Introduction
Hepatocellular carcinoma (HCC) is a life-threatening
malignancy that occurs worldwide; 250,000–1,000,000 new cases are
diagnosed each year worldwide and the life expectancy from the time
of diagnosis is ~6 months (1,2). As
there are different major causative and risk factors, the incidence
of HCC varies by geographical area. HCC is particularly common in
Asia due to the high incidence of hepatitis B virus (HBV) and
hepatitis C virus infection, and the intake of dietary aflatoxin.
In China, HBV infection is the most common cause of HCC (3–5).
Hepatectomy and liver transplantation are potentially curative
approaches in HCC (6). However, as
HCC tends to be diagnosed at the later stage, only a small
proportion of patients are eligible for these treatments (6,7).
Therefore, it is necessary to investigate the genes responsible for
the development and progression of HCC, establish an improved
prognostic model, and identify prognostic biomarkers with higher
sensitivity, accuracy and specificity.
In normal somatic cells, centrosomes complete
duplication precisely during cell division (8–10).
Increased centrosome number or centrosome amplification causes
chromosomal instability, aneuploidy and tumorigenesis, which affect
the cell cycle and apoptosis, and are associated with tumor
differentiation, metastasis and recurrence. However, cancer cells
can evade these conditions and maintain survival by clustering the
extra centrosomes (8–10). In previous studies, centrosome
amplification has been recognized as a hallmark of cancer (9,11,12).
A C-type kinesin of the kinesin-14 family (13), kinesin family member C1 (KIFC1,
also known as HSET), is a minus end-directed motor protein
(14) with a critical role in
centrosome clustering in cancer cells (15). KIFC1 is reported to exert its
function in vesicular and organelle trafficking (16), oocyte development (17), spermatogenesis (18,19)
and double-stranded DNA transportation (20). Previously, investigations have
focused on KIFC1, as it is essential for the bipolar mitotic
division of cancer cells but appears redundant in normal cells
(15,21). KIFC1 is upregulated in ovarian
cancer, breast cancer and non-small cell lung cancer, and it
facilitates cancer initiation and progression (22–24).
In addition, the overexpression of KIFC1 suppresses
docetaxel-mediated apoptosis in breast cancer cells (25). However, the expression of KIFC1 in
HCC has not been investigated. In addition, the effects of KIFC1 on
apoptosis, the cell cycle and epithelial-mesenchymal transition
(EMT) in HCC remain to be fully elucidated. Accordingly, the
present study examined the expression and distribution of KIFC1 in
HCC via immunohistochemistry, and assessed the expression of KIFC1
in HCC specimens, in terms of its association with HCC
clinicopathological characteristics and tumor-free survival rates
of patients. Furthermore, the effects of KIFC1 knockdown on the
regulation of HCC cell malignant behaviors were examined in
vitro.
Materials and methods
Patients and tissue samples
A total of 91 paired tumor tissues and corresponding
normal liver tissues were obtained from patients pathologically
diagnosed with HCC, who underwent surgical resection between 2009
and 2013 at the Second Affiliated Hospital of Nanchang University
(Nanchang, China). These patients had not received chemotherapy or
radiotherapy prior to surgery. The samples were cut into 4–5
mm3 sections, quick-frozen in liquid nitrogen and stored
at −80°C. The clinicopathological data of the patients, including
sex, age, tumor size and tumor stage, were collected concomitantly.
Histologically, tumor stage was classified separately by two
experienced pathologists according to the American Joint Committee
on Cancer (26). The present study
was approved by the Ethics Committee of the Second Affiliated
Hospital of Nanchang University, written consent was obtained from
each participant, and the study was performed in accordance with
the ethical standards of the Declaration of Helsinki.
Immunohistochemistry
A two-step immunohistochemical method was used to
perform the immunohistochemistry. Specifically, the 4-mm-thick
paraffinized sections of the paired tumor and peritumoral tissues
were dewaxed in a 65°C water bath for 90 min, deparaffinized twice
in xylene for 15 min per deparaffinization, rehydrated in gradient
alcohol for 5 min per concentration, and placed in a pressure
cooker at 100°C for 15 min for antigen repair. The sections were
incubated with 3% H2O2 at room temperature
for 15 min to block endogenous peroxidase activity, washed with
phosphate-buffered saline (PBS) three times (5 min per wash), and
blocked with goat serum (Boster, Wuhan, China) for 30 min.
