Introduction
Despite the remarkable progress made in cancer
biology and technologies for cancer treatment over the past 50
years, malignant neoplasms remain highly threatening diseases for
humans. Malignant neoplasms at the initial stage are curable;
however, once they have progressed, they become invasive,
metastatic and highly resistant to multi-disciplinary treatments,
including chemo-, radio- and immunotherapy. Malignant melanoma and
osteosarcoma are representatives of malignant tumors that are
highly resistant to multiple anticancer drugs (1–4).
Tumor necrosis factor-related apoptosis-inducing ligand
(Apo2L/TRAIL), a member of the tumor necrosis factor superfamily,
is a promising cancer-selective anticancer drug, since it exhibits
potent cytotoxicity toward various cancer cell types with minimal
cytotoxicity toward non-transformed cells (5–8).
TRAIL triggers the extrinsic and intrinsic apoptotic pathways by
binding to two death receptors (DRs), TRAIL receptor (TRAIL-R)1/DR4
and TRAIL-R2/DR5 (9,10). Disconcertingly, however, current
clinical trials have revealed that several cancer cell types,
including melanoma and osteosarcoma are resistant to TRAIL, despite
expressing DRs (11–15). In addition to their inherent
resistance, the acquired resistance to the drug dampens effective
TRAIL treatment.
Recently, non-thermal (cold) atmospheric plasma
(CAP) has emerged as a novel promising anticancer tool, since it
has potent antitumor activity. CAP irradiation inhibits cell
proliferation, migration and invasion, and triggers different cell
death modalities, including apoptosis, necrosis and autophagy in
vitro in various cancer cell lines and primary cancerous cells
and tissues (16–23). CAP irradiation also reduces the
growth of xenografted tumors in vivo (24). Moreover, CAP irradiation is
tumor-selective under the optimal conditions (16,17,20).
However, the outreach of CAP is very limited so that its primary
targets may be limited to cancerous surface tissues. More recently,
various types of plasma-stimulated medium (PSM) have been generated
from culture medium, buffers and water. PSM has emerged as an
alternative tool for cancer treatment, since similar to direct CAP
irradiation, it exhibits potent cytotoxicity toward various
malignant cells, such as glioblastoma, ovarian, gastric and
pancreatic cancers, while causing minimal damage to normal cell
counterparts under optimal conditions (25–29).
PSM seems to affect a wider range of cancers than CAP irradiation,
as it can be readily administered systematically or locally to deep
tissues.
Ca2+ is an essential intracellular second
messenger whose level is tightly regulated. The finely and
spatiotemporal tuning of Ca2+ leads to short and
synchronized Ca2+ waves, which are primarily essential
for energy production, cell function and survival (30). However, a significant and
persistent increase in Ca2+ is a master cause of cell
death. An excess rise in the mitochondrial Ca2+
concentration ([Ca2+]mit), so-called
mitochondrial Ca2+ overload, can cause both necrosis and
apoptosis; this results in the increased permeability of the inner
mitochondrial membrane, mitochondrial permeability transition
(MPT). MPT, in turn, leads to a rapid collapse of mitochondrial
membrane potential, the loss of ATP and the osmotic rupture of the
outer mitochondrial membrane. Ultimately, the loss of ATP and the
fall of the mitochondrial integrity lead to necrosis (30,31).
In addition, the rupture of the outer mitochondrial membrane can
result in the release of different pro-apoptotic proteins, such as
cytochrome c and apoptosis-inducing factor (32,33),
thereby leading to apoptosis. Recent evidence suggests that
Ca2+ also plays a regulatory role in other cell death
modalities, such as autophagy and anoikis (34). Furthermore, different cancer cell
types exhibit tumor-specific traits in Ca2+ dynamics,
which contribute to tumorigenesis, malignant phenotypes, drug
resistance, increased proliferation, and evasion from apoptosis and
survival (35). Thus,
Ca2+ is emerging as a novel target for cancer treatment
(36,37).
Mitochondria are highly dynamic organelles with a
reticular network organization that is regulated by the delicate
balance between the fission and fusion of the mitochondrial
membrane. The mitochondrial network is critical for cell function
and apoptosis (38,39), since a defect in either fission or
fusion causes severe mitochondrial and cellular dysfunctions.
Mitochondrial fission helps to eliminate damaged mitochondria
through mitophagy (40).
Accordingly, the disruption of mitochondrial fission leads to an
extensively interconnected and collapsed mitochondrial network and
defects in mitochondrial quality control. Moreover, mitochondrial
fusion facilitates the exchange of mitochondrial DNA and
metabolites required for mitochondrial function. Consequently,
defects in mitochondrial fusion lead to mitochondrial fragmentation
and the loss of mitochondrial DNA, reduced growth, decreased
mitochondrial membrane potential (also known as ΔΨm) and
defective respiration (41,42).
A series of our earlier studies have revealed the importance of the
mitochondrial network dynamics in melanoma and osteosarcoma cells.
We have previously demonstrated that cell killing by TRAIL or PSM,
as well as sensitization to either insult is preceded by
mitochondrial network alterations, such as excessive mitochondrial
fragmentation and clustering or hyperfusion (43–45).
Moreover, we found several critical regulators of mitochondrial
morphology. One key regulator is plasma membrane depolarization
(PMD). Persistent PMD is essential for the progression of
mitochondrial fragmentation and clustering (46). The other regulator is
Ca2+ since mitochondria Ca2+
([Ca2+]mit) overload leads to mitochondrial
fragmentation, while [Ca2+]mit depletion
results in mitochondrial hyperfusion (46,47).
TRAIL and CAP/PSM share several biochemical and
biological properties, including the production of, and regulation
by reactive oxygen/nitrogen species (RONS), the induction of
apoptosis via the intrinsic pathway, and high tumor-selective
cytotoxicity (19,21,45).
The advantages of PSM over TRAIL may provide a significant driving
force in its development as a novel tool for cancer treatment.
However, at present, it is unclear as to whether PSM is more
efficient than TRAIL, since, at least to the best of our knowledge,
there is no available literature comparing their antitumor activity
under given conditions between them. Therefore, in the present
study, we systematically compared their ability to kill cancerous
cells, disrupt the mitochondrial network dynamics, and to modulate
pro-death events between TRAIL and PSM. We found that PSM evoked
caspase-independent cell death and efficiently killed different
cancer cell types that were highly tolerant to TRAIL. We also found
that PSM had a greater capacity to evoke mitochondrial network
aberration, PMD and Ca2+ dynamics dyshomeostasis.
Materials and methods
Materials
Soluble recombinant human TRAIL was obtained from
Enzo Life Sciences (San Diego, CA, USA). The general caspase
inhibitor, z-VAD-FMK, and caspase-3/7-specific inhibitor,
z-DEVD-FMK, were purchased from Merck Japan (Tokyo, Japan).
Glibenclamide, FCCP, antimycin A and U37883A were from
Sigma-Aldrich (St. Louis, MO, USA). Potassium chloride (KCl) was
obtained from Wako Pure Chemicals (Osaka, Japan). Insoluble
reagents (z-VAD-FMK, z-DEVD-FMK, glibenclamide, FCCP, antimycin A
and U37883A) were dissolved in dimethyl sulfoxide (DMSO) and
diluted with high glucose-containing Dulbecco’s modified Eagle’s
medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine
serum (FBS; Sigma-Aldrich) or Hank’s balanced salt solution (HBSS)
(pH 7.4) to a final concentration of <0.1% prior to use. The
manganese-porphyrin superoxide dismutase mimetic MnTBaP (Enzo Life
Sciences) were dissolved in 1 mM NaOH (pH 8.0) and added HBSS to
lower pH 7.4.
Cell culture
The following cell lines were used in the present
study: Human malignant melanoma cells (A2058, cell number
IFO50276), human osteosarcoma cells (MG63, cell number IFO50108)
(both from the Health Science Research Resource Bank, Osaka,
Japan), human neuroblastoma cells (NB-1, cell number RCB1953 and
SK-N-SH, cell number RCB0426), osteosarcoma cells (143B, cell
number RCB0701; SAOS-2, cell number RCB0428; and HOS, cell number
RCB0992), murine osteosarcoma cells (LM8, cell number RCB1450) and
murine osteoblasts (MC3T3-E1, cell number RCB1126) (all from the
Riken BioResource Center, Tukuba, Japan). It should be noted that
the 143B cell line is identical to the osteosarcoma HTK cell line
(http://web.expasy.org/cellosaurus/CVCL_2270). However,
as the HTK cell line is also an osteosarcoma cell line, it was
deemed that this would not affect the outcome of this study. In
addition, human malignant melanoma cells (A375, ATCC®
CRL 1619™) and immortalized osteoblast cells (hFOB1.19,
ATCC® CRL 11372™) were obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA) and the murine
osteosarcoma K7M3 cells, originally derived from a primary
osteosarcoma in a BALB/c mouse were generously provided by Dr
Eugenie Kleinerman (Department of Pediatrics, University of Texas
MD Anderson Cancer Center, Houston, TX, USA). Human dermal
fibroblasts (HDFs) from the facial dermis were obtained from Cell
Applications (San Diego, CA, USA). All cells, apart from the hFOB
were cultured in 10% FBS/DMEM and streptomycin and penicillin (5000
units/ml) and streptomycin (5000 µg/ml) (Pen-Strep; Thermo
Fisher Scientific Japan, Tokyo, Japan) in a 95% air/5%
CO2 humidified atmosphere. The hFOB cell line was
cultured according to the ATCC protocol. The cells were harvested
by incubation with 0.25% trypsin-EDTA (Thermo Fisher Scientific
Japan) for 5 min at 37°C.
PSM preparation
CAP was generated using an originally-developed
low-frequency plasma jet device equipped with an asymmetrical
dielectric barrier discharge, as previously described (45). The typical frequency was 20 kHz,
with a peak voltage of 8 kV, a current of 20 mA and helium flow
rate of 300 ml/min. PSM was prepared by irradiating 1 ml of DMEM
with CAP for 5 min once. The original PSM was diluted to a final
concentration of 12.5–50% with 10% FBS/DMEM (for cell experiments)
or HBSS (for biochemical experiments) and indicated as PSM
(12.5–50%).
Cell viability assay
Cell viability was measured by WST-8 assay, a
colorimetric assay based on the formation of a water-soluble
formazan product assay using the Cell Counting Reagent SF (Nacalai
Tesque, Kyoto, Japan) as previously described (46). The cells were seeded at a density
of 2×103 cells/well in 96-well plates (Corning, New
York, NY, USA) and incubated for 24 or 72 h with 100 ng/ml TRAIL
and PSM (3.1, 6.3, 12.5 and 25.50%) alone or in combination prior
to the addition of 10 µl of cell counting reagent SF and
further incubation for 1 h. The absorbances were measured at 450 nm
using an ARVO MX microplate reader (Perkin-Elmer, Waltham, MA,
USA).
