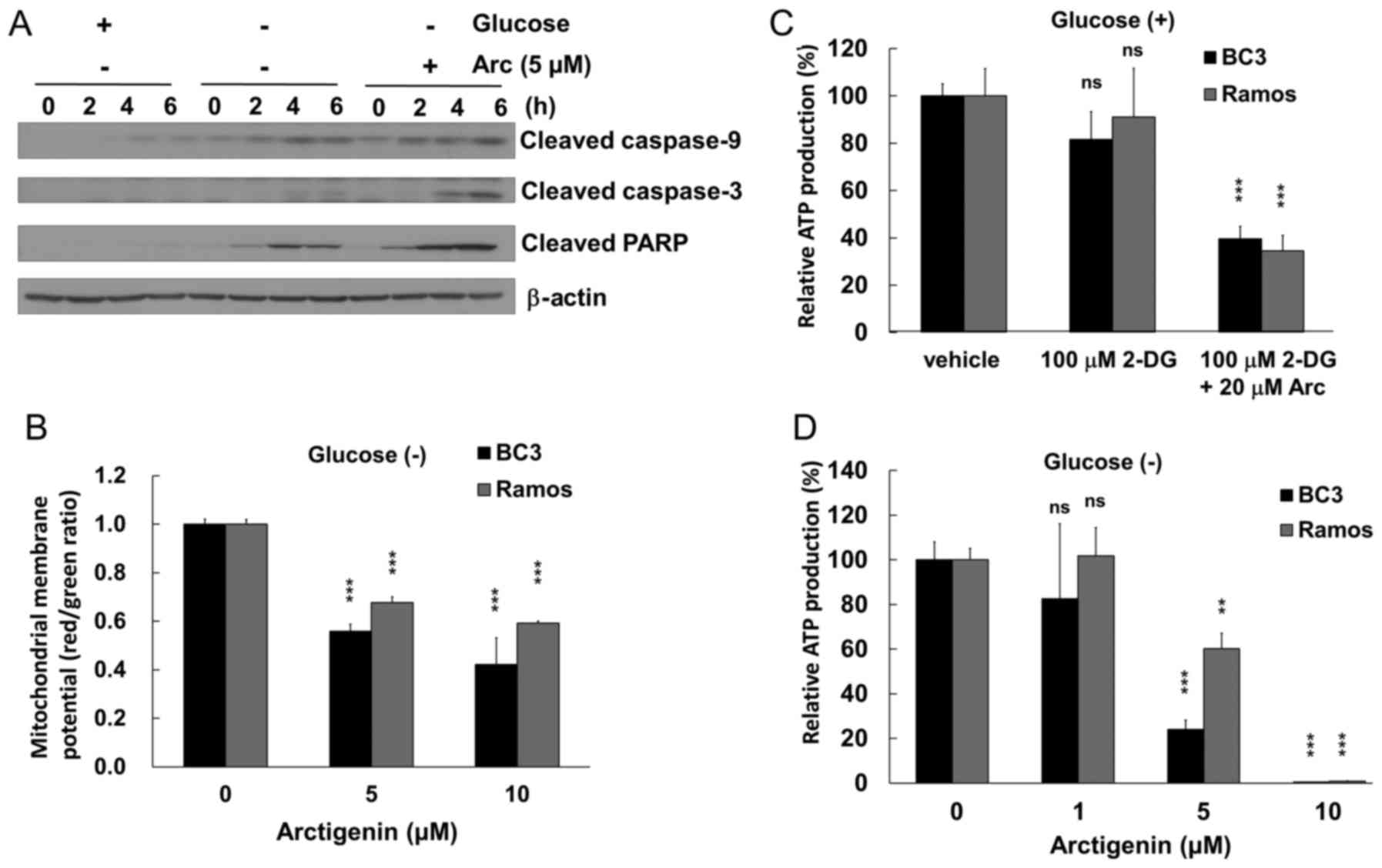
Arctigenin induces the caspase-9-mediated
apoptosis of glucose-starved PEL cells
We then examined whether the anti-proliferative
effects of arctigenin under conditions of glucose deprivation are
due to apoptotic cell death. The PEL cells cultured in glucose-free
medium were treated with/or without 5 µM arctigenin and were
then examined by western blot analysis using cleaved caspase and
PARP antibodies (Fig. 3A). Active
caspase-9 and -3 were detected in the arctigenin-treated BC3 PEL
cells. In a time-dependent manner, the cleaved caspase-9, -3 and
PARP fragments were increased in the glucose-starved BC3 cells
treated with arctigenin, whereas modest cleaved caspase-9 and PARP
were detected in the arctigenin-untreated BC3 cells. However, no
significant cleavage of caspases and PARP was detected in the
untreated BC3 cells cultured in glucose-containing medium. In
addition, cleaved caspase-8 was not detected in the
arctigenin-treated BC3 cells (data not shown).
Arctigenin induces mitochondrial
disruption in glucose-starved PEL cells by decreasing ATP levels
and disrupting the mitochondrial membrane
To gain insight into the molecular mechanisms
through which arctigenin induces apoptosis via caspase-9
activation, we examined whether arctigenin induces mitochondrial
disruption in glucose-starved BC3 PEL cells. The BC3 and
KSHV-uninfected Ramos cells cultured in glucose-free medium were
treated with arctigenin for 30 min, and the inner mitochondrial
membrane potential of the cells was evaluated by JC-1 staining. As
a result, arctigenin preferentially depolarized the mitochondrial
membrane in the BC3 cells compared with the Ramos cells, which was
indicated by a decrease in the red/green ratio (Fig. 3B). To obtain further evidence of
mitochondrial dysfunction induced by arctigenin, the effects of
arctigenin on the cellular ATP concentration in glucose-starved BC3
and Ramos cell lines were examined (Fig. 3C and D). Treatment with
2-deoxyglucose (also termed 2-DG) was used for the
glucose-starvation of the cells. The results revealed that cellular
ATP production decreased to ~30% in both cell lines treated with
arctigenin and 2-deoxyglucose (Fig.
3C), and arctigenin preferentially suppressed ATP production in
BC3 cells cultured in glucose-free medium, compared with the Ramos
cells (Fig. 3D). Thus, our data
demonstrated that arctigenin impaired normal mitochondrial function
and homeostasis in glucose-deprived PEL cells, resulting in the
depolarization of mitochondrial membrane potential and a decrease
in ATP production. Furthermore, mitochondrial disruption can induce
caspase-9-mediated apoptosis. As regards cell viability (Fig. 2B) and ATP production (Fig. 3C) under glucose-supplied
conditions, co-treatment with arctigenin and 2-deoxyglucose
decreased the viability of the Ramos and BC3 cells. However, the
co-treatment led to a decrease in ATP production in both the Ramos
and BC3 cells to a varying extent, which may be due to differences
in the ability of the cells to respond to ATP depletion or a
difference in the inhibitory effect of 2-deoxyglucose on ATP
production between the Ramos and BC3 cells.
Arctigenin suppresses ERK and p38 MAPK
signaling in PEL cells under conditions of glucose-deprivation
Arctigenin is known to exert anticancer effects by
modulating cellular signaling, including p38 MAPK (25,26)
and ERK (24,27). On the other hand, the activation of
the p38 MAPK (39,40) and ERK (14,41,42)
signaling pathways is necessary for KSHV to establish infection,
maintain malignant phenotypes and ensure survival in host cells.
Therefore, in this study, we investigated the effects of arctigenin
treatment on MAPK signaling, including p38 MAPK, ERK and JNK, in
cells cultured in glucose-deprived medium. The phosphorylation
(i.e., activation) level of JNK was not markedly altered in the
glucose-deprived cells by arctigenin (Fig. 4A), while the phosphorylation of p38
MAPK and ERK1/2 was decreased in the PEL cells (BC3 and BCBL1)
treated with arctigenin for 4 and 6 h. In addition, we examined the
phosphorylation of p90RSK, which is an ERK substrate. The
phosphorylation of ERK1/2 was also confirmed by a different
antibody. U0126 treatment was used as a control for the inhibition
of MEK1/2/ERK1/2 signaling. Compared to the KSHV-uninfected Ramos
and DG75 cells, arctigenin markedly decreased the phosphorylation
of p90RSK and ERK1/2 in the glucose-deprived PEL cells in a
dose-dependent manner (Fig. 4B).