Subsequently, the sections were incubated with rabbit anti-KIFC1
antibody (1:200; ABclonal, Wuhan, China) at 4°C overnight. The
following day, the sections were incubated with the secondary
antibody (PV-6000, 1:200; ZSGB-Bio, Beijing, China) for 30 min at
37°C. Following washing with PBS three times, diaminobenzidine and
hematoxylin were used for the color reaction. Pathologists
evaluated the immunostained tissue sections independently in a
blinded manner using a microscope (Olympus, Tokyo, Japan). KIFC1
protein was localized in the nuclei. Staining was scored as 0 (no
staining), 1 (weak staining), 2 (moderate staining) or 3 (strong
staining) according to the staining intensity. Based on the
percentage of staining, the extent was scored as 1 (<10%
staining), 2 (10–40% staining), or 3 (>40% staining). The
overall staining scores were then computed by multiplying the
scores for staining intensity and extent of staining. The final
score ranged between 0 and 9, with scores of 0–1 indicating
negative expression of KIFC1 and scores of 2–9 indicating positive
expression of KIFC1.
Data mining
The data of expression of KIFC1 for liver HCC was
obtained from the Gene Expression Profiling Interactive Analysis
(GEPIA) online database (http://gepia.cancer-pku.cn/), a web server providing
customizable functions (27).
Tumors and normal samples in the GEPIA database were derived from
The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression
(GTEx) projects. The correlations of disease-free survival and
overall survival rates with the expression of KIFC1 in HCC were
also computed by the GEPIA database.
Cell lines and culture
The MHCC-97H, SMMC-7721 and HCC-LM3 human HCC cell
lines, and the HL-7702 human liver cell line (also named LO2) were
obtained from the Shanghai Institute for Biological Sciences,
Chinese Academy of Sciences (Shanghai, China). The HL-7702 cells
were used as the control. The cells were cultured in high-glucose
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (FBS; Gibco, Grand Island, NY, USA) and maintained in a
humidified incubator with 5% carbon dioxide at 37°C. Exponential
growth-phase cells were used for the in vitro assays. The
cells were used for in vitro assays or were subcultured to
~80% confluence when in the logarithmic growth phase.
Small interfering RNA (siRNA) depletion
of KIFC1
Two human KIFC1 siRNA reagents were purchased from
GenePharma (Shanghai, China); the sequences of siRNA1 (KIFC1-s1)
and siRNA2 (KIFC1-s2) were 5′-UAACUGACCCUUUAAGUCCUU-3′ and
5′-UGGUCCAACGUUUGAGUCCUU-3′, respectively. A negative siRNA was
used as the control. The HCC-LM3 and SMMC-7721 cells were
transfected using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol for 48 h
in 6-well plates. The transfected cells were used for protein
extraction or other subsequent assays.
Protein extraction and western blot
analysis
The cells in the 6-well plates were lysed using
radioimmunoprecipitation assay buffer containing 1%
phenylmethanesulfonyl fluoride to extract the total protein, and
quantified using the Bradford method as previously described
(28). Protein concentrations were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and then transferred onto polyvinylidene fluoride
membranes. The membranes were blocked using 5% non-fat milk for 2 h
at room temperature, and subsequently incubated overnight with
anti-KIFC1 (ABclonal) or other primary antibodies at 4°C. The
antibodies against BAX (#2772), Bcl-2 (#4223), p53 (#2527),
E-cadherin (#3195), N-cadherin (#13116), β-catenin (#8480), MMP-2
(#13132), Slug (#9585), ZEB-1 (#3396), p-PI3K (#4228), PI3K
(#4249), AKT (#4691) and p-AKT (#4060), were acquired from Cell
Signaling Technology (Danvers, MA, USA) and the anti-GAPDH (ab8245)
was acquired from Abcam (Cambridge, MA, USA). All these antibodies
were used according to the manufacturers' instructions. Following
three washes with Tris-buffered saline containing Tween-20 at 10
min per wash, the membranes were incubated at room temperature for
1 h with horseradish peroxidase (HRP)-conjugated secondary antibody
(1:10,000; Abcam). The membranes were then washed three times, as
previously, and the target protein immunoreactivity was detected
using an enhanced chemiluminescence system (Clinx, Shanghai,
China). As an endogenous protein, glyceraldehyde-3-phosphate
dehydrogenase was used for normalization. The results of western
blot was quantified by densitometric analysis using ImageJ
software.
Cell viability assay
Cell viability was assayed using Cell Counting Kit-8
(CCK-8; Nanjing KeyGEN Biotech Co., Ltd., Nanjing, China). A total
of 5,000 cells were seeded in each 96-well plate for 24 h following
KIFC1 siRNA-transfection, and incubated for another 24, 48, 72 and
96 h. At the end of each incubation period, 10 µl CCK-8
reagent was added to each well and incubated for 1 h prior to
measurement of the optical density of each well at 450 nm using a
microplate reader.
Analysis of apoptosis
Apoptosis was detected using fluorescence-activated
cell sorting (FACS) with an Annexin V Cell Apoptosis Analysis kit
(Sungene Biotech, Tianjing, China). The cells (~1×105)
were harvested for each assay, washed with cold PBS, and suspended
twice with 1X binding buffer. Annexin V-fluorescein isothiocyanate
was added (5 µl) and the cells were incubated for 10 min in
the dark at room temperature. Subsequently, 5 µl propidium
iodide (PI) solution was added and the samples were incubated for 5
min at room temperature in the dark. Finally, the samples were
analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA)
within 1 h.