Caspase-3/7 activation, membrane
integrity and cell death assay
Caspase-3/7 activation, membrane integrity and cell
death were simultaneously measured using a Muse™ Cell Analyzer
(Merck Millipore, Darmstadt, Germany) with the Muse™ Caspase-3/7
kit as previously described (46).
Briefly, the cells (1×105/ml) in 24-well plates were
treated with the agents to be tested 100 ng/ml TRAIL and PSM (25
and 100%) alone or in combination in the absence or presence of 10
µM Z-VAD-FMK or 30 µM MnTBaP for 24 h in 10% FBS/DMEM
at 37°C and then stained with a novel Caspase-3/7 reagent NucView™
and 7-amino-actinomycin D (7-AAD). 7-AAD is a dead cell marker that
is excluded from healthy and early apoptotic cells, while it
permeates late apoptotic and dead cells. Accordingly, 4 cell
populations can be distinguished by this method: Live cells,
caspase−/7-AAD−; early apoptotic cells,
caspase+/7-AAD−; late apoptotic/dead cells,
caspase+/7-AAD+; necrotic cells,
caspase-/7-AAD+.
Live-cell mitochondrial network
imaging
The mitochondrial network was analyzed as previously
described (44). Briefly, cells in
FBS/DMEM (3×104/well) adherent on an 8-well chambered
coverslips were treated with the agents to be tested 100 ng/ml
TRAIL and PSM (25%) alone or in combination in the absence or
presence of 10 µM Z-VAD-FMK or 30 µM MnTBaP for 24 h
at 37°C in a 5% CO2 incubator. After removing the medium
by aspiration, the cells were washed with fresh FBS/DMEM and
stained with 20 nM MitoTracker Red CMXRos (Thermo Fisher Scientific
Japan) and 1 µg/ml Hoechst 33342 (Dojindo Laboratories,
Kumamoto, Kumamoto, Japan) for 1 h at 37°C in the dark in a 5%
CO2 incubator. The cells were then washed with and
immersed in FluoroBrite™ DMEM (Thermo Fisher Scientific Japan).
Images were obtained using a BZ X-700 Fluorescence Microscope
(Keyence, Osaka, Japan) equipped with a X100, 1.40 n.a. UPlanSApo
Super-Apochromat, coverslip-corrected oil objective (Olympus,
Tokyo, Japan). Images were analyzed using BZ-H3A application
software (Keyence) and free NIH ImageJ software (NIH, Bethesda, MD,
USA).
Measurument of PMD
PMD was measured using an anionic bis-oxonol
voltage-sensitive dye DiBAC4(3). Briefly, the cells (1×106
cells/ml) suspended in HBSS were incubated with 5 µM
DiBAC4(3) (Dojindo
Laboratories) for 40 min at 37°C for loading. The cells were
washed, resuspended in HBSS, and measured for their fluorescence
for 1, 3, 5 and 10 min using a microplate fluorescence reader
(Fluoroskan Ascent; Thermo Fisher Scientific Japan) with excitation
and emission at 485 and 538 nm. The data are expressed as the
fluorescence intensity normalized (per 1×106 cells).
Measurment of Ca2+
concentrations
Changes in the cytosolic Ca2+
([Ca2+]cyt) and
[Ca2+]mit levels were measured using Fluo
4-AM and rhod 2-AM (Dojindo Laboratories), respectively as
previously described (46,47). For the improvement of mitochondrial
localization of rhod 2-AM, it was reduced to the colorless,
non-fluorescent dihydrorhod 2-AM by sodium borohydride, according
to the manufacturer’s instructions. The cells were loaded with 4
µM each of Fluo 4-AM or dihydrorhod 2-AM for 40 min at 37°C,
washed with HBSS. Subsequently, the cells (1×106/ml)
were resuspended in HBSS in 96-well plates. The cells were then
manually supplemented with the agents to be tested PSM (12.5–50%),
100 ng/ml TRAIL, 5 mM KCl, 100 µM glibenclamide, 100
µM U37883A alone or in combination for 1, 3, 5 and 10 min.
The cells were then measured for fluorescence using a microplate
reader (Fluoroskan Ascent) with excitation and emission at 485 and
538 nm (for Fluo 4-AM) and 542 and 592 nm (for rhod 2-AM),
respectively. For the analyses of Ca2+ release and
store-operated Ca2+ entry (SOCE), Fluo4-AM-loaded cells
were suspended in a Ca2+-free medium (HBSS supplemented
with 1 mM CaCl2) and treated with 1 µM
thapsigargin (Tg) for 10 min and then added with 2 mM
CaCl2. The fluorescence was measured as described above
with excitation and emission at 485 and 538 nm.
Statistical analysis
Data were analyzed by one-way analysis of variance
followed by Tukey’s post hoc test using an add-in software for
Excel 2016 for Windows (SSRI, Tokyo, Japan). All values are
expressed as the means ± SD and a value of P<0.05 was considered
to indicate a statistically significant difference.
Results
The PSM kills TRAIL-resistant human
malignant cells with different origins
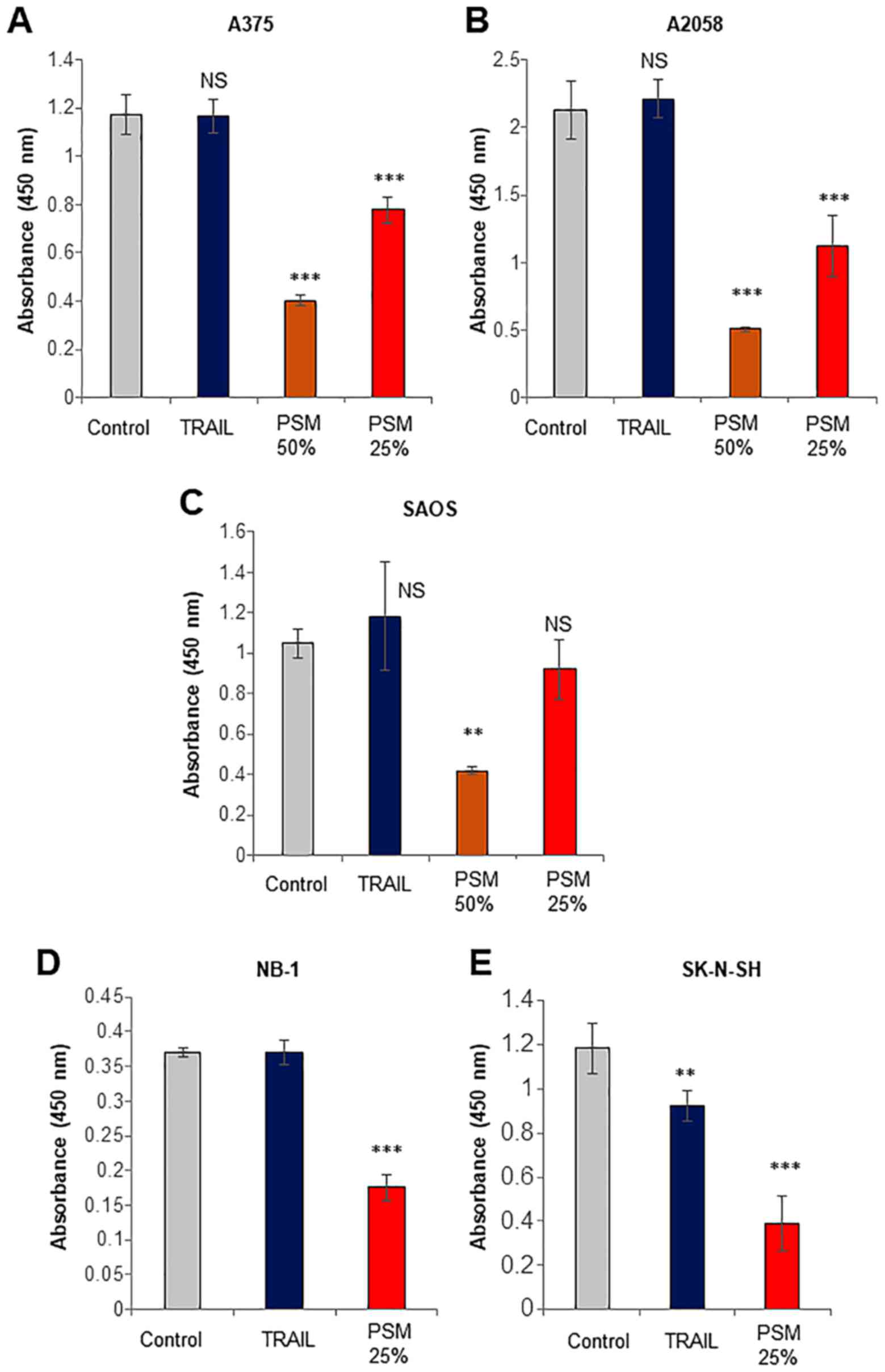
First, we systematically compared the capacity of
the PSM to kill cancerous cells with that of TRAIL. TRAIL primarily
causes cell death by apoptosis in various tumor cell types,
including malignant melanoma and osteosarcoma cells (9–15).
Treatment with TRAIL at ≤100 ng/ml for 72 h resulted in a minimal
decrease in the viability of A375, A2058, SAOS-2, NB-1 and SK-N-SH
cells (Fig. 1), whereas, PSM
(≥25%) decreased the viability of the cells. The effect of the PSM
was observed as early as 24 h (data not shown). These results
indicate that PSM can kill TRAIL-resistant human malignant cells
with different origins.