These results indicate that the apoptotic effects of arctigenin are
related to the modulation of the ERK and p38 MAPK signaling
pathways.
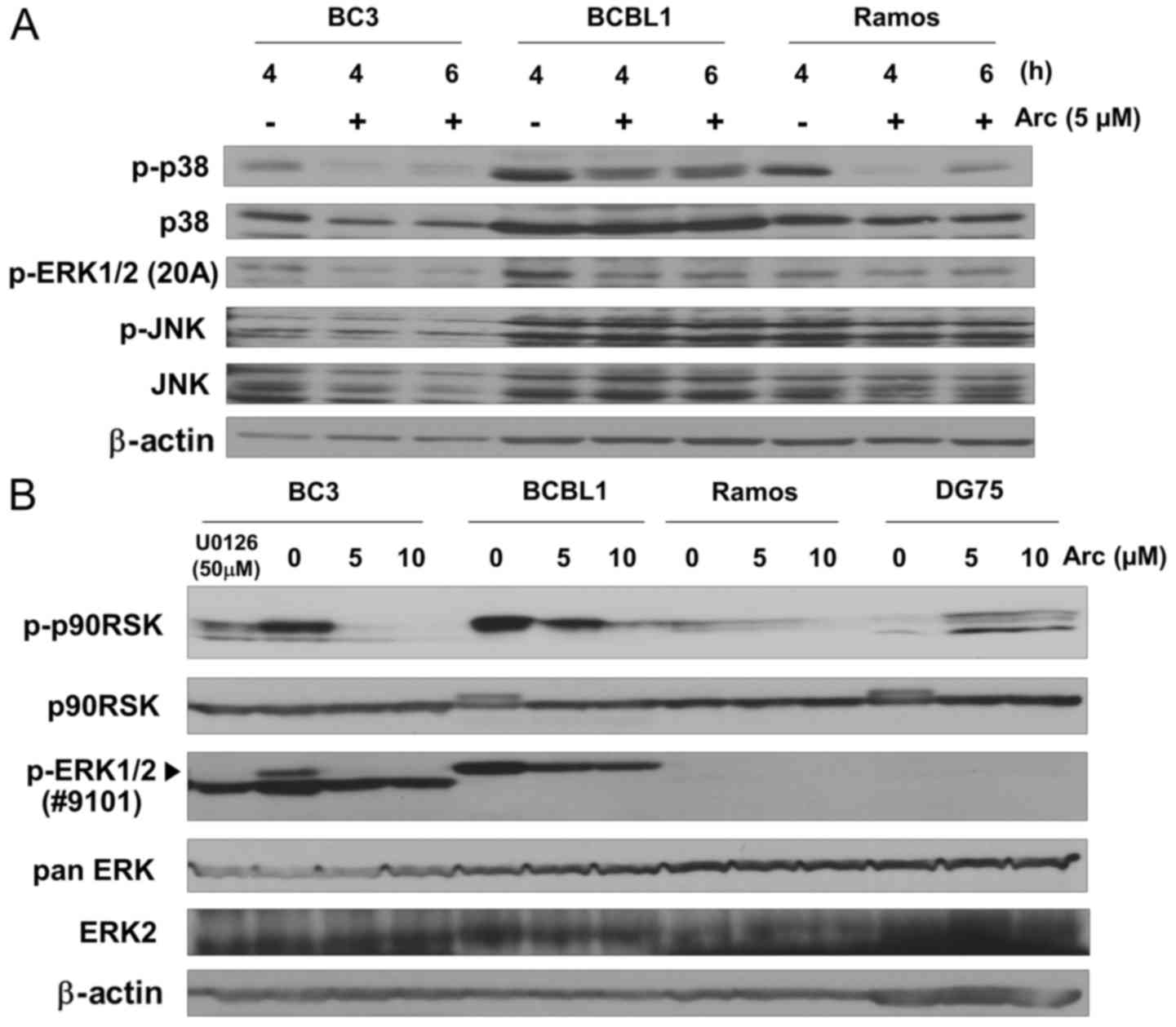 | Figure 4Arctigenin suppresses ERK and p38
MAPK signaling in glucose-deprived PEL cells. (A) Western blot
analysis of the phosphorylation of p38, ERK1/2, and JNK in
arctigenin-treated B-lymphoma cells. PEL cells (BC3 and BCBL1) and
infected Ramos cells were cultured in glucose-free medium with 5
µM arctigenin (Arc) for 4 or 6 h, or cultured without
arctigenin for 4 h. Whole-cell lysates were subjected to western
blot analysis with an anti Thr202/Tyr204-phospho-ERK1/2 (20A),
Thr180/Tyr182-phospho-p38, p38, Thr183/Tyr185-phospho-JNK or JNK
antibodies. (B) Western blot analysis of phosphorylated p90RSK in
arctigenin-treated PEL cells. PEL and uninfected cells (Ramos and
DG75) were cultured in glucose-free medium with 0, 5 or 10
µM arctigenin for 3 h, or BC3 cells were cultured in
glucose-free medium with 50 µM U0126 for 3 h. U0126 was used
as an inhibitor of MEK1/2-ERK1/2 signaling. Cell extracts were
subjected to blotting with anti Ser380-phospho-p90RSK, p90RSK,
Thr202/Tyr204-phospho-ERK1/2 (#9101), pan ERK, or ERK2 antibodies.
The pan ERK antibody recognizes ERK1, 2 and 3. The arrowhead
indicates phosphorylated ERK1/2. |
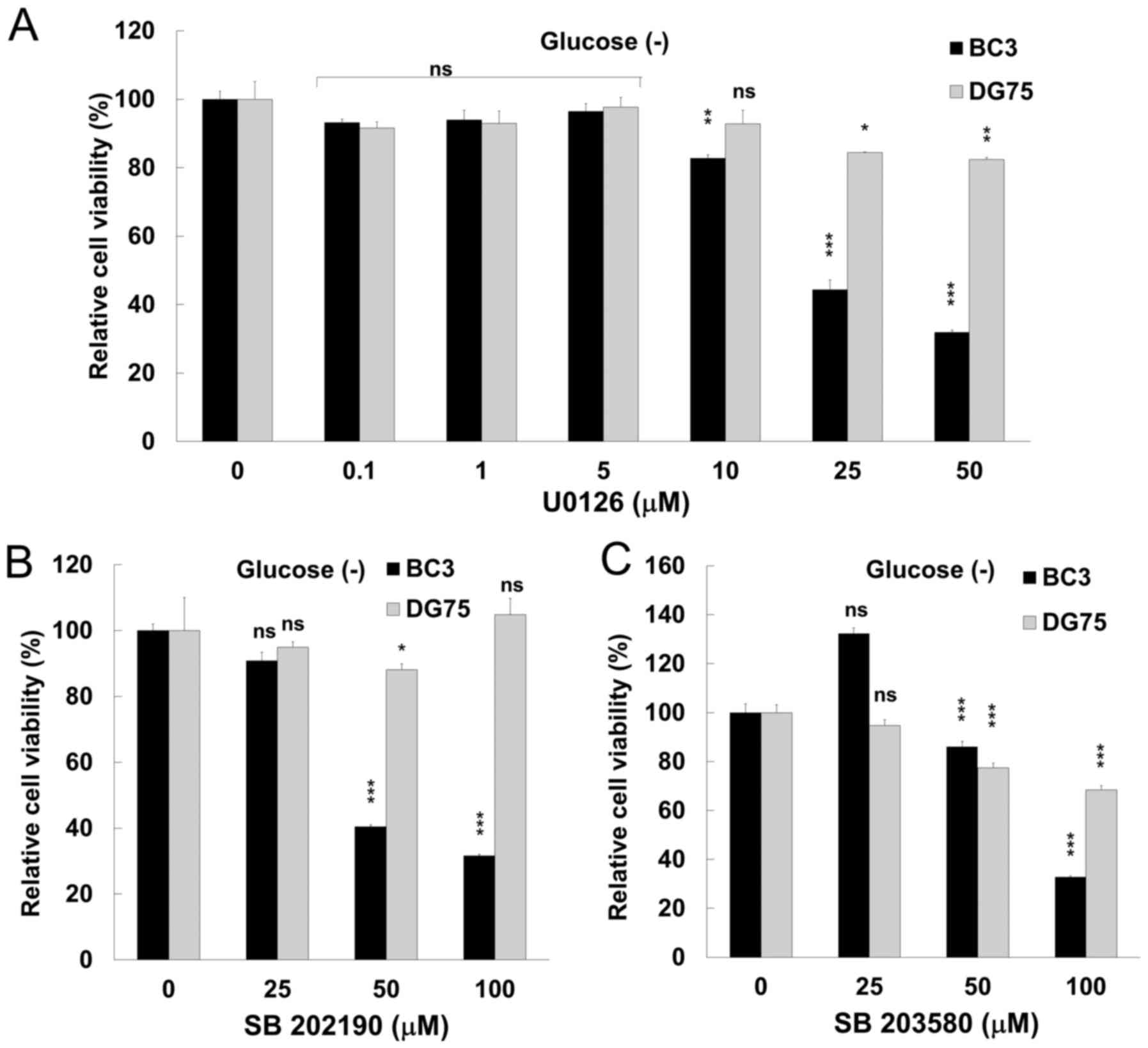
The inhibition of either ERK or p38 MAPK
signaling suppresseses the proliferation of glucose-deprived PEL
cells
Arctigenin led to a decrease in PEL cell
proliferation through apoptosis (Figs.