Cell cycle analysis
Cell cycle distribution was analyzed via FACS. The
cells were harvested and the cell numbers adjusted to
1×106. The cells were then fixed overnight in cold 70%
ethanol at 2–8°C, washed twice with cold PBS, resuspended with 0.5
ml PI/RNase (Sungene Biotech) staining solution, and incubated for
30 min at 37°C in dark prior to analysis by flow cytometry.
Transwell assay
The migration and invasive capabilities of the
HCC-LM3 and SMMC-7721 cells were detected using Transwell assays.
In the invasion assay, 60 µl diluted Matrigel (BD
Biosciences) was added to Transwell chambers for 24 h at 37°C. The
cells were resuspended in serum-free DMEM, the cell number was
adjusted to 1×104, and the cells were added to the
Transwell chambers. The chambers were placed in a 24-well plate and
600 µl DMEM containing 20% FBS was added to the lower
surface of the chambers. Following incubation for 24 h, the
non-invaded cells and Matrigel were wiped away carefully, and the
invaded cells were fixed in 4% paraformaldehyde for 30 min, and
stained with 0.1% crystal violet for 30 min. The number of invaded
cells was calculated under a microscope (Olympus). The same steps
were used in the cell migration assay; however, Matrigel was not
used.
Statistical analysis
SPSS software (version 17.0) was used for all
statistical analyses. The Mann-Whitney U test was used to compare
the expression of KIFC1 between HCC tissues and paired normal
tissues; the expression of KIFC1 and clinicopathological data were
examined using the χ2 test. Tumor-free survival rates
were evaluated using Kaplan-Meier curves, and the Cox proportional
hazards regression model was used to evaluate the hazard ratio (HR)
and to confirm independent prognostic predictor factors. The in
vitro data are expressed as the means ± standard error and
performed using one-way analysis of variance. Multiple comparison
between the groups was performed using a Student-Newman-Keuls
(S-N-K) test. P-values were based on a two-sided statistical
analysis. All assays were performed independently three times.
P≤0.05 was considered to indicate a statistically significant
difference.
Results
KIFC1 is overexpressed in HCC clinical
samples
Using immunohistochemistry, the present study
evaluated the protein expression of KIFC1 in 91 paired HCC clinical
samples and corresponding normal tissues for the first time, to the
best of our knowledge. 78/91 normal tissue samples (Fig. 1A-a) and 41/91 HCC tissue samples
(Fig. 1A-b) had negligible
expression of KIFC1. There was positive KIFC1 expression in 50/91
samples (Fig. 1A-c and d), and
KIFC1 was mainly localized to the nuclei in the HCC samples.
Therefore, KIFC1 was expressed at high levels in HCC tissues,
compared with the peritumoral tissues (54.9 vs. 14.3%, P<0.01).
Furthermore, it was found that the mRNA level of KIFC1 was
significantly upregulated in HCC clinical samples, compared with
that in the normal tissues via the online GEPIA database, the
samples of which were from TCGA and the GTEx projects. (P<0.05)
(Fig. 1B and C).
Overexpression of KIFC1 is associated
with HCC clinicopathological characteristics and tumor-free
survival rates
The association between the protein expression of
KIFC1 and clinicopathological data in 91 HCC clinical samples is
shown in Table I. Specifically, a
high level of KIFC1 was significantly associated with tumor emboli
(P=0.033), metastasis (P=0.001), recurrence (P=0.015) and time of
recurrence (P=0.015). However, the expression of KIFC1 was not
associated with sex, age, HBV, cirrhosis, α-fetoprotein (AFP),
tumor size, tumor amount, differentiation, tumor capsule,
tumor-node-metastasis (TNM) stage or Barcelona Clinic Liver Cancer
(BCLC) stage. The Kaplan-Meier analysis showed that the expression
of KIFC1 was significantly associated with tumor-free survival rate
(P=0.004) (Fig. 1D). Furthermore,
univariate analysis showed a significant association between
tumor-free survival rate and tumor differentiation [HR=0.453; 95%
confidence interval (95% CI)=0.210–0.974; P=0.043), tumor emboli
(HR=0.365; 95% CI=0.188–0.709; P=0.003), TNM stage (HR=0.375; 95%
CI=0.207–1.680; P=0.001) and expression of KIFC1 (HR=0.438; 95%
CI=0.242–0.792; P=0.006). However, there was no statistical
significance between tumor-free survival rate and sex, age, HBV,
cirrhosis, AFP, tumor size, amount, tumor capsule or BCLC stage
(Table II). The multivariate
analysis also showed that the expression of KIFC1 (HR=0.518; 95%
CI=0.281–0.953; P=0.035) was an independent predictive marker in
patients with HCC (Table II). To
confirm the above results, it was also found that the mRNA
expression of KIFC1 was significantly associated with disease-free
survival rate (P=0.00064) (Fig.