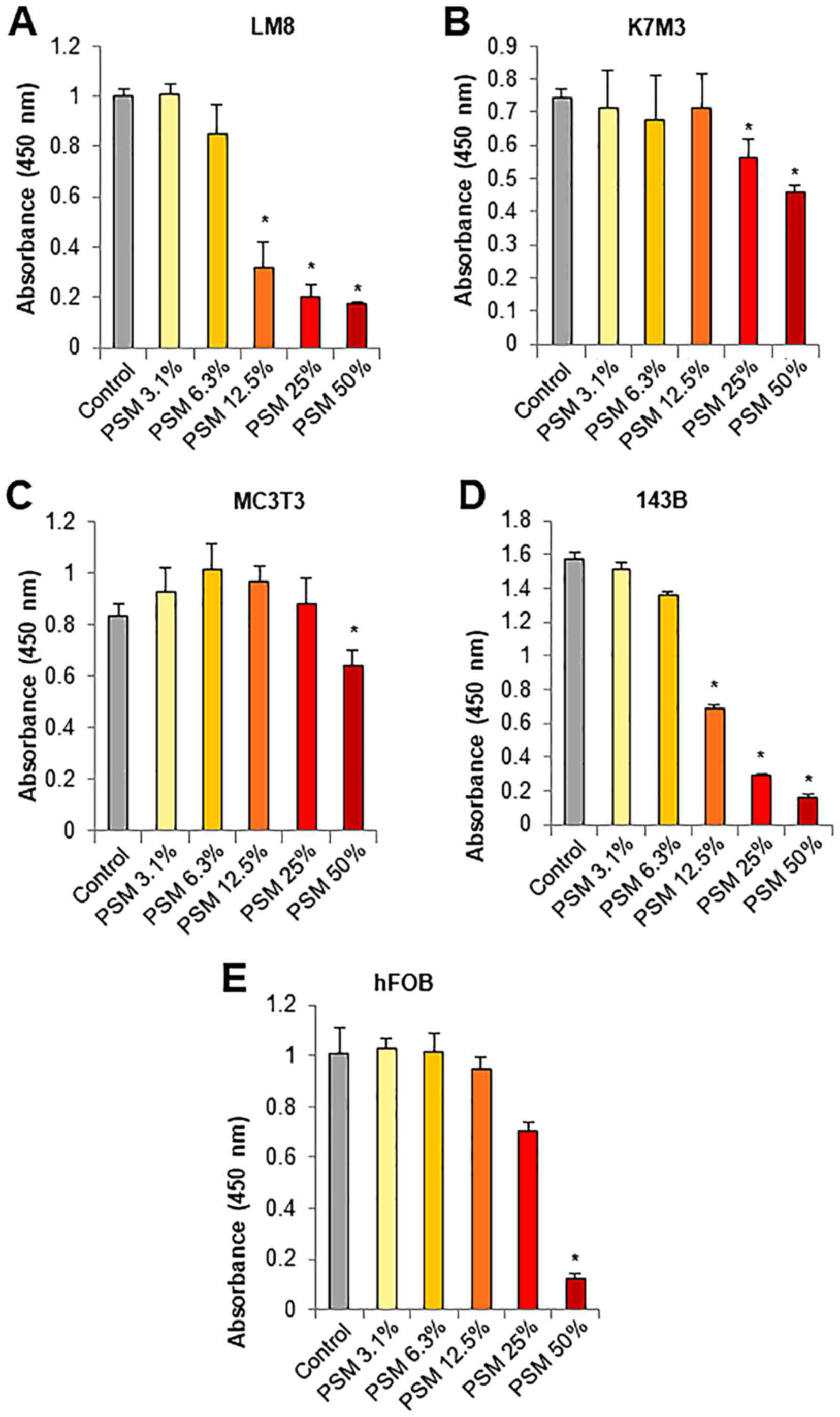
PSM preferentially causes injuries in
transformed cells
The most attractive properties of PSM is its high
tumor-selective activity. To confirm the properties of PSM, we
compared its effect on cell viability between osteosarcoma cells
and their normal counterparts, osteoblasts. PSM at concentrations
of ≥12.5% decreased the viability of mouse LM8 osteosarcoma cells
in a dose-dependent manner (≥70% reduction) (Fig. 2A). Moreover, PSM at concentrations
ranging from 12.5–50% had no or smaller effects on the viability of
mouse K7M3 and MC3T3 osteoblasts, although the former was more
sensitive than the latter (Fig. 2B and
C). Similarly, PSM at concentrations of ≥12.5% decreased the
viability of human 143B osteosarcoma cells in a dose-dependent
manner, while it had less potent effects on the viability of human
hFOB osteoblasts (Fig. 2D and E).
These results expand our previous findings on melanocytes and
fibroblasts (45) and indicate
that PSM preferentially causes injuries in transformed cells.
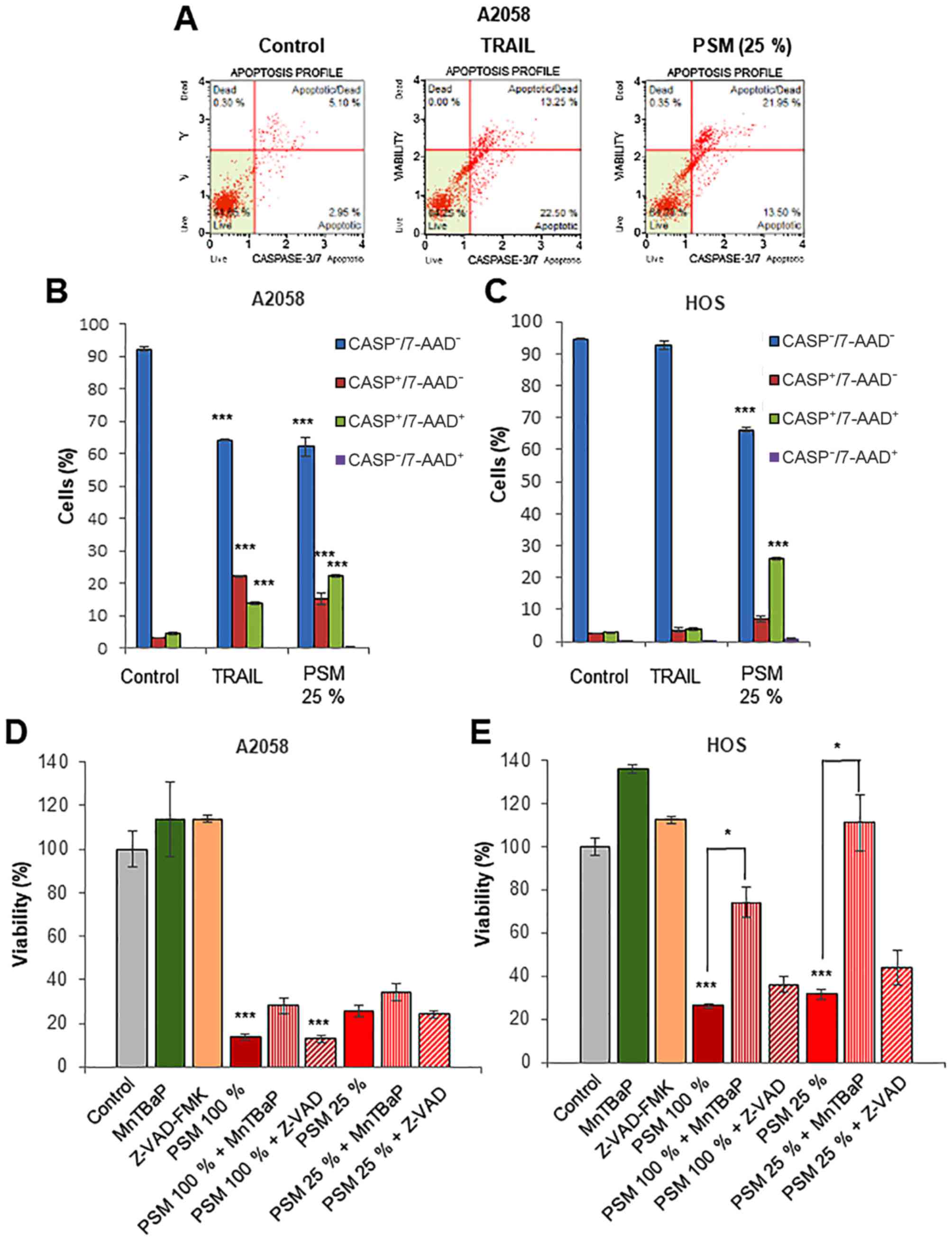
PSM cytotoxicity is caspase-independent
and superoxide-dependent, depending on the cell type
Previous studies have demonstrated that apoptosis is
the primary mechanism of PSM-induced in vitro killing of
different types of tumors, including colon ovarian, lung, cancer
and glioblastoma (48–51). On the other hand, we previously
demonstrated that PSM cytotoxicity toward melanoma cells was not
inhibited by the caspase-3/7-specific inhibitor, z-DEVD-FMK, while
PSM increased the number of caspase-3/7-activated cells. Moreover,
PSM caused the minimal cleavage of caspase-3/7 and poly ADP-ribose
polymerase, a substrate of caspase-3/7 (45). These findings suggest that the cell
death caused by PSM differs from canonical apoptosis. To test this
view, in this study, we monitored caspase-3/7 activation and cell
death concomitantly using NucView™ and 7-AAD staining. NucView™ is
a novel caspase-3/7 substrate and 7-AAD is a dead cell marker,
which is excluded from healthy and early apoptotic cells, while
permeates late apoptotic and dead cells. Accordingly, we could
distinguish 4 cell populations by this method: Live cells,
caspase-3/7-inactivated, 7-AAD-negative
(caspase−/7-AAD−); early apoptotic cells,
caspase-3/7-activated, 7-AAD-negative
(caspase+/7-AAD−); late apoptotic/dead cells,
caspase-3/7-activated, 7-AAD-positive
(caspase+/7-AAD+); and necrotic cells,
caspase-3/7-inactivated, 7-AAD-positive
(caspase-/7-AAD+). Treatment of the A2058 cells with
TRAIL or PSM (25%) for 24 h resulted in a comparable decrease in
the number of live cells (35% reduction). However, TRAIL
preferentially increased the number of
caspase+/7-AAD− cells than that of
caspase+/7-AAD+ cells, while PSM
preferentially increased the number of
caspase+/7-AAD+ cells than that of
caspase+/7-AAD− cells (Fig. 3A and B). Moreover, PSM, but not
TRAIL, caused a robust decrease in the number of live HOS cells
(30%), in association with an increased number of
caspase+/7-AAD+ cells (Fig. 3C). When applied for 72 h, the
cytotoxicity of PSM (25 and 100%) toward the HOS, but not the A2058
cells, was significantly inhibited by the treatment with superoxide
mimetic, MnTBaP (30 µM) for 72 h, and these effects were
more pronounced for the low concentration of PSM (Fig. 3D and E). By contrast, treatment
with the pan-caspase-inhibitor, z-VAD-FMK (10 µM) for 72 h,
had no inhibitory effect regardless of the cell type, and the
concentration of PSM employed. These results indicate that despite
its ability to evoke caspase-3/7 activation, PSM cytotoxicity is
primarily caspase-independent and is superoxide-dependent,
depending on the cell type.