1 and 3), and moreover
suppressed ERK and p38 MAPK signaling (Fig. 4) in PEL cells under conditions of
glucose deprivation. We therefore wished to examine the cytotoxic
effects of a MEK inhibitor and p38 MAPK inhibitors on PEL cells to
confirm that ERK and JNK signaling contribute to the survival and
growth of PEL cells under conditions of glucose deprivation. U0126
is known to inhibit MEK1 and MEK2, which phosphorylate and activate
ERK1/2. SB202190 and SB203580 inhibit p38 MAPK activity by
competing with ATP for binding in the ATP-binding pocket of p38,
respectively. When the BC3 PEL and KSHV-uninfected DG75 cells were
treated with 50 µM U0126, 50 µM SB202190 or 100
µM SB203580 for 24 h, the viability of the glucose-deprived
BC3 cells was decreased compared to that of the DG75 cells
(Fig. 5). These data indicate that
ERK and p38 MAPK signaling strongly contribute to the survival and
proliferation of PEL cells. In addition, we evaluated the
anti-proliferative effects of the inhibitors under glucose-supplied
conditions. SB202190 and SB203580 did not exert any
anti-proliferative effects on the PEL cells cultured in
glucose-containing medium (data not shown). We have also previously
reported the anti-proliferative effects of U1026 (13,14).
The viability of the BC3 cells cultured with glucose was decreased
by 60 and 25% following treatment with 100 and 50 µM of
U1026, respectively (13,14).
Arctigenin and p38 MAPK inhibitor
suppress the transcriptional expression of GRP78/Bip and ATF6α in
glucose-deprived PEL cells
Arctigenin inhibited ERK and p38 MAPK signaling in
glucose-deprived PEL cells (Fig.
4). Cytokines and forms of cellular stress, such as UV
irradiation, heat shock and osmotic changes, have been known to
activate p38 MAPK signaling, which correlates with the upregulation
of the UPR including GRP78/BiP, CHOP and ATF6α (43,44).
Furthermore, it has been reported that arctigenin blocks the UPR
and the expression of GRP78, CHOP, ATF4 and XBP1 (28,29).
Therefore, in this study, we investigated whether arctigenin
downregulates the mRNA expression of ER stress-related molecules in
PEL cells under conditions of glucose deprivation. GRP78 and GRP94
are ER stress-inducible ER chaperones. GRP78, also known as BiP,
plays an essential role in the UPR (45). ER degradation enhancing
α-mannosidase-like proteins (EDEMs) are ER membrane proteins and
are involved in ER-associated degradation (ERAD). EDEM1/2 targets
misfolded glycoproteins for degradation in an N-glycan-dependent
manner (45). ATF6α is a
UPR-related transcription factor and activates the transcriptional
expression of GRP78. CHOP is also an UPR-related transcription
factor and transcriptionally upregulates GADD34 which induces a
translation block (46). In this
study, the mRNA expression levels of these molecules in cells
cultured in glucose-free medium with (or without) arctigenin were
measured by RT-PCR (Fig. 6A).
Arctigenin increased the mRNA levels of GRP94 and CHOP in the
glucose-deprived PEL and uninfected cells. The expression of GADD34
was slightly upregulated in the arctigenin-treated BC3 and
uninfected cells. In addition, GRP78 and ATF6α exhibited very low
levels of transcription in all cell types in the absence of
glucose; however, arctigenin decreased the transcription of GRP78
and ATF6α. By contrast, EDEM1 and EDEM2 exhibited high levels of
transcription in all cell types, while arctigenin did not affect
the transcription of these molecules.
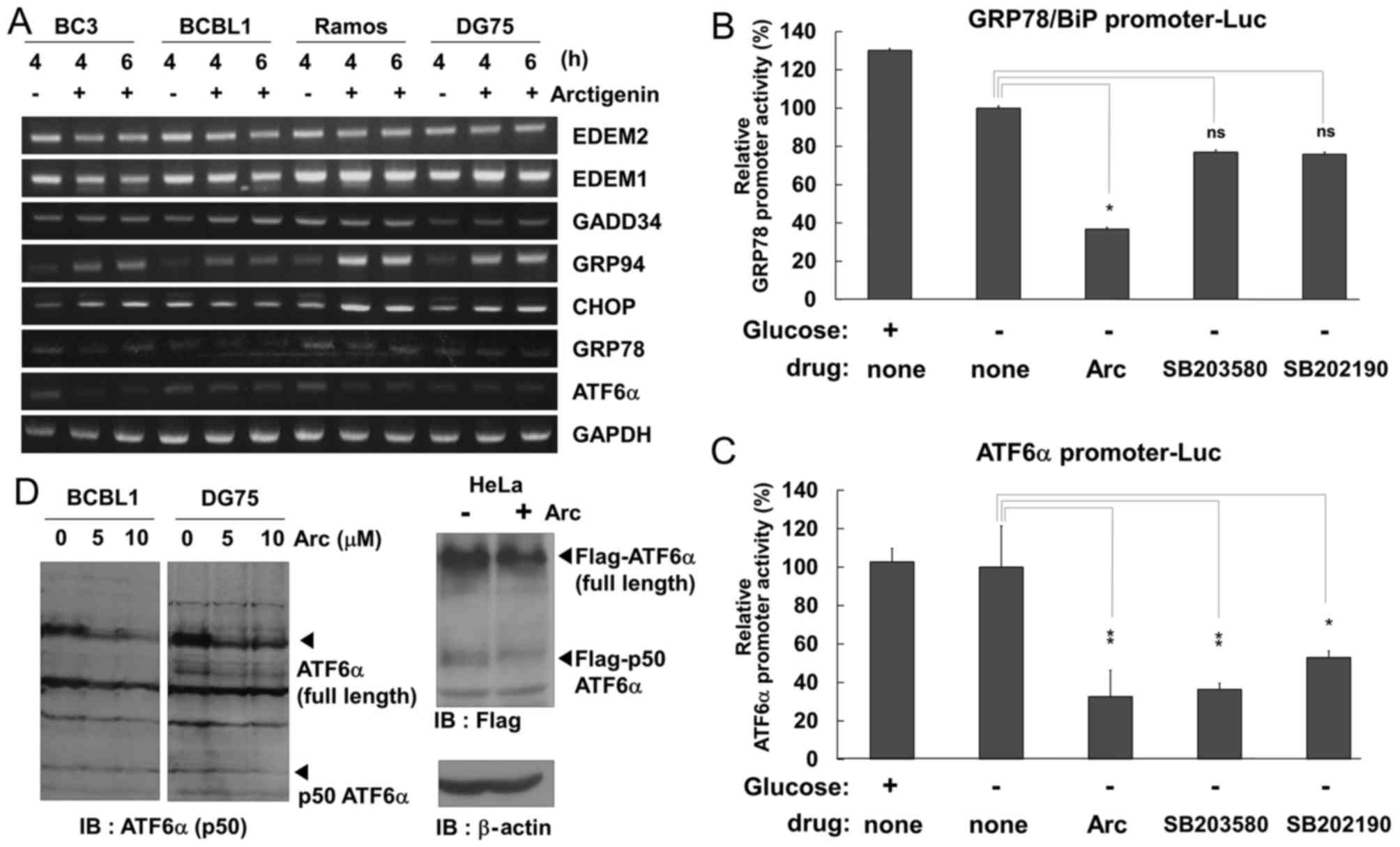 | Figure 6Arctigenin and p38 MAPK inhibitor
suppress the transcriptional expression of GRP78 and ATF6α in
glucose-deprived PEL cells. (A) Effects of arctigenin on the mRNA
expression of the UPR- and ER-stress related molecules. PEL cells
(BC3 and BCBL1) and uninfected cells (Ramos and DG75) cells were
treated with or without 5 µM arctigenin (Arc) for 4 or 6 h,
and harvested. Total RNA was extracted from the cells and subjected
to RT-PCR to detect the mRNA expression of EDEM2, EDEM1, GADD34,
GRP94, CHOP, GRP78/BiP, ATF6α and GAPDH. (B and C) Effects of
arctigenin and p38 MAPK inhibitor on the promoter activities of
GRP78/BiP and ATF6α genes. HeLa cells transfected with the
luciferase reporter plasmids containing the GRP78 or ATF6α promoter
were cultured in glucose-free medium with 10 µM arctigenin,
100 µM SB203580 or 100 µM SB202190 for 4 h, and lysed
in buffer for luciferase analysis. The promoter activities of GRP78
and ATF6α in drug-untreated and glucose-deprived cells is defined
as 100% relative activity. *P<0.01 and
**P<0.001 indicate a statistically significantly
difference compared with the untreated cell; ns, not significant.