1E) and overall survival rate (P=1.9e-0) (Fig. 1F) by GEPIA.
 | Table IAssociation between protein
expression of KIFC1 and clinicopathological parameters in patients
with hepatocellular carcinoma. |
Table I
Association between protein
expression of KIFC1 and clinicopathological parameters in patients
with hepatocellular carcinoma.
| Characteristic | Patients (n) | KIFC1 status
| χ2 | P-value |
|---|
| Negative | Positive |
|---|
| Sex | | | | | |
| Male | 76 | 35 | 41 | 0.185 | 0.667 |
| Female | 15 | 6 | 9 | | |
| Age (years) | | | | | |
| <60 | 78 | 34 | 44 | 0.473 | 0.491 |
| ≥60 | 13 | 7 | 6 | | |
| Hepatitis B
status | | | | | |
| Negative | 13 | 8 | 5 | 1.665 | 0.197 |
| Positive | 78 | 33 | 45 | | |
| Cirrhosis | | | | | |
| No | 33 | 14 | 19 | 0.145 | 0.704 |
| Yes | 58 | 33 | 45 | | |
| AFP
(µg/l) | | | | | |
| <400 | 60 | 29 | 31 | 0.765 | 0.382 |
| ≥400 | 31 | 12 | 19 | | |
| Tumor size
(cm) | | | | | |
| ≤3 | 16 | 5 | 11 | 1.495 | 0.222 |
| >3 | 75 | 36 | 39 | | |
| Tumor number | | | | | |
| 1 | 75 | 34 | 41 | 0.013 | 0.908 |
| >1 | 16 | 7 | 9 | | |
| Tumor capsule | | | | | |
| No | 49 | 22 | 27 | 0.001 | 0.974 |
| Yes | 42 | 19 | 23 | | |
|
Differentiation | | | | | |
| High | 21 | 13 | 8 | 3.131 | 0.077 |
| Low to medium | 70 | 28 | 42 | | |
| Tumor emboli | | | | | |
| No | 76 | 38 | 38 | 4.554 | 0.033b |
| Yes | 15 | 3 | 12 | | |
| Metastasis | | | | | |
| No | 51 | 31 | 20 11.596 | | 0.001b |
| Yes | 40 | 10 | 30 | | |
| TNM stage | | | | | |
| T1-2 | 69 | 34 | 35 | 2.054 | 0.152 |
| T3-4 | 22 | 7 | 15 | | |
| BCLC stage | | | | | |
| A | 62 | 30 | 32 | 1.138 | 0.566 |
| B | 14 | 6 | 8 | | |
| C | 15 | 5 | 10 | | |
| Recurrencea | | | | | |
| No | 37 | 22 | 15 | 5.931 | 0.015b |
| Yes | 51 | 17 | 34 | | |
| Recurrence time
(months)a | | | | | |
| <6 | 34 | 9 | 25 | 8.393 | 0.015b |
| 6≤ to <12 | 8 | 3 | 5 | | |
| ≥12 | 46 | 27 | 19 | | |
| Table IIUnivariate and multivariate analyses
of relapse-free survival rates in hepatocellular carcinoma. |
Table II
Univariate and multivariate analyses
of relapse-free survival rates in hepatocellular carcinoma.
| Variable | Univariate analysis
| Multivariate
analysis
|
|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Sex
(male/female) | 2.098 | 0.893–4.930 | 0.089 | | | |
| Age (<60/≥60
years) | 1.468 | 0.583–3.700 | 0.415 | | | |
| Hepatitis B
(no/yes) | 0.746 | 0.336–1.661 | 0.474 | | | |
| Cirrhosis
(no/yes) | 1.458 | 0.834–2.549 | 0.186 | | | |
| AFP (<400/≥400
µg/l) | 0.757 | 0.428–1.336 | 0.337 | | | |
| Tumor size | 0.615 | 0.277–1.367 | 0.233 | | | |
| Tumor number | 0.651 | 0.333–1.271 | 0.209 | | | |
| Tumor capsule
(no/yes) | 0.801 | 0.458–1.400 | 0.436 | | | |
| Differentiation
(high/low to medium) | 0.453 | 0.210–0.974 | 0.043a | 0.463 | 0.205–1.045 | 0.064 |
| Tumor emboli
(no/yes) | 0.365 | 0.188–0.709 | 0.003a | 0.597 | 0.234–1.524 | 0.281 |
| TNM stage
(T1-2/T3-4) | 0.375 | 0.207–1.680 | 0.001a | 0.367 | 0.193–0.696 | 0.002a |
| BCLC stage A
(A/C) | 0.577 | 0.283–1.176 | 0.130 | | | |
| B (B/C) | 0.908 | 0.358–2.304 | 0.839 | | | |
| KIFC1 status
(negative/positive) | 0.438 | 0.242–0.792 | 0.006a | 0.518 | 0.281–0.953 | 0.035a |
Expression of KIFC1 in HCC cell
lines
To further validate the role of KIFC1 in HCC,
western blot analysis was performed to evaluate the expression of
KIFC1 in human HCC cell lines and human normal liver epithelial
cells. The expression of KIFC1 was high in MHCC-97H, SMMC-7721 and
HCC-LM3 cells, but was absent in LO2 cells (Fig. 2A). As the HCC-LM3 and SMMC-7721
cell lines had high protein levels of KIFC1, these two cell lines
were selected for the subsequent cell function investigation.