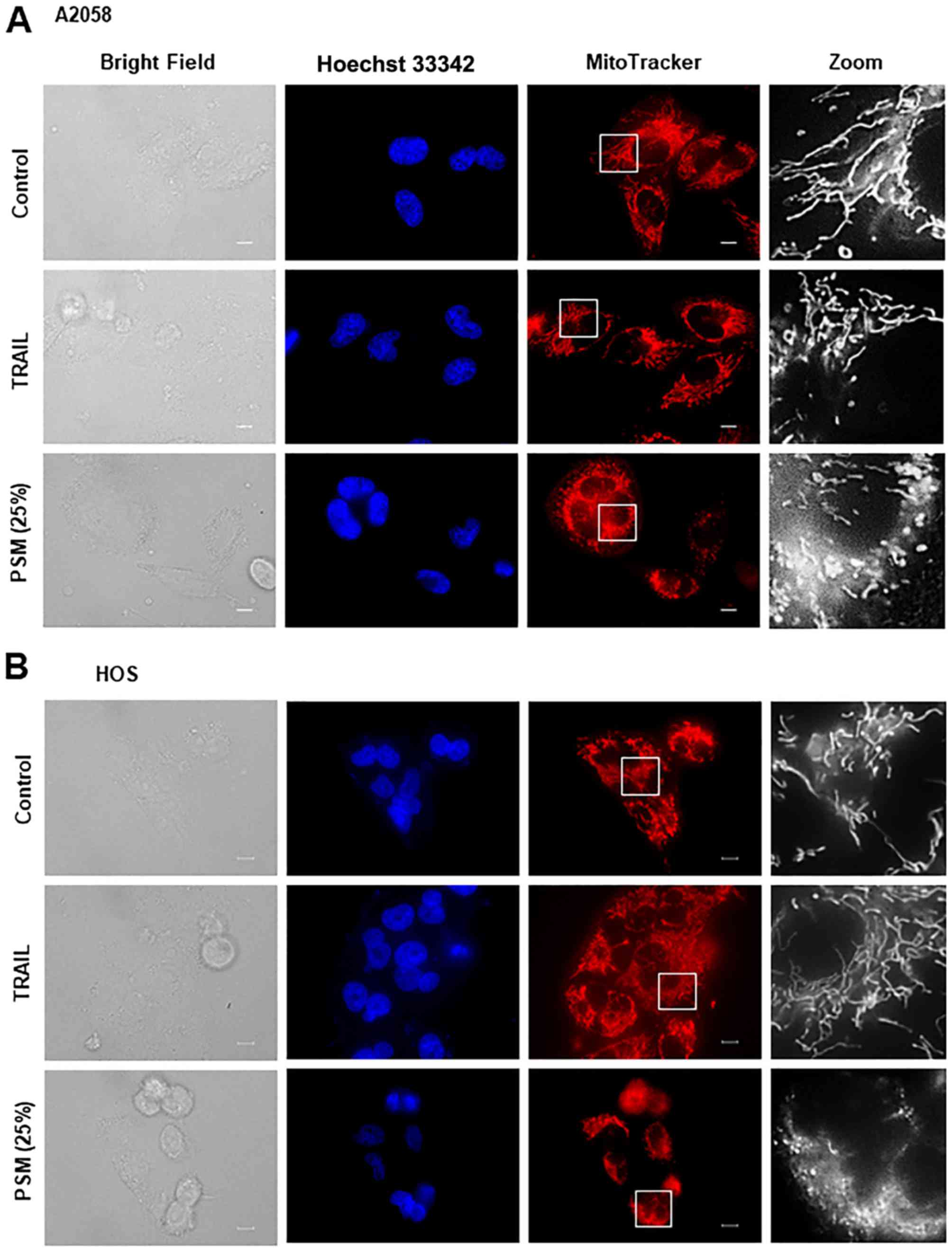
PSM is more potent than TRAIL in inducing
mitochondrial network aberration
We then directly compared the effects of TRAIL and
the PSM on mitochondrial morphology in different tumor cell types.
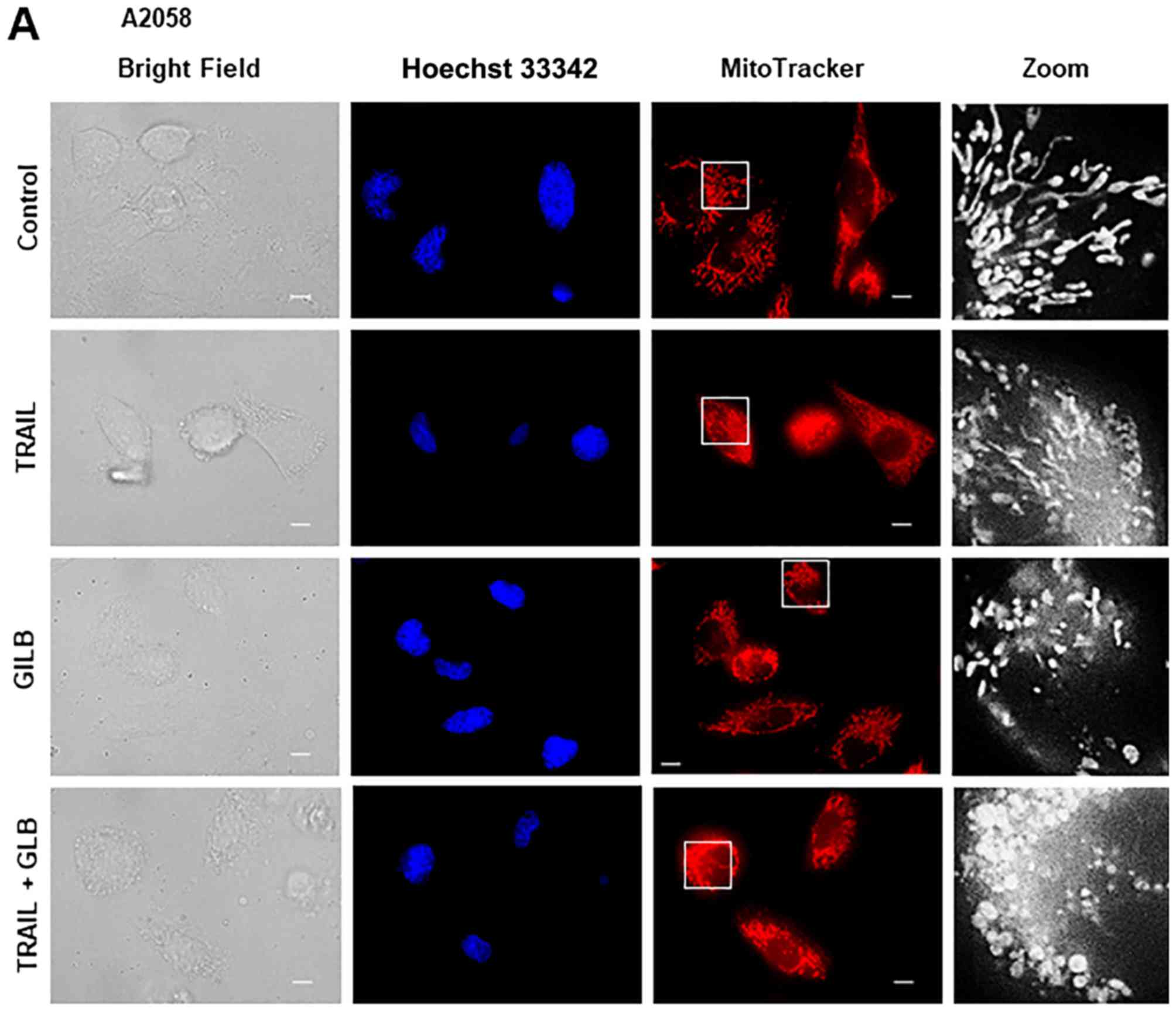
After treating the cells for 24 h, the mitochondria were stained
with the mitochondrial-targeting dye, MitoTracker Red CMXRos, and
the nuclei were counterstained with Hoechst 33342. The A2058, HOS,
SAOS-2, SK-N-SH cells were all highly adherent spindle cells that
displayed tubular mitochondrial morphology around healthy nuclei in
the absence of an insult (Fig.
4A–D, upper panels). TRAIL treatment resulted in minimal
cellular and nuclear morphological changes, and a modest
mitochondrial fragmentation in all cell types apart from the SAOS-2
cells (Fig. 4A–D, middle panels).
We found that this cell type was frequently considerably damaged by
TRAIL treatment. In this case, a substantial amount of cells became
round in shape and eventually became detached from the coverslips
(Fig. 4C). In these cells, the
mitochondria became heavily fragmented and clustered. In all cell
types tested, PSM (25%) caused a more profound impact than TRAIL
(≤100 ng/ml) on cellular and mitochondrial morphology. PSM
treatment led to a considerable increase in the amount of round,
damaged cells that possessed punctate and clustered mitochondria,
as well as broken nuclei (Fig.
4A–D, lower panels). These results indicate that PSM is more
potent than TRAIL in inducing mitochondrial network aberration.
PSM, but not TRAIL, evokes rapid and
persistent PMD
Previously, we demonstrated that PMD is a critical
cellular event in TRAIL-induced apoptosis and is essential for the
mitochondrial network aberration in different malignant cell types
(52,53). This finding led us to compare the
ability evoked PMD between PSM and TRAIL. In this study, analysis
using the voltage-sensitive fluorescence probe
DiBAC4(3) revealed that
in all cell lines tested (A2058, HOS, SAOS-2 and NB-1), PSM
treatment resulted in robust PMD within 1 min, which lasted at
least for 10 min (Fig. 5A–D). PSM
(≥25%) exerted its effect in a dose-dependent manner (data not
shown). By contrast, TRAIL (≤100 ng/ml) caused minimal PMD until
the 10-min time point, regardless of the cell types tested. We
noted that in parallel with the cellular sensitivity to PSM, the
degree of PMD varied considerably in different experiments.
Accordingly, in some cases, PSM (≤50%) induced minimal PMD or
membrane hyperpolarization instead (Fig. 5E and F). The ATP-sensitive
potassium channel (KATP) antagonist, glibenclamide,
constantly evoked persistent PMD. These results indicate that PSM,
but not TRAIL, elicits rapid, persistent PMD in malignant cells
with different origins.