(D) The level of ATF6α protein in arctigenin-treated cells. BCBL1
and DG75 cells were incubated with glucose-free medium with 5 or 10
µM arctigenin for 6 h, and cell lysates were probed by
blotting using an antibody that recognizes ATF6α N-terminal region
and ATF6α cleaved form, p50 ATF6α (left panels). HeLa cells were
transfected with N-terminal FLAG-tagged ATF6α by the Chen and
Okayama calcium-phosphate method (37) and were treated with 5 µM
arctigenin for 6 h. Cell lysates were subjected to blotting using
Flag antibody (right panel). |
As the mRNA expression levels of GRP78 and ATF6α
were decreased in the arctigenin-treated cells, we examined whether
arctigenin inhibits the transcription of these genes by a
luciferase reporter assay using the promoter sequences of GRP78 or
ATF6α. HeLa cells transfected with GRP78 or ATF6α promoter-driven
luciferase reporter plasmids were incubated in glucose-free medium
with arctigenin or p38 MAPK inhibitors (SB203580 and SB202190) for
4 h, and cell extracts were subjected to measure luciferase
activities. The results revealed that arctigenin markedly
suppressed GRP78 and ATF6α promoter activities in the HeLa cells
cultured in glucose-free medium (Fig.
6B and C). In addition to arctigenin, GRP78 promoter activity
was decreased by ~40% by glucose depletion and either SB203580 or
SB202190 treatment (Fig. 6B),
while ATF6α promoter activity was decreased by ~70 and 50% by
SB203580 or SB202190 treatment, respectively (Fig. 6C). These data indicate that
arctigenin and p38 MAPK inhibition downregulate the mRNA expression
of GRP78 and ATF6α under conditions of glucose deprivation. We then
wished to elucidate whether arctigenin treatment alters the protein
expression of ATF6α. The BCBL1 and DG75 cells were incubated in
glucose-free medium with arctigenin, and the amount of ATF6α
protein was detected by western blot analysis using an antibody
that recognizes the N-terminal region of both full-length ATF6α and
p50 ATF6α, which is the cleaved form of ATF6α containing the
transcriptional activation domain (Fig. 6D, left panel). HeLa cells
transfected with N-terminal Flag-tagged ATF6α were treated with
arctigenin, and were then examined by western blot analysis using a
Flag antibody (Fig. 6D, right
panel). As a result, the amount of ATF6α protein was decreased in
the arctigenin-treated BCBL1 and DG75 cells, and the amount of p50
ATF6α was also decreased in the arctigenin-treated DG75 and HeLa
cells. These data suggest that arctigenin suppressed not only the
expression of ATF6α, but also the production of p50 ATF6α.
Effects of arctigenin on KSHV replication
in PEL
Previous studies have indicated that cellular
stress, such as ER stress, oxidative stress and hypoxia,
reactivates latent KSHV infection and induces lytic replication in
PEL cells (47,48). Therefore, in this study, we
evaluated whether arctigenin enhances (or suppresses) de
novo KSHV production in BC3 PEL cells cultured in normal
medium. BC3 cells were treated with 3 mM NaB in the presence or
absence of arctigenin for 48 h, and the culture media were
harvested. Viral DNA purified from culture media was quantified by
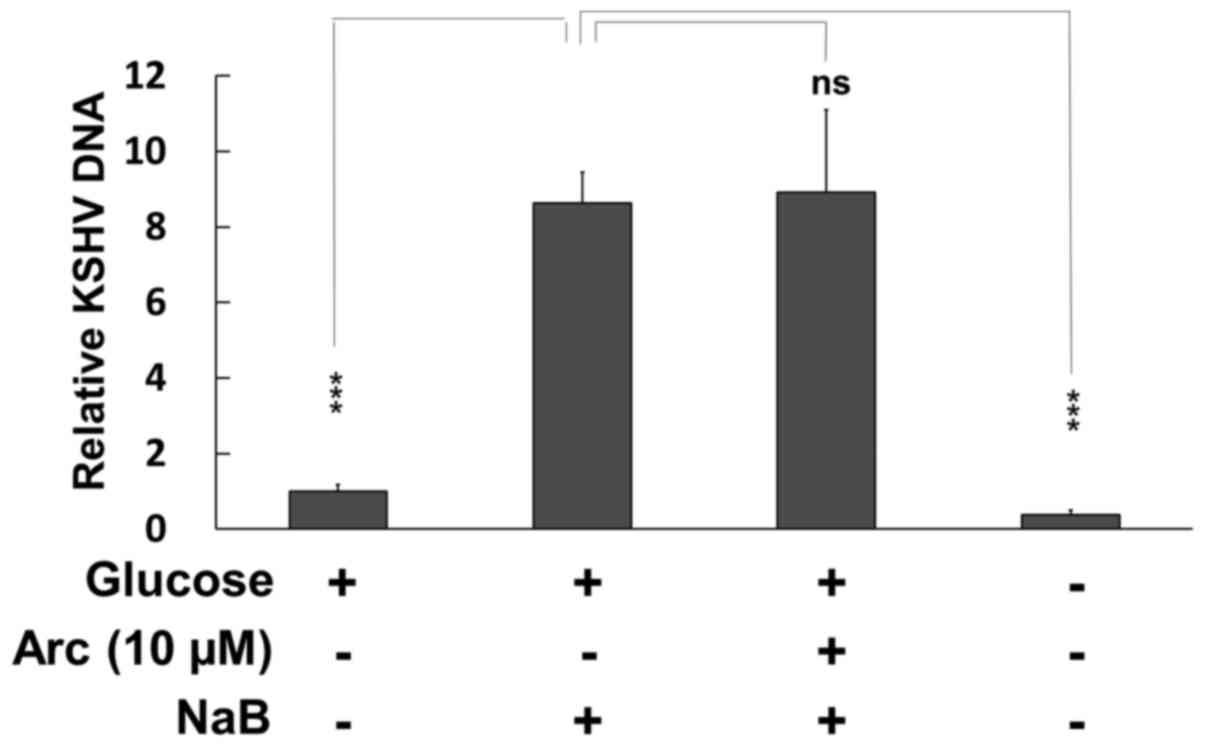
real-time PCR (Fig. 7). NaB
treatment was used for the induction of lytic replication. As a
result, NaB prominently increased viral production by the BC3 cells
cultured in culture medium containing glucose; however, arctigenin
neither enhanced nor suppressed viral production at 10 µM,
which also did not affect BC3 cell growth (Fig. 1A). These results indicate that
arctigenin may kill PEL cells without the risk of de novo
KSHV infection and production.