Effects of the knockdown of KIFC1 on cell
viability, apoptosis and cell cycle in HCC-LM3 and SMMC-7721
cells
Two independent siRNAs targeting KIFC1 were used to
examine the role of KIFC1 in the development and progression of
HCC, with that the mRNA and protein expression of KIFC1 shown to be
significantly silenced by RT-qPCR and western blot analyses at 48 h
post-siRNA transfection (Fig. 2B and
C). The cell viability of the two cell lines was significantly
suppressed following KIFC1 knockdown (Fig. 2D). In addition, the KIFC1
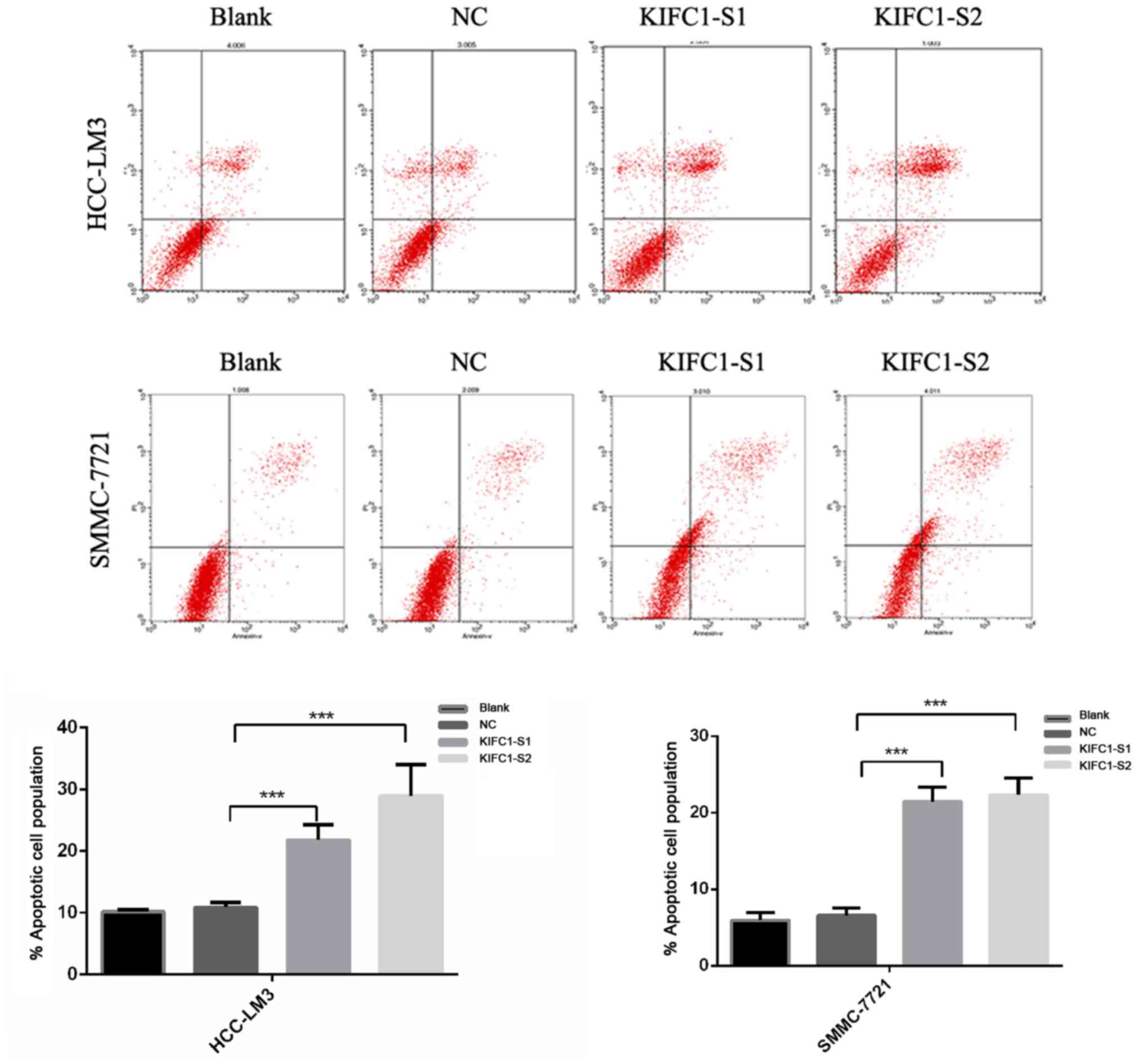
siRNA-treated cells showed a marked increase in the rate of
apoptosis, compared to the control (P<0.001) (Fig. 3). In confirming these results, it
was demonstrated that the apoptosis-related protein Bcl-2 was
downregulated, whereas Bax and p53 were upregulated (Fig. 4A). Furthermore, it was found that
the expression levels of phosphorylated (p-)phosphoinositide
3-kinase (PI3K) and p-AKT were decreased significantly when KIFC1
was silenced (Fig. 4B). However,
when DNA content was analyzed to assess cell cycle distribution,
there was minimal change in the KIFC1 siRNA-treated group, compared
with the control (P=0.254) (Fig.