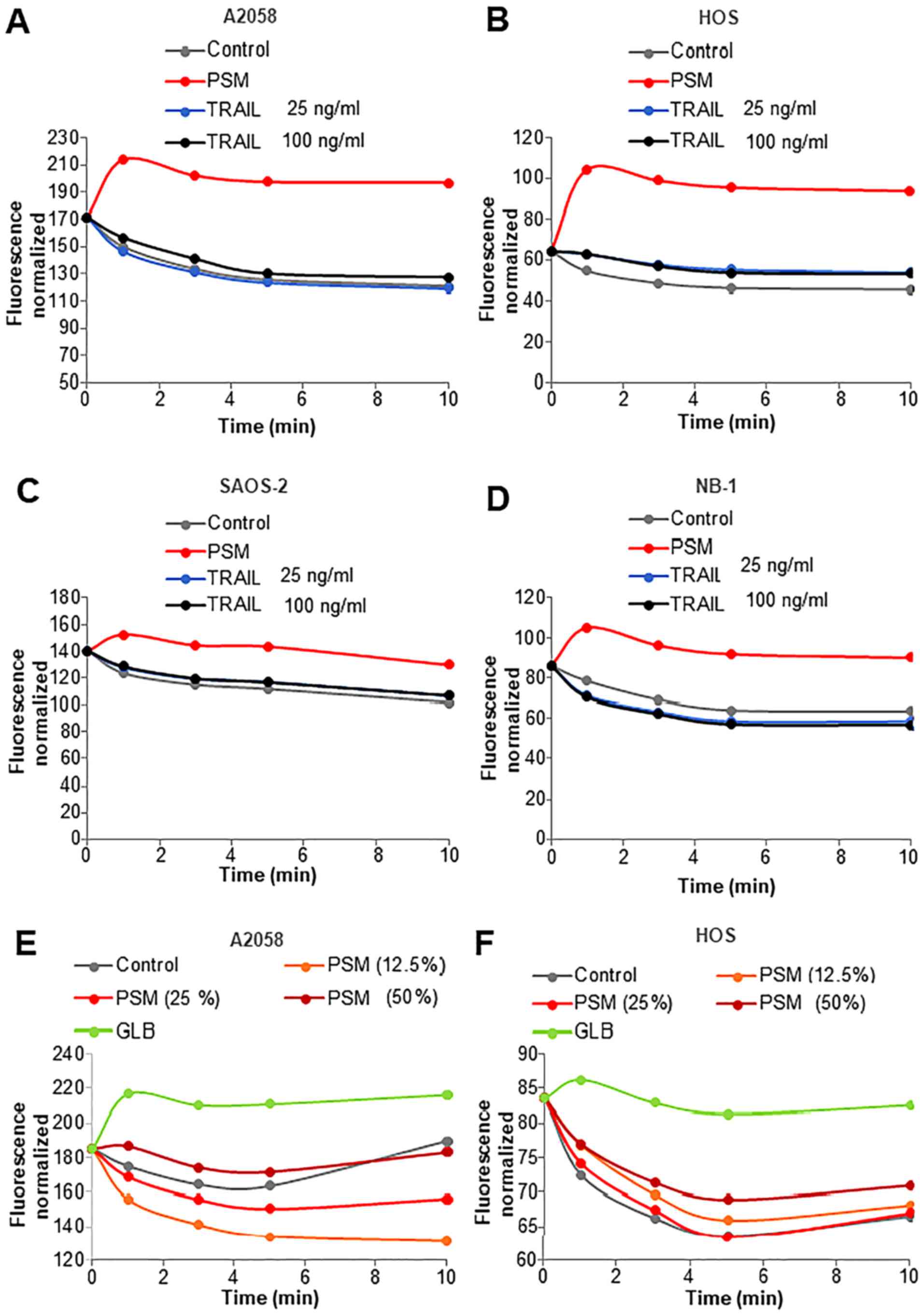 | Figure 5Plasma-stimulated medium (PSM) evokes
rapid and persistent plasma membrane depolarization (PMD). (A)
A2058 and (B) HOS (C) SAOS-2, and (D) NB-1 cells, were loaded with
5 M 5 µM DiBAC4(3) for 40 min at 37°C. The probe-loaded
cells were washed, resuspended in HBSS, treated with PSM (25%) or
25, 100 ng/ml TRAIL, and measured for their fluorescence for 1, 3,
5 and 10 min in a microplate fluorescence reader with excitation
and emission at 485 and 538 nm. (E) A2058 and (F) HOS cells were
loaded with DiBAC4(3)
as shown above and treated with PSM (12.5–50%) or 100 M
glibenclamide (GLB), and measured for their fluorescence for 1, 3,
5 and 10 min in a microplate fluorescence reader with excitation
and emission at 485 and 538 nm. The data are expressed as the
fluorescence intensity normalized (per 1×106 cells). |
Enforced PMD promotes TRAIL-induced
caspase-independent cell killing and mitochondrial network
aberration
To explore the link between PMD and cell killing, we
examined whether enforced PMD enhances caspase-independent cell
killing by TRAIL. While TRAIL (100 ng/ml) and glibenclamide (100
µM) alone had only a modest effect on cell viability (≤20%
decrease), their combined use profoundly decreased the viability of
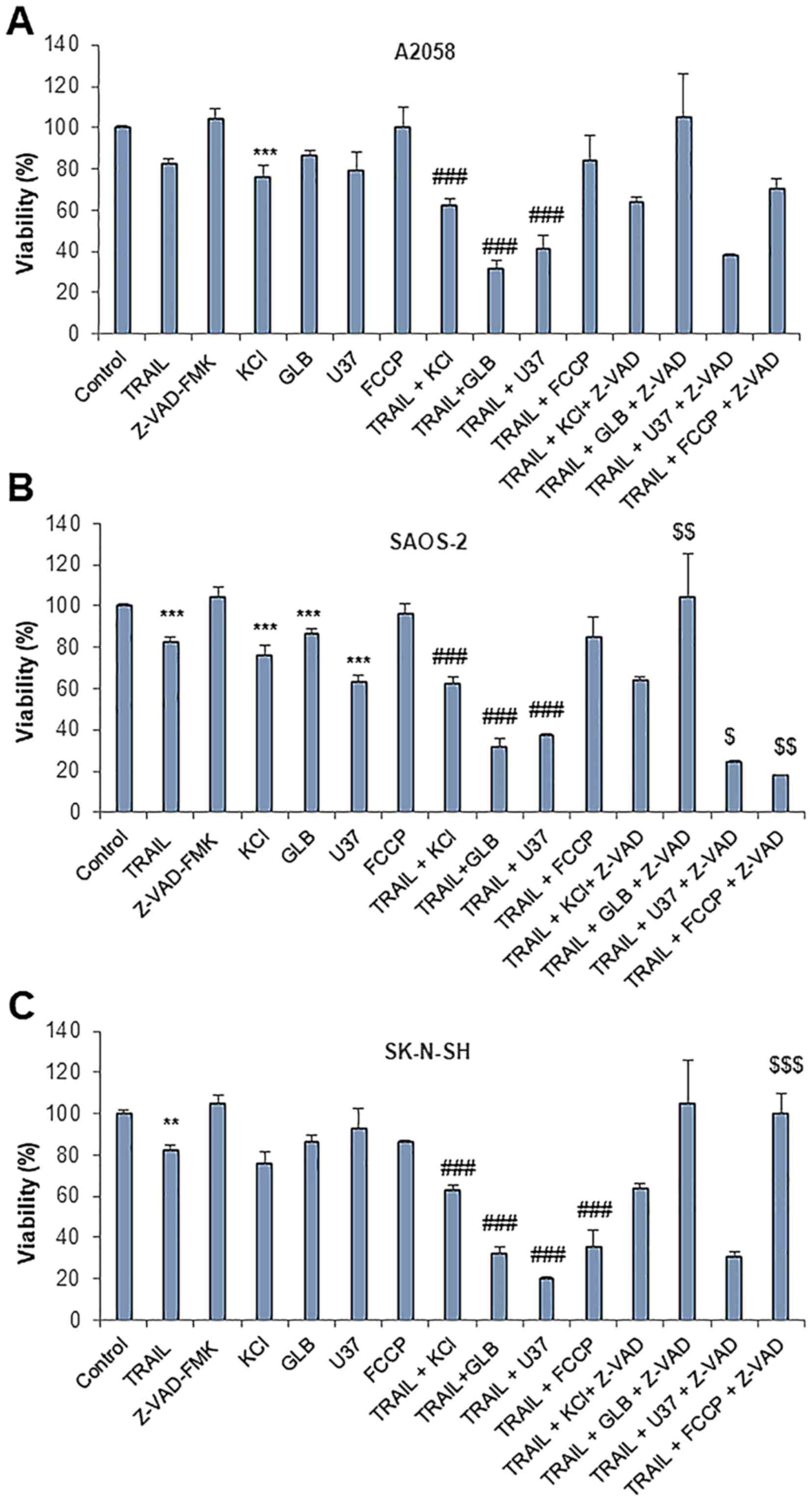
the A2058, SAOS-2 and SK-N-SH cells (≥70% reduction) (Fig. 6A). The other membrane-depolarizing
agents, such as potassium chloride (KCl) (50 mM) and another type
of KATP inhibitor, U37883A, also caused a synergistic
cytotoxic effect with TRAIL. By contrast, the oxidative
phosphorylation (OXOPHOS) inhibitor, FCCP (5 µM), which
sensitizes melanoma and osteosarcoma cells to TRAIL-induced
apoptosis (47,53) was generally ineffective in the
cells tested, aparat from the SK-N-SH cells. In the A2058 and
SAOS-2 cells, the cell death caused by TRAIL and either
membrane-depolarizing agents was not inhibited by z-VAD-FMK,
indicating that it is caspase-independent, as that produced by PSM.
Moreover, in the SK-N-SH cells, the cell death induced by TRAIL and
glibenclamide or FCCP was entirely blocked by z-VAD-FMK (Fig. 6C), indicating that it is
caspase-dependent. We then examined the effects on mitochondrial
morphology. Glibenclamide alone caused substantial mitochondrial
fragmentation (Fig. 7A and B,
third panels), and when applied in conjunction with TRAIL, led to
the clustering of punctate, swollen mitochondria in A2058 and
SAOS-2 cells (Fig. 7A and B, fifth
panels). The combined use of TRAIL and GLB also resulted in cell
damage and nuclear fragmentation. U37883A exerted essentially
similar effects (Fig. 7B, fourth
and bottom panels).
PSM is more potent than TRAIL in
disrupting Ca2+ homeostasis
Recently, we demonstrated that Ca2+
dynamics, particularly those in the mitochondrial matrix, are a
critical regulator of mitochondrial morphology and death in
melanoma and osteosarcoma cells. Specifically, increasing
[Ca2+]mit resulted in mitochondrial
fragmentation, while decreasing [Ca2+]mit led
to mitochondrial hyperfusion (46,47).
These observations led us to compare their ability to modulate
Ca2+ dynamics between TRAIL and PSM. In this study, we
first examined whether these two insults have any affect on
intracellular Ca2+ stores and extracellular
Ca2+ influx. Treatment with the Ca2+-ATPase
inhibitor, thapsigargin (Tg), in a Ca2+-free medium
followed by the addition of Ca2+ resulted in a rapid
small rise and a higher degree of elevation in
[Ca2+]cyt (Fig.