Discussion
PEL is a rare subtype of large B-cell lymphoma
associated with KSHV and it has the highest incidence rate in
HIV-positive individuals. In the majority of cases, PEL is present
as a malignant pleural, peritoneal or pericardial effusion without
a detectable solid mass. However, in rare cases, solid variants of
PEL are present as solid tumor masses or extracavitary solid tumors
(49,50). In general, chemotherapeutic drugs
targeting the proliferation machinery of tumor cells also damage
proliferating normal cells, such as hematopoietic stem cells.
Hence, the development of tumor-specific anticancer agents is
required for safe and effective cancer therapy. The tumor tissue
environment is known to lack oxygen and glucose compared with
normal tissues. Thus, selectively targeting metabolically stressed
(glucose-deprived) tumor cells, including PEL, may represent a
promising strategy with which to inhibit tumor cell proliferation
without affecting normal cells. In this regard, arctigenin has been
shown to preferentially induce tumor growth suppression and
apoptosis in glucose-deprived tumor cells (34–36).
In this study, we examined a novel biological function of
arctigenin and the molecular mechanisms through which it
preferentially induces the apoptosis of glucose-starved PEL cells.
We propose a model in which arctigenin downregulates MAPK signaling
and induces mitochondrial disruption, leading to PEL cell death
(Fig. 8).
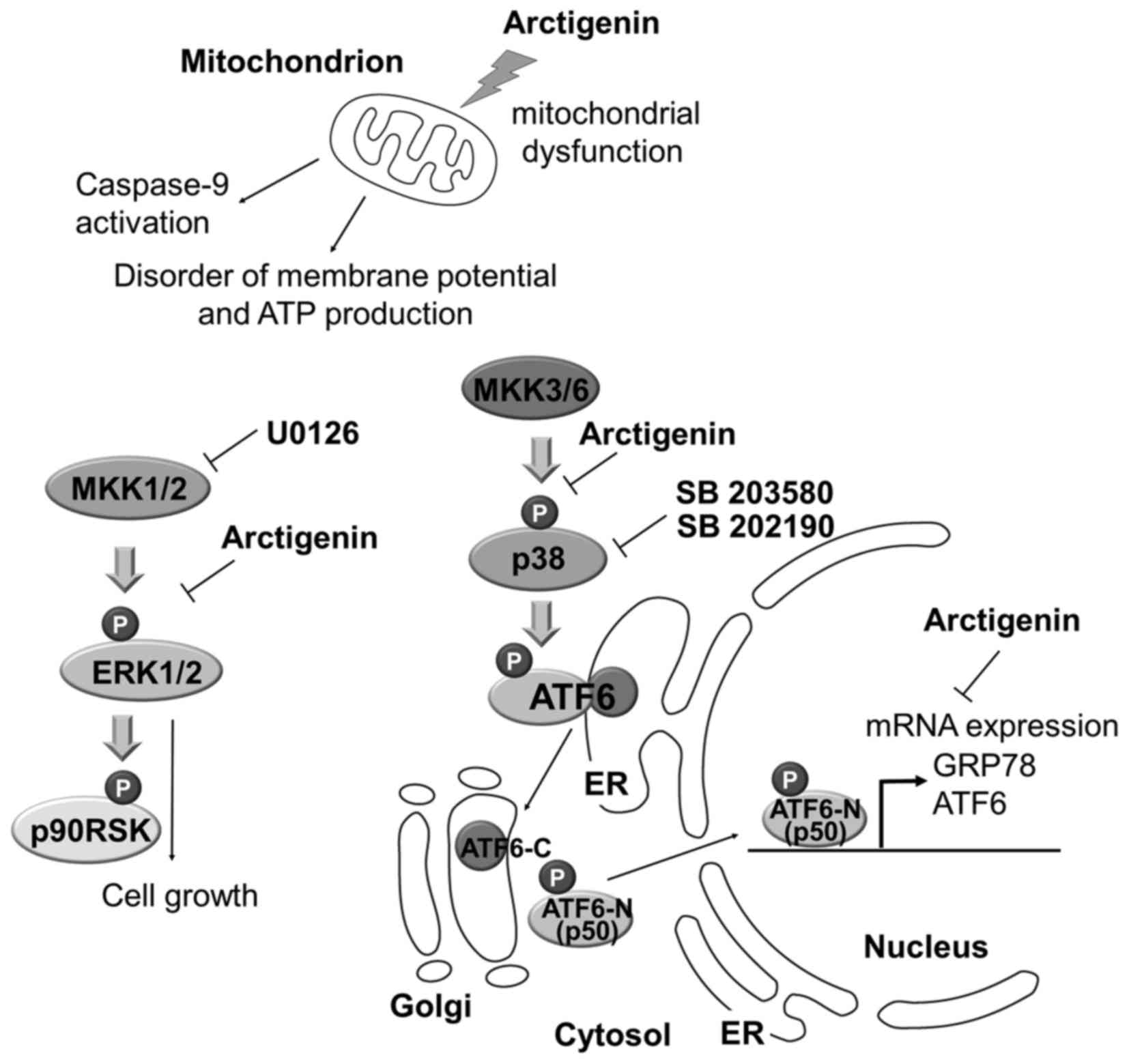 | Figure 8Model of arctigenin-mediated
inhibition of PEL cell growth. Arctigenin induced a decrease in ATP
levels, the disruption of mitochondrial membrane potential and
apoptosis via caspase-9 activation in PEL cells under conditions of
glucose deprivation. Arctigenin-induced mitochondrial disruption
correlates with caspase-9-mediated apoptosis. The ERK and p38 MAPK
signaling pathways are known to be required for KSHV to establish
infection, maintain malignant phenotypes and cell survival in a
host cell. In addition to mitochondrial disruption, arctigenin
suppresses the ERK and p38 MAPK pathways in glucose-deprived PEL
cells, resulting in the inhibition of cell proliferation. On the
other hand, various cellular stresses activate p38 MAPK, which
enhances the UPR, including the ATF6α along with the p38
MAPK-mediated phosphorylation and cleavage of ATF6α. Cleaved ATF6α
(p50ATF6/ATF6-N) induces transcriptional activation of GRP78 and
ATF6α itself. Arctigenin inhibits not only the p38 MAPK pathway,
but also the expression of GRP78, ATF6α and p50 ATF6α. Hence, the
suppression may be due to an increase in ER stress, which can cause
the apoptosis of PEL cells. |
Based on our understanding of KSHV-infected PEL and
the biochemical properties of arctigenin, we hypothesize 3 possible
mechanisms through which arctigenin induces the apoptosis of PEL
cells under conditions of glucose deprivation: i) mitochondrial
disruption; ii) suppression of the ERK and p38 MAPK pathways which
are involved in the proliferation and survival of PEL cells; and
iii) the suppression of ATF6α and GRP78 expression. We can surmise
that the cytotoxic effects of arctigenin in glucose-deprived PEL
cells may be mediated via a combination of these mechanisms.
Arctigenin has been shown to induce a decrease in
ATP production (36,30,51)
and mitochondrial membrane disruption (21,25,30)
in glucose-deprived tumor cells. Previous studies have suggested
that arctigenin inhibits complex I, II and IV of the mitochondrial
respiratory chain (21,36,30)
and consequently causes mitochondrial membrane disruption, which
results in an increase in cytochrome c release and cytochrome
c-related reactive oxygen species (ROS) production, thereby
decreasing intracellular ATP levels. These studies are in agreement
with the findings of the present study on arctigenin-treated PEL
cells. We found that arctigenin led to a loss of mitochondrial
membrane potential (Fig. 3B),
decreased intracellular ATP levels (Fig. 3C and D) and increased caspase-9
activation (Fig. 3A) in
glucose-deprived PEL cells, suggesting that the arctigenin-induced
apoptosis was mitochondrial dependent.
In addition to mitochondrial damage, arctigenin has
been known to cause cell cycle arrest or apoptosis by modulating
various signaling pathways, including STAT3 (22), Akt (23,24,34),
NF-κB (24), p38 MAPK (25,26)
and ERK (24,27). MAPKs, including ERK, p38 MAPK and
JNK, are involved in the development, proliferation and progression
of cancer. In addition, p38 MAPK and JNK are also associated with
various stress responses. ERK is mainly activated by growth
factors, while p38 and JNK are activated by cytokines, growth
factors and cellular stresses, such as ER stress, UV irradiation
and heat shock (52). It has been
reported that arctigenin activates p38 MAPK signaling, as well as
ROS production (25,26) and inhibits ERK signaling (24,27).