5). These results demonstrated that the knockdown of KIFC1 be
an effective strategy for inhibiting HCC growth.
 | Figure 4Expression of apoptosis-related
proteins. (A) Western blots showing downregulated Bcl-2, and
upregulated Bax and p53 following KIFC1 knockdown in HCC-LM3 and
SMMC-7721 cells. *P<0.05. (B) Western blot analysis
was used to determine the PI3K/AKT pathway proteins.
*P<0.05. KIFC1, kinesin family member C1; Bcl-2,
B-cell lymphoma-2; Bax, Bcl-2-associated X protein; PI3K,
phosphoinositide 3-kinase, p-, phosphorylated; NC, negative
control; si-1/S1, small interfering RNA 1; si-2/S2, small
interfering RNA 2. |
Effects of KIFC1 knockdown on inhibition
of HCC-LM3 and SMMC-7721 cell migration and invasion
To investigate the effects of the inhibition of
KIFC1 on HCC cell migration and invasion, Transwell assays were
performed following transfection of KIFC1 siRNA or control siRNA
into the two HCC cell lines. KIFC1 siRNA-treated cells had markedly
impaired migration ability, compared with the blank and control
groups (P<0.001) (Fig. 6), and
there was a parallel downward trend in invasive ability
(P<0.001) (Fig. 6). In
addition, the expression of EMT-related proteins, including
N-cadherin, matrix metalloproteinase-2 (MMP-2), β-catenin, Slug,
Zinc finger E-box-binding homeobox 1 (ZEB1) and E-cadherin, were
examined using western blot analysis. Compared with the negative
control group, N-cadherin, MMP-2, β-catenin, Slug and ZEB1 were
downregulated, whereas E-cadherin was upregulated (Fig. 7). These results indicated that the
expression of KIFC1 may be associated with tumor invasion and
metastasis in HCC.
Discussion
HCC is a life-threatening malignancy worldwide
(1). Although traditional TNM
classification systems provide a basic prognostic model, difficulty
remains in differentiating outcomes considering the asymptomatic
nature of early-stage HCC (29).
Therefore, it is imperative to clarify the molecular mechanism
underlying the progression of HCC and identify a novel HCC marker
and therapeutic target.
KIFC1, a minus end-directed motor protein (14), is critical in centrosome clustering
in cancer cells and is essential for cancer cell survival (15). KIFC1 predicts poor prognosis and
shorter overall survival rate, and may serve as a potential marker
of metastasis in ovarian cancer (22). In non-small cell lung cancer,
together with Alzheimer antigen and N-cadherin, the overexpression
of KIFC1 predicts brain metastasis in patients with early and
advanced disease (23). KIFC1 is
also overexpressed in breast cancer tissues and cell lines, and
KIFC1 knockdown reduces breast cancer cell viability (24). However, the correlation between the
expression of KIFC1 and HCC has not been reported. KIFC1 is
essential for bipolar mitotic division of cancer cells, but appears
redundant in normal cells (15,21),
suggesting that KIFC1 is a promising thera peutic target in
investigations on highly selective anticancer agents. The present
study reported for the first time, to the best of our knowledge,
that KIFC1 was expressed at a high level in HCC tissues, compared
with that in peritumoral tissues, and its expression was correlated
with tumor emboli, metastasis, recurrence and time of recurrence.
Kaplan-Meier analysis showed that the expression of KIFC1 was
significantly associated with tumor-free survival rates. In
addition, multivariate analyses revealed that the overexpression of
KIFC1 was an independent predictive marker in patients with HCC.
Consistently, data derived from the TCGA and the GTEx projects via
the online GEPIA database confirmed that the expression of KIFC1
was upregulated and significantly associated with disease-free
survival and overall survival rates in HCC. These results indicated
that a high expression of KIFC1 may be a crucial prognostic
indicator for HCC.