8A). It is established that the former represents
Ca2+ release from the intracellular stores mainly the
endoplasmic reticulum (ER), while the latter depicts the SOCE.
Treatment with PSM (≥25%) led to a marked increase in
Ca2+ release in a dose-dependent manner (Fig. 8B). By contrast, treatment with
TRAIL at up to 100 ng/ml had no significant effect on
Ca2+ release (Fig. 8C).
Morever, treatment with PSM (≥25%), but not TRAIL, decreased SOCE
(Fig. 8D and E). The combined use
of TRAIL and 100 µM each of glibenclamide or U37883A led to
reduced SOCE, as did the combined application of TRAIL and the
OXOPHOS inhibitors, FCCP (5 µM) and antimycin A (5
µg/ml) for 10 min. However, none of these agents resulted in
an increased Ca2+ release from the intracellular stores
(Fig. 8F).
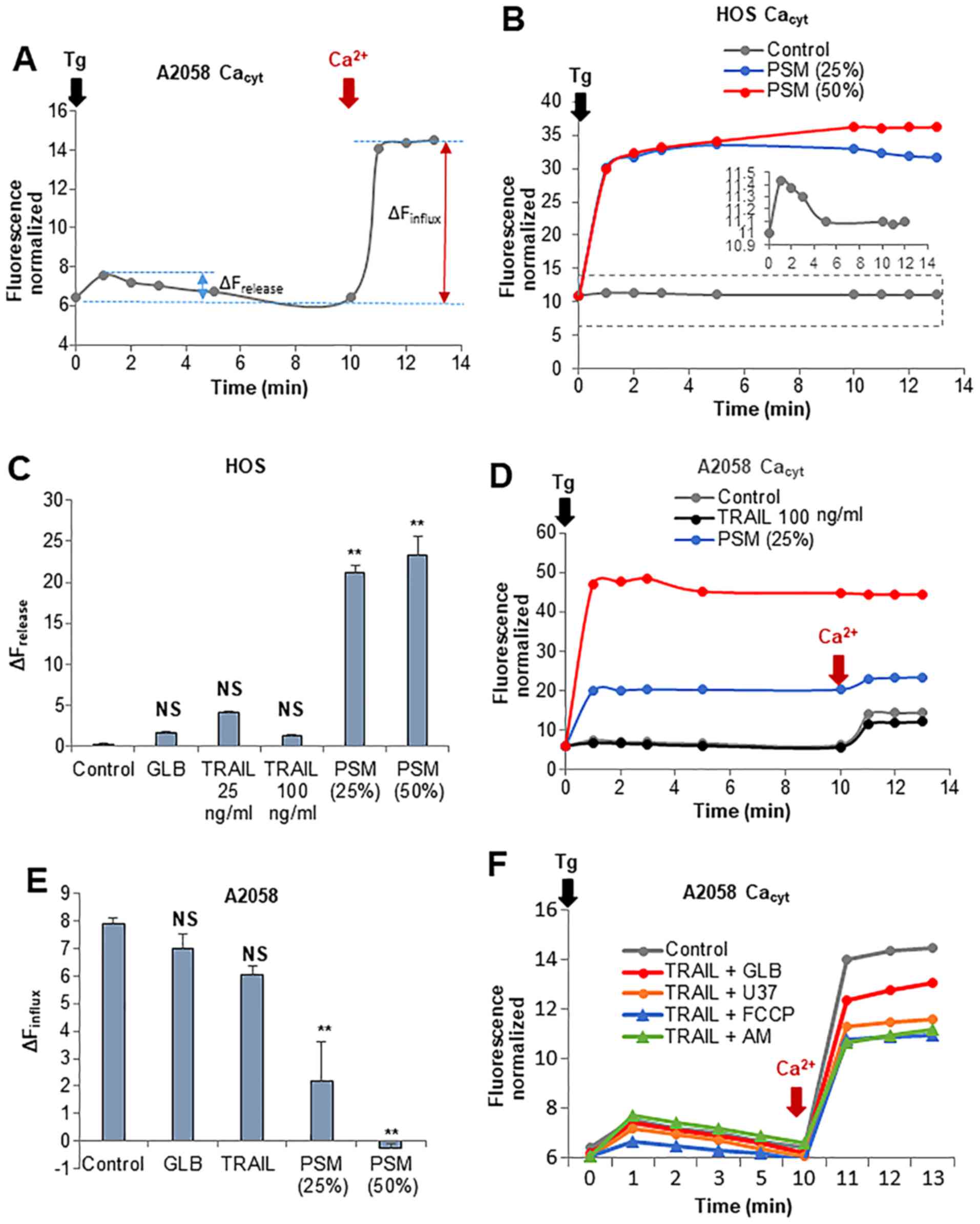 | Figure 8Plasma-stimulated medium (PSM) is
more potent than TRAIL in disrupting Ca2+ homeostasis.
(A, D, E and F) A2058 and (B and C) HOS cells were suspended in a
Ca2+-free buffer (HBSS supplemented with 1 mM EGTA) and
were loaded with Fluo4-AM. The probe-loaded cells were treated with
100 ng/ml TRAIL, the PSM (25,50%), 100 µM glibenclamide
(GLB), 100 µM U37883A (U37), and 5 µM FCCP alone or
in combination. The cells were immediately supplemented with 2
µM thapsigargin (Tg, black arrow) and incubated for 10 min
to deplete the intracellular Ca2+ stores. Subsequently,
2 mM Ca2+ was added to the cells (dark red arrow).
Following the addition of Tg, fluorescence was monitored at 1, 2,
3, 5, 10, 11, 12 and 13 min with excitation and emission at 485 and
538 nm. (A) Representative [Ca2+]cyt changes
in the Ca2+ readdition experiments. The data in (C and
E) represent the means ± SD (n=3) of ΔFrelease and
ΔFinflux, respectively. Data were analyzed by ANOVA
followed by Tukey’s post hoc test; **P<0.01; NS, not
significant vs. control. ΔFrelease, fluorecence
intensity changes in Ca2+ release; ΔFinflux,
fluorecence intensity changes in Ca2+ influx. |
Enforced PMD enables TRAIL to mimic the
effects of PSM on [Ca2+]mit
An excess, persistent elevation in
[Ca2+]cyt is the primary cause of cell death.
Specifically, mitochondrial Ca2+ overload triggers
necrosis and apoptosis (31–34).
Thus, to further explore the link between Ca2+
dyshomeostasis and tumor cell death, we examined the effect of the
insults on [Ca2+]cyt and
[Ca2+]mit. For this purpose, we concomitantly
measured Ca2+ concentrations in the two different
intracellular sites using the site-specific Ca2+ probes,
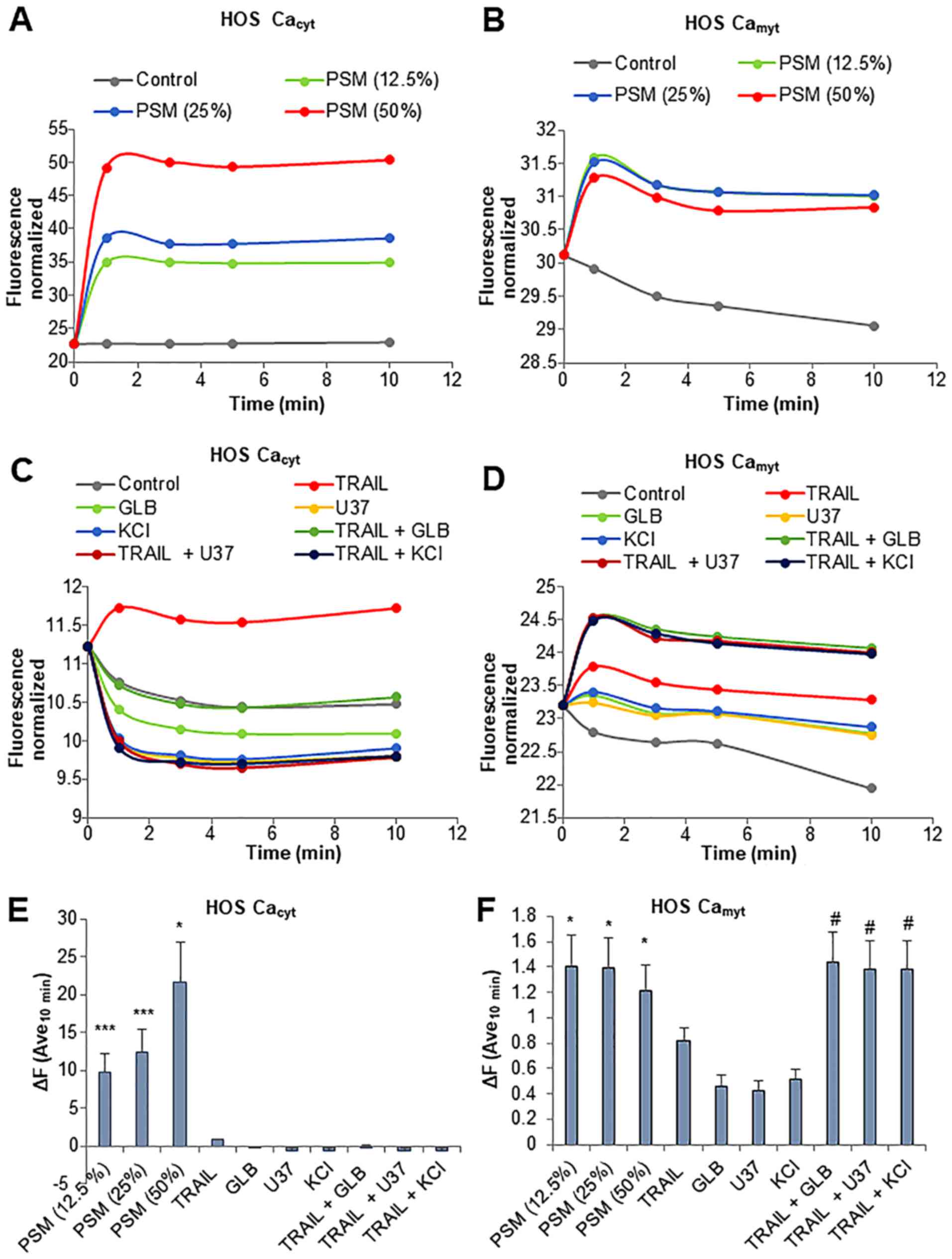
Fluo4-AM and rhod-2-AM. PSM led to a rapid increase in
[Ca2+]cyt in the HOS cells. The effect was
observed within 1 min and lasted at least 10 min. PSM exerted its
effect in a dose-dependent manner with the minimal effective
concentration of 12.5% (Fig. 9A and
E). Concomitantly, the levels of
[Ca2+]mit also increased with similar
kinetics, although the effects of PSM at concentrations of 12.5 to
50% were comparable (Fig. 9B and
F). TRAIL resulted in a much smaller increase in the levels of
[Ca2+]cyt while glibenclamide, U37883A and
KCl alone led to a decrease in the levels of
[Ca2+]cyt (Fig.