By contrast, other studies have found that arctigenin suppresses
p38 MAPK signaling in lipopolysaccharide (LPS)-treated RAW264.7
cells (53,54). In this study, our data are in line
with these findings, in that ERK and p38 MAPK activation were
suppressed by arctigenin in PEL cells under glucose-deprived
conditions (Fig. 4). We
hypothesize that these conflicting results of different studies as
mentioned above may be due to differences in the genetic background
of the tested cancer cells, KSHV infection, glucose conditions
during viability assays, or the assayed arctigenin
concentrations.
As described above, arctigenin treatment strongly
perturbs cellular signaling cascades that are essential for KSHV to
maintain a malignant phenotype and ensure survival in an infected
cell, including PEL and Kaposi's sarcoma. The constitutive and/or
transient activation of several signaling pathways, such as NF-κB,
Akt, Wnt and ERK, is known to be necessary for KSHV to establish
and maintain infection, cell proliferation, viral lytic replication
and cell survival in a host cell (7–10).
In fact, we have previously demonstrated that the suppression of
NF-κB, Akt and ERK signaling by diallyl trisulfide (12), pyrrolidinium fullerene derivatives
(13) and sangivamycin (14), respectively, inhibits the growth
and the apoptosis of PEL cells. In particular, the activation of
p38 MAPK is required for primary infection (39,42),
persistent infection (40,55), tumorigenicity and angiogenic
potential (56), reactivation
(i.e., lytic replication) (42,57)
and ROS stress response (47,58).
In addition, ERK signaling plays an important role in the
establishment of infection (41,42,59),
the expression of viral genes (41,42),
survival (14), lytic replication
(42) and the activation of
cellular transcription factors, such as c-Fos, c-Jun, STAT1α and
c-Myc (41). These observations
are consistent with our data. Namely, arctigenin reduced the
proliferation of PEL cells and suppressed the phosphory-lation of
p38 MAPK, ERK and p90RSK under conditions of glucose deprivation
(Fig. 4). Furthermore, the
suppression of p38 MAPK and ERK by a specific inhibitor decreased
the proliferation of the PEL cells (Fig. 5). Therefore, it can by hypothesized
that p38 MAPK inhibition or/and ERK inhibition by arctigenin may
disrupt the function of these signaling pathways (i.e., persistent
infection, tumorigenicity, stress response and gene expression),
thus resulting in growth inhibition and the apoptosis of PEL cells.
It can also be hypothesized that the arctigenin-induced inhibitory
effects on these kinases may lead to antitumor activities against
KSHV-infected PEL, but not KSHV-uninfected B-lymphoma.
ER stress, such as ROS, misfolded proteins and
denatured protein generation, leads to the UPR, which induces the
expression of ER chaperones and ER-related molecules to facilitate
the refolding or the degradation of misfolded proteins and to
suppress the translation of de novo proteins. UPR is
primarily regulated by GRP78, and consists of 3 pathways involving
ATF6α, IRE1α and PERK (45). Under
normal conditions, GRP78 interacts with and inactivates ATF6α,
IRE1α and PERK, but dissociates from these during ER stress, which
activates these pathways. In an ER stress-dependent manner,
ER-localized ATF6α is translocated into the Golgi apparatus and is
cleaved. Cleaved ATF6α (p50 ATF6α) is then translocated to the
nucleus, which in turn activates the transcriptional expression of
ER chaperones, such as GRP78, GRP94, XBP1, EDEM1/2 and ATF6α itself
(45,46). IRE1 induces the splicing of
immature XBP1 mRNA, and the translated XBP1 protein activates the
transcription of GRP78 and XBP1 itself. PERK phosphorylates eIF2α,
and induces a translation block. However, in the presence of severe
ER stress, pro-apoptotic UPR is activated. Pro-apoptotic UPR
activates the expression of the transcription factor, CHOP, which
transcriptionally upregulates GADD34 and pro-apoptotic Bcl-2
proteins, leading to apoptosis (45,46).
As for the association between arctigenin and UPR, arctigenin has
been shown to induce ROS production (25,26,36)
and ER stress along with the UPR (30). By contrast, arctigenin has been
reported to inhibit UPR and alleviate brefeldin A-induced ER stress
by activating AMP-activated protein kinase (51). Furthermore, arctigenin has been
known to inhibit the UPR by the transcriptional suppression of
GRP78, GRP94 or CHOP, resulting in a reduction of ER stress,
pro-apoptotic UPR or apoptosis (28,29,51).
These observations are partially in agreement with the findings of
the present study on glucose-deprived PEL cells, as we observed an
upregulation in GRP94, GADD34 and CHOP expression, and a
downregulation in GRP78 and ATF6α expression. These discrepancies
may be due to differences in KSHV infection or genetic backgrounds
of the tested cells. Indeed, we have previously found that the mRNA
expression levels of IRE1α and PERK are decreased in PEL cells
compared with KSHV-uninfected cells under normal culture
conditions, and KSHV-encoded LANA and v-cyclin D suppressed IRE1α
transcription (15). In this
study, we found that arctigenin suppressed the phosphorylation
(i.e., the activation) of p38 MAPK and the expression of GRP78,
ATF6α and p50 ATF6α in glucose-deprived PEL cells (Figs. 4 and 6). In addition, a p38 MAPK inhibitor also
suppressed cell growth (Fig. 5B and
C) and the promoter activities of the GRP78 and ATF6α genes
(Fig. 6B and C) in
glucose-deprived cells. These findings are consistent with previous
studies, which indicate that the activation of p38 MAPK upregulates
GRP78 expression (60,61), induces the phosphorylation and
cleavage of ATF6α, and then activates p50 ATF6α in an
ATF6α-phosphorylation-dependent manner (43,44).
It can thus be hypothesized that arctigenin suppresses the p38
MAPK-mediated ATF6α activation (or cleavage), which in turn
suppresses the ATF6α-mediated transcription of GRP78 and ATF6α
itself. These disruptions of UPR can evoke severe ER stress and
subsequent apoptosis through pro-apoptotic UPR activation,
including CHOP and GADD34 expression in PEL cells (Fig. 6A).
In conclusion, the findings of this study
demonstrate that arctigenin treatment induces mitochondrial
disruption, the suppression of the ERK and p38 MAPK pathways, and
the downregulation of ATF6α and GRP78 expression in
glucose-deprived PEL cells. These phenotypes of arctigenin can
serve as cytotoxic effectors in glucose-deprived PEL cells.
Therefore, PEL cells are more sensitive to the anti-proliferative
effects of arctigenin than KSHV-uninfected cells, thereby raising
the possibility that arctigenin may serve as a novel therapeutic
agent in the treatment of KSHV-associated lymphoma.
Acknowledgments
This study was supported in part by a Grant-in-Aid
for Scientific Research (C) (15K07952), Young Scientists (B)
(16K18925) and Strategic Research Foundation at Private
Universities (S1311035) from MEXT of Japan, and by the Japan Agency
for Medical Research and Development (16fk0410107h0201). We would
like to thank Dr Peter Gee (Kyoto University, Kyoto, Japan) for
critically proofreading the manuscript.