In the present study, KIFC1 knockdown significantly
suppressed HCC cell viability and markedly increased the rate of
apoptosis. In addition, Bcl-2 was downregulated, whereas Bax and
p53 were upregulated; these proteins act as regulatory proteins in
the process of apoptosis (30).
Apoptosis is one of the most characteristic hallmarks of the cell
death process (31). Dysregulation
of the balance between cell death and proliferation has been
identified as a protumorigenic process during hepatic tumorigenesis
(32). These results provide novel
clues to the development and progression of HCC, and indicate that
silencing KIFC1 can be critical in the anti-apoptotic process and
correlate closely with apoptosis-related proteins. In addition, a
previous study indicated that the PI3K/Akt signal pathway is
involved in cell proliferation and apoptosis (33). In the present study, KIFC1
knockdown inhibited the expression of p-PI3K and p-PAKT, which
indicated that the downregulation of KIFC1 in HCC cells induced
apoptosis via the PI3K/AKT pathway. Investigations in the future
aim to elucidate the mechanisms more directly.
In cancer, metastasis is considered the leading
cause of mortality (34). The poor
prognosis of patients with HCC is mainly due to intrahepatic and
distant metastasis. Metastasis involves a series of interdependent
events, including cancer cell proliferation, migration and invasion
(35). EMT promotes HCC cell
migration and invasive ability (36). Evidence suggests that EMT confers
tumor cells with various malignant characteristics, including
increased mobility, aggressiveness, evasion from apoptosis and a
stem-like phenotype (37).
Accordingly, the present study investigated the effects of the
inhibition of KIFC1 on HCC cell migration and invasion via a
Transwell assay, and the resulting data indicated that KIFC1 was
involved in tumor metastasis. In addition, the present study
examined the expression of the EMT-related proteins, N-cadherin,
MMP-2, β-catenin, Slug, ZEB1 and E-cadherin, using western blot
analysis. The results of the western blot analysis were in
agreement with those of the Transwell assay, and also showed that
KIFC1 mediated HCC metastasis by modulating EMT. Therefore, KIFC1
knockdown may be an ideal therapeutic strategy for metastatic
HCC.
In conclusion, the present study is the first, to
the best of our knowledge, to show that KIFC1 was overexpressed in
HCC tissues compared with peritumoral tissues. Based on the above
results, it is possible to conclude that the overexpression of
KIFC1 is correlated closely with the progression of HCC and poor
prognosis, and the expression level of KIFC1 may be a potential
prognostic biomarker and therapeutic target for HCC.
Acknowledgments
Not applicable.
Funding
This study was supported in part by grants from the
Key Research Project of Jiangxi Province, China (grant no.
20171BBG70063), the Natural Science Foundation of Jiangxi Province,
China (grant no. 20171BAB215038), the Health Department of Jiangxi
Province, China (grant no. 20131077), the Natural Science
Foundation of nation, China (grant no. 81760438) and the Innovation
Research Foundation of Graduate School of Nanchang University
(grant no. cx2016330).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
XF, BL and LF were involved in the conception and
design of the study; XF, YZh, BZ, YZo, CW, PW and JW were involved
in data acquisition; XF, YZh and BL were involved in data analysis
and interpretation; HC and PD were invovled in patient follow-up;
XF, YZh and BL performed the statistical analysis; XF and LF
drafted and edited the manuscript; XF, YZh, BL and LF revised and
reviewed the manuscript. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Second Affiliated Hospital of Nanchang University,
written consent was obtained from each participant, and the study
was performed in accordance with the ethical standards of the
Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Llovet JM and Bruix J: Molecular targeted
therapies in hepatocellular carcinoma. Hepatology. 48:1312–1327.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feo F, Pascale RM, Simile MM, De Miglio
MR, Muroni MR and Calvisi D: Genetic alterations in liver
carcinogenesis: Implications for new preventive and therapeutic
strategies. Crit Rev Oncog. 11:19–62. 2000. View Article : Google Scholar
|
|
3
|
Michielsen PP, Francque SM and van Dongen
JL: Viral hepatitis and hepatocellular carcinoma. World J Surg
Oncol. 3:272005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun W, Xing B, Sun Y, Du X, Lu M, Hao C,
Lu Z, Mi W, Wu S, Wei H, et al: Proteome analysis of hepatocellular
carcinoma by two-dimensional difference gel electrophoresis: Novel
protein markers in hepatocellular carcinoma tissues. Mol Cell
Proteomics. 6:1798–1808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tang ZY: Hepatocellular carcinoma - cause,
treatment and metastasis. World J Gastroenterol. 7:445–454. 2001.
View Article : Google Scholar
|
|
6
|
Sherman M: Modern approach to
hepatocellular carcinoma. Curr Gastroenterol Rep. 13:49–55. 2011.