9C). Even when used together, no significant increase in
[Ca2+]cyt levels was observed (Fig. 9C and E). Moreover, either
membrane-depolarizing agent alone promptly resulted in a small, but
persistent increase in [Ca2+]mit (Fig. 9D and F). The combined use of TRAIL
and the membrane-depolarizing agent led to a greater extent of
[Ca2+]mit rise comparable to that induced by
PSM at concentrations of 12.5 to 50% (Fig. 9F). We obtained substantially the
same results for SAOS-2 cells (data not shown). These results
indicate that the PSM is a potent inducer of the increase in
[Ca2+]cyt and
[Ca2+]mit, and the combined use of TRAIL and
the membrane-depolarizing agent can mimic the effect of PSM on
[Ca2+]mit, but not
[Ca2+]cyt levels.
Discussion
CAP/PSM has emerged as a promising tool for the
treatment of aggressive tumors. In the present study, we
systematically compared the antitumor activity of PSM with that of
TRAIL. PSM prepared by irradiating cold plasma to DMEM killed
different malignant cell types (malignant melanoma, osteosarcoma,
and neuroblastoma) that were highly resistant to TRAIL (Fig. 1), whereas, PSM had little cytotoxic
effects on murine and human osteoblasts (Fig. 2). The findings expand our previous
observations on melanocytes and fibroblasts (28), and indicate that PSM preferentially
causes injuries to malignant cells. PSM prepared from different
types of gases and media has been shown to kill an array of
malignant cells, including colon cancer ovarian cancer, lung cancer
and glioblastoma (48–51). Most of these types of PSM elicit
their antitumor activity by primarily stimulating apoptosis. The
PSM in our study induced a robust caspase-3/7 activation in A2058
and HOS cells prior to cell death (Fig. 3), indicating that apoptosis occurs
in our cell system. Nevertheless, cell death was minimally affected
by z-VAD-FMK (Fig. 3). The results
expand our previous observations (45) on melanoma cells in that the
caspase-3/7-specific inhibitor, z-DEVD-FMK, did not block the cell
death induced by PSM. Collectively, these findings suggest that PSM
primarily leads to caspase-independent cell death in melanoma and
osteosarcoma cells, while apoptosis plays a minor role.
It is widely accepted that RONS play a pivotal role
in mediating the antitumor activity of CAP and PSM (19,21,45).
In agreement with this view, N-acetylcysteine (NAC), a
broad-spectrum antioxidant has been shown to markedly inhibited
cell death caused by PSM in melanoma, osteosarcoma and lung cancer
cells (45). Moreover, in that
study, PSM led to a rapid mitochondrial superoxide generation in
all cell types tested, as detected by the mitochondrial-targeting
superoxide probe, MitoSOX. These observations suggest that RONS
originated from superoxide are critical mediators of antitumor
activity. Nevertheless, we noted that MnTBaP, a superoxide-mimetic
failed to block the antitumor activity against melanoma cells such
as A375 and A2058 (45).
Consistent with the findings of our previous study, the present
study revealed that MnTBaP actively blocked HOS cell death, while
it minimally inhibited A2058 cell death (Fig. 3). Of note, there was a tendency
that MnTBaP was effective when the concentration of PSM was low.
The findings strongly suggest that the actual RONS that are
responsible for the antitumor activity of PSM may be differ
depending on the cell type and the concentration of PSM used.
We have previously demonstrated that the
mitochondrial network dynamics is another common target for the two
insults in damaging cells (44,45).
The direct comparison of their effects on the mitochondrial
morphology revealed that PSM was more potent than TRAIL in evoking
mitochondrial network aberration, regardless of the tumor cell
types examined. Even the cellular conditions where TRAIL led to
only a modest mitochondrial fragmentation, PSM always led to an
excessive mitochondrial fragmentation and the clustering of the
fragmented mitochondria (Fig. 4).
The intense mitochondrial network aberration well-correlated with
cell damage, as indicated by the cellular and nuclear morphological
changes. These findings suggest that PSM is advantageous over TRAIL
in inducing the pro-death mitochondrial network aberration. Our
previous study demonstrated that persistent PMD plays a critical
role in the progression from a modest mitochondrial fragmentation
to an excessive mitochondrial fragmentation and clustering
(44). Strikingly, in the present
study, we found that PSM was more potent than TRAIL in inducing PMD
(Fig. 5). In agreement with our
previous findings (52), TRAIL led
to a substantial PMD with a delay of 2–4 h. Moreover, PSM caused
robust PMD very rapidly (within 1 min) and persistently (at least
for 10 min). We also observed significant mitochondrial
morphological change at as early as 5 min after PSM treatment,
while it occurred with a delay of 2–4 h following TRAIL treatment
(data not shown). These findings are consistent with the view that
PMD is a critical promoter of the mitochondrial network aberration.
Furthermore, we found that enforced PMD by KCl and KATP
antagonists enhanced the mitochondrial network aberration and cell
killing caused by TRAIL (Figs. 6
and 7). PMD has been considered to
be linked to apoptosis primarily. Apoptosis is characterized by
cell shrinkage, which is caused by the disruption of the
maintenance of physiological concentrations of K+ and
Na+ and intracellular ion homeostasis (55,56).
The loss of these monovalent ions has been shown to facilitate
caspase-3 activation (56). The
loss also leads to PMD. Indeed, PMD is an early event in apoptosis
induced by diverse insults including TRAIL, Fas, arsenic trioxide
and rotenone (57–59). Consistent with this view,
glibenclamide, which caused persistent PMD, amplified TRAIL-induced
apoptosis in different tumor cell types (Fig. 6). By contrast, KCl and U37883A,
another type of KATP inhibitor, which also caused
persistent PMD (52,53), enhanced nonapoptotic cell death all
cell types tested. These observations indicate that persistent PMD
can promote non-apoptotic cell death, as well as canonical
apoptosis. Since the enhancement of the two modes of cell death was
proceeded by mitochondrial network aberration (Fig. 7), these findings suggest that the
mitochondrial network aberration leads to different modes of cell
death.
It is noteworthy that PSM was more potent than
TRAIL in disrupting the Ca2+ dynamics. PSM increased
both [Ca2+]cyt and
[Ca2+]mit levels (Fig. 8), and persistent PMD could mimic
the effect of the latter, but not the former (Fig. 9), suggesting that distinct
Ca2+ entry pathways with different sensitivities to PMD
may contribute to these Ca2+ responses. PSM seemed to
increase the ER Ca2+ stores in cancer cells, thereby
compromising SOCE, as SOCE is activated in response to
Ca2+ depletion in the stores. Since persistent PMD can
also lead to SOCE inactivation (Fig.
8), PSM may elicit the effect through PMD. An increasing body
of evidence suggests that SOCE plays a pivotal role in malignant
phenotypes, including the evasion of cell death (59). Thus, the inactivation of SOCE may
play a role in the antitumor activity of the PSM. Given that is a
critical regulator of the mitochondrial network dynamics in these
cancer cells (46,47), the effects may also contribute to
the mitochondrial network aberration. Further analyses of the
mechanisms underlying Ca2+ dynamics modulation by the
PSM are ongoing.
In conclusion, in this study, and to the best of
our knowledge, we demonstrate for the first time that the PSM has a
significant advantage over TRAIL in killing TRAIL-resistant cancer
cells from different origins owing to its capacity to evoke a
Ca2+-dependent, caspase-independent cell death. The
findings may provide fundamentals for the development of PSM as a
novel approach for the treatment of TRAIL-resistant cancer
cells.
Acknowledgments
The authors would like to thank the Health Science
Research Resource Bank (Osaka, Japan), Riken BioResource Center,
and Dr Eugenie Kleinerman for providing the cell lines. This study
was supported in part by JSPS KAKENHI grant nos. 16K10851 to T.A.,
15K09792 to T.O., 15K06883 to M.S. and 15K09750 to Y.S.-K.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Cerchia C and Lavecchia A: Small molecule
drugs and targeted therapy for melanoma: Current atrategies and
future directions. Curr Med Chem. 24:2312–2344. 2017. View Article : Google Scholar
|
|
2
|
Kalal BS, Upadhya D and Pai VR:
Chemotherapy resistance mechanisms in advanced skin cancer. Oncol
Rev. 11:3262017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li S, Sun W, Wang H, Zuo D, Hua Y and Cai
Z: Research progress on the multidrug resistance mechanisms of
osteosarcoma chemotherapy and reversal. Tumour Biol. 36:1329–1338.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
He H, Ni J and Huang J: Molecular
mechanisms of chemoresistance in osteosarcoma (Review). Oncol Lett.
7:1352–1362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Almasan A and Ashkenazi A: Apo2L/TRAIL:
Apoptosis signaling, biology, and potential for cancer therapy.
Cytokine Growth Factor Rev. 14:337–348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gonzalvez F and Ashkenazi A: New insights
into apoptosis signaling by Apo2L/TRAIL. Oncogene. 29:4752–4765.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25:4505–4506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar
|
|
14
|
Guiho R, Biteau K, Heymann D and Redini F:
TRAIL-based therapy in pediatric bone tumors: How to overcome
resistance. Future Oncol. 11:535–542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Miguel D, Lemke J, Anel A, Walczak H
and Martinez-Lostao L: Onto better TRAILs for cancer treatment.
Cell Death Differ. 23:733–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Keidar M, Walk R, Shashurin A, Srinivasan
P, Sandler A, Dasgupta S, Ravi R, Guerrero-Preston R and Trink B:
Cold plasma selectivity and the possibility of a paradigm shift in
cancer therapy. Br J Cancer. 105:1295–1301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zucker SN, Zirnheld J, Bagati A, DiSanto
TM, Des Soye B, Wawrzyniak JA, Etemadi K, Nikiforov M and Berezney
R: Preferential induction of apoptotic cell death in melanoma cells
as compared with normal keratinocytes using a non-thermal plasma
torch. Cancer Biol Ther. 13:1299–1306. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ishaq M, Evans MM and Ostrikov KK: Effect
of atmospheric gas plasmas on cancer cell signaling. Int J Cancer.