Abbreviations:
|
ATF6α
|
activating transcription factor
6α
|
|
ATP
|
adenosine triphosphate
|
|
EDEM
|
ER degradation enhancing
α-mannosidase-like protein
|
|
ER
|
endoplasmic reticulum
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
GRP78
|
glucose-regulated protein 78
|
|
BiP
|
immunoglobulin heavy-chain-binding
protein
|
|
JNK
|
c-Jun N-terminal kinase
|
|
KSHV
|
Kaposi's sarcoma-associated
herpesvirus
|
|
NaB
|
sodium butyrate
|
|
p38 MAPK
|
p38 mitogen-activated protein
kinase
|
|
PEL
|
primary effusion lymphoma
|
|
UPR
|
unfolded protein response
|
References
|
1
|
Russo JJ, Bohenzky RA, Chien MC, Chen J,
Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, et
al: Nucleotide sequence of the Kaposi sarcoma-associated
herpesvirus (HHV8). Proc Natl Acad Sci USA. 93:14862–14867. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nador RG, Cesarman E, Chadburn A, Dawson
DB, Ansari MQ, Sald J and Knowles DM: Primary effusion lymphoma: A
distinct clinicopathologic entity associated with the Kaposi's
sarcoma-associated herpes virus. Blood. 88:645–656. 1996.PubMed/NCBI
|
|
3
|
Antar A, El Hajj H, Jabbour M, Khalifeh I,
El-Merhi F, Mahfouz R and Bazarbachi A: Primary effusion lymphoma
in an elderly patient effectively treated by lenalidomide: Case
report and review of literature. Blood Cancer J. 4:e1902014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boulanger E, Gérard L, Gabarre J, Molina
JM, Rapp C, Abino JF, Cadranel J, Chevret S and Oksenhendler E:
Prognostic factors and outcome of human herpesvirus 8-associated
primary effusion lymphoma in patients with AIDS. J Clin Oncol.
23:4372–4380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carbone A and Gloghini A:
KSHV/HHV8-associated lymphomas. Br J Haematol. 140:13–24. 2008.
|
|
6
|
Okada S, Goto H and Yotsumoto M: Current
status of treatment for primary effusion lymphoma. Intractable Rare
Dis Res. 3:65–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Damania B and Cesarman E: Kaposi's
sarcoma-associated herpesvirus. Fields Virology. Knipe DM and
Howley PM: 2. 6th edition. Lippincott Williams & Wilkins;
Philadelphia: pp. 2080–2128. 2013
|
|
8
|
Schulz TF and Cesarman E: Kaposi
Sarcoma-associated Herpesvirus: Mechanisms of oncogenesis. Curr
Opin Virol. 14:116–128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fujimuro M, Wu FY, ApRhys C, Kajumbula H,
Young DB, Hayward GS and Hayward SD: A novel viral mechanism for
dysregulation of beta-catenin in Kaposi's sarcoma-associated
herpesvirus latency. Nat Med. 9:300–306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ashizawa A, Higashi C, Masuda K, Ohga R,
Taira T and Fujimuro M: The Ubiquitin system and Kaposi's
sarcoma-associated herpesvirus. Front Microbiol. 3:662012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F
and Miller G: A viral gene that activates lytic cycle expression of
Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci USA.
95:10866–10871. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shigemi Z, Furukawa Y, Hosokawa K, Minami
S, Matsuhiro J, Nakata S, Watanabe T, Kagawa H, Nakagawa K, Takeda
H, et al: Diallyl trisulfide induces apoptosis by suppressing NF-κB
signaling through destabilization of TRAF6 in primary effusion
lymphoma. Int J Oncol. 48:293–304. 2016. View Article : Google Scholar
|
|
13
|
Watanabe T, Nakamura S, Ono T, Ui S, Yagi
S, Kagawa H, Watanabe H, Ohe T, Mashino T and Fujimuro M:
Pyrrolidinium fullerene induces apoptosis by activation of
procaspase-9 via suppression of Akt in primary effusion lymphoma.
Biochem Biophys Res Commun. 451:93–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wakao K, Watanabe T, Takadama T, Ui S,
Shigemi Z, Kagawa H, Higashi C, Ohga R, Taira T and Fujimuro M:
Sangivamycin induces apoptosis by suppressing Erk signaling in
primary effusion lymphoma cells. Biochem Biophys Res Commun.
444:135–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shigemi Z, Baba Y, Hara N, Matsuhiro J,
Kagawa H, Watanabe T and Fujimuro M: Effects of ER stress on
unfolded protein responses, cell survival, and viral replication in
primary effusion lymphoma. Biochem Biophys Res Commun. 469:565–572.
2016. View Article : Google Scholar
|
|
16
|
Shigemi Z, Manabe K, Hara N, Baba Y,
Hosokawa K, Kagawa H, Watanabe T and Fujimuro M: Methylseleninic
acid and sodium selenite induce severe ER stress and subsequent
apoptosis through UPR activation in PEL cells. Chem Biol Interact.
266:28–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bastos JK, Carvalho JC, de Souza GH,
Pedrazzi AH and Sarti SJ: Anti-inflammatory activity of cubebin, a
lignan from the leaves of Zanthoxyllum naranjillo Griseb. J
Ethnopharmacol. 75:279–282. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Swarup V, Ghosh J, Mishra MK and Basu A:
Novel strategy for treatment of Japanese encephalitis using
arctigenin, a plant lignan. J Antimicrob Chemother. 61:679–688.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayashi K, Narutaki K, Nagaoka Y, Hayashi
T and Uesato S: Therapeutic effect of arctiin and arctigenin in
immunocompetent and immunocompromised mice infected with influenza
A virus. Biol Pharm Bull. 33:1199–1205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jang YP, Kim SR, Choi YH, Kim J, Kim SG,
Markelonis GJ, Oh TH and Kim YC: Arctigenin protects cultured
cortical neurons from glutamate-induced neurodegeneration by
binding to kainate receptor. J Neurosci Res. 68:233–240. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang SL, Yu RT, Gong J, Feng Y, Dai YL,
Hu F, Hu YH, Tao YD and Leng Y: Arctigenin, a natural compound,
activates AMP-activated protein kinase via inhibition of
mitochondria complex I and ameliorates metabolic disorders in ob/ob
mice. Diabetologia. 55:1469–1481. 2012. View Article : Google Scholar
|
|
22
|
Yao X, Zhu F, Zhao Z, Liu C, Luo L and Yin
Z: Arctigenin enhances chemosensitivity of cancer cells to
cisplatin through inhibition of the STAT3 signaling pathway. J Cell
Biochem. 112:2837–2849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang X, Zeng L, Huang J, Zhou H and Liu
Y: Arctigenin, a natural lignan compound, induces apoptotic death
of hepatocellular carcinoma cells via suppression of PI3-K/Akt
signaling. J Biochem Mol Toxicol. 29:458–464. 2015. View Article : Google Scholar
|
|
24
|
Maxwell T, Chun SY, Lee KS, Kim S and Nam
KS: The anti-metastatic effects of the phytoestrogen arctigenin on
human breast cancer cell lines regardless of the status of ER
expression. Int J Oncol. 50:727–735. 2017. View Article : Google Scholar
|
|
25
|
Li QC, Liang Y, Tian Y and Hu GR:
Arctigenin induces apoptosis in colon cancer cells through
ROS/p38MAPK pathway. J BUON. 21:87–94. 2016.PubMed/NCBI
|
|
26
|
Hsieh CJ, Kuo PL, Hsu YC, Huang YF, Tsai
EM and Hsu YL: Arctigenin, a dietary phytoestrogen, induces
apoptosis of estrogen receptor-negative breast cancer cells through
the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic Biol
Med. 67:159–170. 2014. View Article : Google Scholar
|
|
27
|
Zhang M, Cai S, Zuo B, Gong W, Tang Z,
Zhou D, Weng M, Qin Y, Wang S, Liu J, et al: Arctigenin induced
gallbladder cancer senescence through modulating epidermal growth
factor receptor pathway. Tumour Biol.
39:10104283176983592017.PubMed/NCBI
|
|
28
|
Kim JY, Hwang JH, Cha MR, Yoon MY, Son ES,
Tomida A, Ko B, Song SW, Shin-ya K, Hwang YI, et al: Arctigenin
blocks the unfolded protein response and shows therapeutic
antitumor activity. J Cell Physiol. 224:33–40. 2010.PubMed/NCBI
|
|
29
|
Sun S, Wang X, Wang C, Nawaz A, Wei W, Li
J, Wang L and Yu DH: Arctigenin suppresses unfolded protein
response and sensitizes glucose deprivation-mediated cytotoxicity
of cancer cells. Planta Med. 77:141–145. 2011. View Article : Google Scholar
|
|
30
|
Brecht K, Riebel V, Couttet P, Paech F,
Wolf A, Chibout SD, Pognan F, Krähenbühl S and Uteng M: Mechanistic
insights into selective killing of OXPHOS-dependent cancer cells by
arctigenin. Toxicol In Vitro. 40:55–65. 2017. View Article : Google Scholar
|
|
31
|
Jeong JB, Hong SC, Jeong HJ and Koo JS:
Arctigenin induces cell cycle arrest by blocking the
phosphorylation of Rb via the modulation of cell cycle regulatory
proteins in human gastric cancer cells. Int Immunopharmacol.
11:1573–1577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kudou N, Taniguchi A, Sugimoto K, Matsuya
Y, Kawasaki M, Toyooka N, Miyoshi C, Awale S, Dibwe DF, Esumi H, et
al: Synthesis and antitumor evaluation of arctigenin derivatives
based on antiausterity strategy. Eur J Med Chem. 60:76–88. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang K, Li LA, Meng YG, You YQ, Fu XY and
Song L: Arctigenin promotes apoptosis in ovarian cancer cells via
the iNOS/NO/STAT3/survivin signalling. Basic Clin Pharmacol
Toxicol. 115:507–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Awale S, Lu J, Kalauni SK, Kurashima Y,
Tezuka Y, Kadota S and Esumi H: Identification of arctigenin as an
antitumor agent having the ability to eliminate the tolerance of
cancer cells to nutrient starvation. Cancer Res. 66:1751–1757.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ikeda M, Sato A, Mochizuki N, Toyosaki K,
Miyoshi C, Fujioka R, Mitsunaga S, Ohno I, Hashimoto Y, Takahashi
H, et al: Phase I trial of GBS-01 for advanced pancreatic cancer
refractory to gemcitabine. Cancer Sci. 107:1818–1824. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gu Y, Qi C, Sun X, Ma X, Zhang H, Hu L,
Yuan J and Yu Q: Arctigenin preferentially induces tumor cell death
under glucose deprivation by inhibiting cellular energy metabolism.
Biochem Pharmacol. 84:468–476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen C and Okayama H: High-efficiency
transformation of mammalian cells by plasmid DNA. Mol Cell Biol.
7:2745–2752. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Takayanagi S, Fukuda R, Takeuchi Y,
Tsukada S and Yoshida K: Gene regulatory network of unfolded
protein response genes in endoplasmic reticulum stress. Cell Stress
Chaperones. 18:11–23. 2013. View Article : Google Scholar :
|
|
39
|
Xie J, Pan H, Yoo S and Gao SJ: Kaposi's
sarcoma-associated herpesvirus induction of AP-1 and interleukin 6
during primary infection mediated by multiple mitogen-activated
protein kinase pathways. J Virol. 79:15027–15037. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sharma-Walia N, Patel K, Chandran K,
Marginean A, Bottero V, Kerur N and Paul AG: COX-2/GE2: Molecular
ambassadors of Kaposi's sarcoma-associated herpes virus
oncoprotein-v-FLIP. Oncogenesis. 1:e52012. View Article : Google Scholar
|
|
41
|
Sharma-Walia N, Krishnan HH, Naranatt PP,
Zeng L, Smith MS and Chandran B: ERK1/2 and MEK1/2 induced by
Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early
during infection of target cells are essential for expression of
viral genes and for establishment of infection. J Virol.
79:10308–10329. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pan H, Xie J, Ye F and Gao SJ: Modulation
of Kaposi's sarcoma-associated herpesvirus infection and
replication by MEK/ERK, JNK, and p38 multiple mitogen-activated
protein kinase pathways during primary infection. J Virol.
80:5371–5382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gade P, Manjegowda SB, Nallar SC, Maachani
UB, Cross AS and Kalvakolanu DV: Regulation of the death-associated
protein kinase 1 expression and autophagy via ATF6 requires
apoptosis signal-regulating kinase 1. Mol Cell Biol. 34:4033–4048.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Egawa N, Yamamoto K, Inoue H, Hikawa R,
Nishi K, Mori K and Takahashi R: The endoplasmic reticulum stress
sensor, ATF6α, protects against neurotoxin-induced dopaminergic
neuronal death. J Biol Chem. 286:7947–7957. 2011. View Article : Google Scholar
|
|
45
|
Wang M and Kaufman RJ: Protein misfolding
in the endoplasmic reticulum as a conduit to human disease. Nature.
529:326–335. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sovolyova N, Healy S, Samali A and Logue
SE: Stressed to death - mechanisms of ER stress-induced cell death.
Biol Chem. 395:1–13. 2014. View Article : Google Scholar
|
|
47
|
Li X, Feng J and Sun R: Oxidative stress
induces reactivation of Kaposi's sarcoma-associated herpesvirus and
death of primary effusion lymphoma cells. J Virol. 85:715–724.
2011. View Article : Google Scholar :
|
|
48
|
Leung HJ, Duran EM, Kurtoglu M, Andreansky
S, Lampidis TJ and Mesri EA: Activation of the unfolded protein
response by 2-deoxy-D-glucose inhibits Kaposi's sarcoma-associated
herpesvirus replication and gene expression. Antimicrob Agents
Chemother. 56:5794–5803. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liao G, Cai J, Yue C and Qing X:
Extracavitary/solid variant of primary effusion lymphoma presenting
as a gastric mass. Exp Mol Pathol. 99:445–448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Katz H, Rose C, Rivera NT and Bray N:
HIV-associated primary effusion lymphoma presenting as a
paracardial mass. BMJ Case Rep. 2015.bcr20142087182015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gu Y, Sun XX, Ye JM, He L, Yan SS, Zhang
HH, Hu LH, Yuan JY and Yu Q: Arctigenin alleviates ER stress via
activating AMPK. Acta Pharmacol Sin. 33:941–952. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ichijo H: From receptors to
stress-activated MAP kinases. Oncogene. 18:6087–6093. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cho MK, Jang YP, Kim YC and Kim SG:
Arctigenin, a phenylpropanoid dibenzylbutyrolactone lignan,
inhibits MAP kinases and AP-1 activation via potent MKK inhibition:
The role in TNF-alpha inhibition. Int Immunopharmacol. 4:1419–1429.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee JY, Cho BJ, Park TW, Park BE, Kim SJ,
Sim SS and Kim CJ: Dibenzylbutyrolactone lignans from Forsythia
koreana fruits attenuate lipopolysaccharide-induced inducible
nitric oxide synthetase and cyclooxygenase-2 expressions through
activation of nuclear factor-κB and mitogen-activated protein
kinase in RAW264.7 cells. Biol Pharm Bull. 33:1847–1853. 2010.
View Article : Google Scholar
|
|
55
|
Li X, Chen S, Feng J, Deng H and Sun R:
Myc is required for the maintenance of Kaposi's sarcoma-associated
herpesvirus latency. J Virol. 84:8945–8948. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bais C, Santomasso B, Coso O, Arvanitakis
L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gershengorn MC and
Mesri EA: G-protein-coupled receptor of Kaposi's sarcoma-associated
herpesvirus is a viral oncogene and angiogenesis activator. Nature.
391:86–89. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sadagopan S, Sharma-Walia N, Veettil MV,
Raghu H, Sivakumar R, Bottero V and Chandran B: Kaposi's
sarcoma-associated herpesvirus induces sustained NF-kappaB
activation during de novo infection of primary human dermal
microvascular endothelial cells that is essential for viral gene
expression. J Virol. 81:3949–3968. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ye F, Zhou F, Bedolla RG, Jones T, Lei X,
Kang T, Guadalupe M and Gao SJ: Reactive oxygen species hydrogen
peroxide mediates Kaposi's sarcoma-associated herpesvirus
reactivation from latency. PLoS Pathog. 7:e10020542011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Naranatt PP, Akula SM, Zien CA, Krishnan
HH and Chandran B: Kaposi's sarcoma-associated herpesvirus induces
the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling
pathway in target cells early during infection: Implications for
infectivity. J Virol. 77:1524–1539. 2003. View Article : Google Scholar :
|
|
60
|
Ranganathan AC, Zhang L, Adam AP and
Aguirre-Ghiso JA: Functional coupling of p38-induced up-regulation
of BiP and activation of RNA-dependent protein kinase-like
endoplasmic reticulum kinase to drug resistance of dormant
carcinoma cells. Cancer Res. 66:1702–1711. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
He B, Luo B, Chen Q and Zhang L: Cigarette
smoke extract induces the expression of GRP78 in A549 cells via the
p38/MAPK pathway. Mol Med Rep. 8:1683–1688. 2013. View Article : Google Scholar : PubMed/NCBI
|