134:1517–1528. 2014. View Article : Google Scholar
|
|
19
|
Vandamme M, Robert E, Lerondel S, Sarron
V, Ries D, Dozias S, Sobilo J, Gosset D, Kieda C, Legrain B, et al:
ROS implication in a new antitumor strategy based on non-thermal
plasma. Int J Cancer. 130:2185–2194. 2012. View Article : Google Scholar
|
|
20
|
Guerrero-Preston R, Ogawa T, Uemura M,
Shumulinsky G, Valle BL, Pirini F, Ravi R, Sidransky D, Keidar M
and Trink B: Cold atmospheric plasma treatment selectively targets
head and neck squamous cell carcinoma cells. Int J Mol Med.
34:941–946. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ishaq M, Kumar S, Varinli H, Han ZJ, Rider
AE, Evans MD, Murphy AB and Ostrikov K: Atmospheric gas
plasma-induced ROS production activates TNF-ASK1 pathway for the
induction of melanoma cancer cell apoptosis. Mol Biol Cell.
25:1523–1531. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hirst AM, Simms MS, Mann VM, Maitland NJ,
O’Connell D and Frame FM: Low-temperature plasma treatment induces
DNA damage leading to necrotic cell death in primary prostate
epithelial cells. Br J Cancer. 112:1536–1545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang M, Holmes B, Cheng X, Zhu W, Keidar M
and Zhang LG: Cold atmospheric plasma for selectively ablating
metastatic breast cancer cells. PLoS One. 8:e737412013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vandamme M, Robert E, Lerondel S, Sarron
V, Ries D, Dozias S, Sobilo J, Gosset D, Kieda C, Legrain B, et al:
ROS implication in a new antitumor strategy based on non-thermal
plasma. Int J Cancer. 130:2185–2194. 2012. View Article : Google Scholar
|
|
25
|
Utsumi F, Kajiyama H, Nakamura K, Tanaka
H, Mizuno M, Ishikawa K, Kondo H, Kano H, Hori M and Kikkawa F:
Effect of indirect nonequilibrium atmospheric pressure plasma on
anti-proliferative activity against chronic chemo-resistant ovarian
cancer cells in vitro and in vivo. PLoS One. 8:e815762013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Torii K, Yamada S, Nakamura K, Tanaka H,
Kajiyama H, Tanahashi K, Iwata N, Kanda M, Kobayashi D, Tanaka C,
et al: Effectiveness of plasma treatment on gastric cancer cells.
Gastric Cancer. 18:635–643. 2015. View Article : Google Scholar
|
|
27
|
Hattori N, Yamada S, Torii K, Takeda S,
Nakamura K, Tanaka H, Kajiyama H, Kanda M, Fujii T, Nakayama G, et
al: Effectiveness of plasma treatment on pancreatic cancer cells.
Int J Oncol. 47:1655–1662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adachi T, Tanaka H, Nonomura S, Hara H,
Kondo S and Hori M: Plasma-activated medium induces A549 cell
injury via a spiral apoptotic cascade involving the
mitochondrial-nuclear network. Free Radic Biol Med. 79:28–44. 2015.
View Article : Google Scholar
|
|
29
|
Kurake N, Tanaka H, Ishikawa K, Kondo T,
Sekine M, Nakamura K, Kajiyama H, Kikkawa F, Mizuno M and Hori M:
Cell survival of glioblastoma grown in medium containing hydrogen
peroxide and/or nitrite, or in plasma-activated medium. Arch
Biochem Biophys. 605:102–108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Elustondo PA, Nichols M, Robertson GS and
Pavlov EV: Mitochondrial Ca2+ uptake pathways. J
Bioenerg Biomembr. 49:113–119. 2017. View Article : Google Scholar
|
|
31
|
Bonora M, Wieckowski MR, Chinopoulos C,
Kepp O, Kroemer G, Galluzzi L and Pinton P: Molecular mechanisms of
cell death: Central implication of ATP synthase in mitochondrial
permeability transition. Oncogene. 34:1475–1486. 2015. View Article : Google Scholar
|
|
32
|
Izzo V, Bravo-San Pedro JM, Sica V,
Kroemer G and Galluzzi L: Mitochondrial permeability transition:
New findings and persisting uncertainties. Trends Cell Biol.
26:655–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Galluzzi L, Bravo-San Pedro JM, Kepp O and
Kroemer G: Regulated cell death and adaptive stress responses. Cell
Mol Life Sci. 73:2405–2410. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Calcium and mitochondria in the regulation of cell death. Biochem
Biophys Res Commun. 460:72–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Danese A, Patergnani S, Bonora M,
Wieckowski MR, Previati M, Giorgi C and Pinton P: Calcium regulates
cell death in cancer: Roles of the mitochondria and
mitochondria-associated membranes (MAMs). Biochim Biophys Acta.
1858:615–627. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Marchi S and Pinton P: Alterations of
calcium homeostasis in cancer cells. Curr Opin Pharmacol. 29:1–6.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Monteith GR, Prevarskaya N and
Roberts-Thomson SJ: The calcium-cancer signalling nexus. Nat Rev
Cancer. 17:367–380. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Landes T and Martinou JC: Mitochondrial
outer membrane permeabilization during apoptosis: The role of
mitochondrial fission. Biochim Biophys Acta. 1813:540–545. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Elgass K, Pakay J, Ryan MT and Palmer CS:
Recent advances into the understanding of mitochondrial fission.
Biochim Biophys Acta. 1833:150–161. 2013. View Article : Google Scholar
|
|
40
|
Twig G and Shirihai OS: The interplay
between mitochondrial dynamics and mitophagy. Antioxid Redox
Signal. 14:1939–1951. 2011. View Article : Google Scholar :
|
|
41
|
Chen H, Chomyn A and Chan DC: Disruption
of fusion results in mitochondrial heterogeneity and dysfunction. J
Biol Chem. 280:26185–26192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hoppins S, Lackner L and Nunnari J: The
machines that divide and fuse mitochondria. Annu Rev Biochem.
76:751–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Akita M, Suzuki-Karasaki M, Fujiwara K,
Nakagawa C, Soma M, Yoshida Y, Ochiai T, Tokuhashi Y and
Suzuki-Karasaki Y: Mitochondrial division inhibitor-1 induces
mitochondrial hyper-fusion and sensitizes human cancer cells to
TRAIL-induced apoptosis. Int J Oncol. 45:1901–1912. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Suzuki-Karasaki Y, Fujiwara K, Saito K,
Suzuki-Karasaki M, Ochiai T and Soma M: Distinct effects of TRAIL
on the mitochondrial network in human cancer cells and normal
cells: Role of plasma membrane depolarization. Oncotarget.
6:21572–21588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saito K, Asai T, Fujiwara K, Sahara J,
Koguchi H, Fukuda N, Suzuki-Karasaki M, Soma M and Suzuki-Karasaki
Y: Tumor-selective mitochondrial network collapse induced by
atmospheric gas plasma-activated medium. Oncotarget. 7:19910–19927.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Takata N, Ohshima Y, Suzuki-Karasaki M,
Yoshida Y, Tokuhashi Y and Suzuki-Karasaki Y: Mitochondrial
Ca2+ removal amplifies TRAIL cytotoxicity toward
apoptosis-resistant tumor cells via promotion of multiple cell
death modalities. Int J Oncol. 51:193–203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ohshima Y, Takata N, Suzuki-Karasaki M,
Yoshida Y, Tokuhashi Y and Suzuki-Karasaki Y: Disrupting
mitochondrial Ca2+ homeostasis causes tumor-selective
TRAIL sensitization through mitochondrial network abnormalities.
Int J Oncol. 51:1146–1158. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Judée F, Fongia C, Ducommun B, Yousfi M,
Lobjois V and Merbahi N: Short and long time effects of low
temperature Plasma Activated Media on 3D multicellular tumor
spheroids. Sci Rep. 6:214212016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Utsumi F, Kajiyama H, Nakamura K, Tanaka
H, Mizuno M, Toyokuni S, Hori M and Kikkawa F: Variable
susceptibility of ovarian cancer cells to non-thermal
plasma-activated medium. Oncol Rep. 35:3169–3177. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Adachi T, Kano A, Nonomura S, Kamiya T and
Hara H: Histone deacetylase inhibitors stimulate the susceptibility
of A549 cells to a plasma-activated medium treatment. Arch Biochem
Biophys. 606:120–127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kurake N, Tanaka H, Ishikawa K, Kondo T,
Sekine M, Nakamura K, Kajiyama H, Kikkawa F, Mizuno M and Hori M:
Cell survival of glioblastoma grown in medium containing hydrogen
peroxide and/or nitrite, or in plasma-activated medium. Arch
Biochem Biophys. 605:102–108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Suzuki Y, Inoue T, Murai M,
Suzuki-Karasaki M, Ochiai T and Ra C: Depolarization potentiates
TRAIL-induced apoptosis in human melanoma cells: Role for
ATP-sensitive K+ channels and endoplasmic reticulum
stress. Int J Oncol. 41:465–475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Suzuki-Karasaki M, Ochiai T and
Suzuki-Karasaki Y: Crosstalk between mitochondrial ROS and
depolarization in the potentiation of TRAIL-induced apoptosis in
human tumor cells. Int J Oncol. 44:616–628. 2014. View Article : Google Scholar
|
|
54
|
McCarthy JV and Cotter TG: Cell shrinkage
and apoptosis: A role for potassium and sodium ion efflux. Cell
Death Differ. 4:756–770. 1997. View Article : Google Scholar
|
|
55
|
Lang F, Föller M, Lang K, Lang P, Ritter
M, Vereninov A, Szabo I, Huber SM and Gulbins E: Cell volume
regulatory ion channels in cell proliferation and cell death.
Methods Enzymol. 428:209–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bortner CD, Gomez-Angelats M and Cidlowski
JA: Plasma membrane depolarization without repolarization is an
early molecular event in anti-Fas-induced apoptosis. J Biol Chem.
276:4304–4314. 2001. View Article : Google Scholar
|
|
57
|
Nolte F, Friedrich O, Rojewski M, Fink RH,
Schrezenmeier H and Körper S: Depolarisation of the plasma membrane
in the arsenic trioxide (As2O3)-and anti-CD95-induced apoptosis in
myeloid cells. FEBS Lett. 578:85–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yin W, Li X, Feng S, Cheng W, Tang B, Shi
YL and Hua ZC: Plasma membrane depolarization and Na, K-ATPase
impairment induced by mitochondrial toxins augment leukemia cell
apoptosis via a novel mitochondrial amplification mechanism.
Biochem Pharmacol. 78:191–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jardin I and Rosado JA: STIM and calcium
channel complexes in cancer. Biochim Biophys Acta. 1863:1418–1426.
2016. View Article : Google Scholar
|