View Article : Google Scholar
|
|
7
|
Calvisi DF, Pascale RM and Feo F:
Dissection of signal transduction pathways as a tool for the
development of targeted therapies of hepatocellular carcinoma. Rev
Recent Clin Trials. 2:217–236. 2007. View Article : Google Scholar
|
|
8
|
Fukasawa K: Oncogenes and tumour
suppressors take on centrosomes. Nat Rev Cancer. 7:911–924. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogden A, Rida PC and Aneja R: Let's huddle
to prevent a muddle: Centrosome declustering as an attractive
anticancer strategy. Cell Death Differ. 19:1255–1267. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anderhub SJ, Krämer A and Maier B:
Centrosome amplification in tumorigenesis. Cancer Lett. 322:8–17.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ogden A, Rida PC and Aneja R: Heading off
with the herd: How cancer cells might maneuver supernumerary
centrosomes for directional migration. Cancer Metastasis Rev.
32:269–287. 2013. View Article : Google Scholar :
|
|
12
|
Pannu V, Rida PC, Ogden A, Clewley R,
Cheng A, Karna P, Lopus M, Mishra RC, Zhou J and Aneja R: Induction
of robust de novo centrosome amplification, high-grade spindle
multipolarity and metaphase catastrophe: A novel chemotherapeutic
approach. Cell Death Dis. 3:e3462012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ando A, Kikuti YY, Kawata H, Okamoto N,
Imai T, Eki T, Yokoyama K, Soeda E, Ikemura T, Abe K, et al:
Cloning of a new kinesin-related gene located at the centromeric
end of the human MHC region. Immunogenetics. 39:194–200. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeLuca JG, Newton CN, Himes RH, Jordan MA
and Wilson L: Purification and characterization of native
conventional kinesin, HSET, and CENP-E from mitotic hela cells. J
Biol Chem. 276:28014–28021. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kleylein-Sohn J, Pöllinger B, Ohmer M,
Hofmann F, Nigg EA, Hemmings BA and Wartmann M: Acentrosomal
spindle organization renders cancer cells dependent on the kinesin
HSET. J Cell Sci. 125:5391–5402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nath S, Bananis E, Sarkar S, Stockert RJ,
Sperry AO, Murray JW and Wolkoff AW: Kif5B and Kifc1 interact and
are required for motility and fission of early endocytic vesicles
in mouse liver. Mol Biol Cell. 18:1839–1849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hall VJ, Compton D, Stojkovic P, Nesbitt
M, Herbert M, Murdoch A and Stojkovic M: Developmental competence
of human in vitro aged oocytes as host cells for nuclear transfer.
Hum Reprod. 22:52–62. 2007. View Article : Google Scholar
|
|
18
|
Yang WX and Sperry AO: C-terminal kinesin
motor KIFC1 participates in acrosome biogenesis and vesicle
transport. Biol Reprod. 69:1719–1729. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang WX, Jefferson H and Sperry AO: The
molecular motor KIFC1 associates with a complex containing
nucleoporin NUP62 that is regulated during development and by the
small GTPase RAN. Biol Reprod. 74:684–690. 2006. View Article : Google Scholar
|
|
20
|
Farina F, Pierobon P, Delevoye C, Monnet
J, Dingli F, Loew D, Quanz M, Dutreix M and Cappello G: Kinesin
KIFC1 actively transports bare double-stranded DNA. Nucleic Acids
Res. 41:4926–4937. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kwon M, Godinho SA, Chandhok NS, Ganem NJ,
Azioune A, Thery M and Pellman D: Mechanisms to suppress multipolar
divisions in cancer cells with extra centrosomes. Genes Dev.
22:2189–2203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pawar S, Donthamsetty S, Pannu V, Rida P,
Ogden A, Bowen N, Osan R, Cantuaria G and Aneja R: KIFCI, a novel
putative prognostic biomarker for ovarian adenocarcinomas:
Delineating protein interaction networks and signaling circuitries.
J Ovarian Res. 7:532014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grinberg-Rashi H, Ofek E, Perelman M,
Skarda J, Yaron P, Hajdúch M, Jacob-Hirsch J, Amariglio N, Krupsky
M, Simansky DA, et al: The expression of three genes in primary
non-small cell lung cancer is associated with metastatic spread to
the brain. Clin Cancer Res. 15:1755–1761. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Lu W, Chen D, Boohaker RJ, Zhai L,
Padmalayam I, Wennerberg K, Xu B and Zhang W: KIFC1 is a novel
potential therapeutic target for breast cancer. Cancer Biol Ther.
16:1316–1322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De S, Cipriano R, Jackson MW and Stark GR:
Overexpression of kinesins mediates docetaxel resistance in breast
cancer cells. Cancer Res. 69:8035–8042. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chun YH, Kim SU, Park JY, Kim DY, Han KH,
Chon CY, Kim BK, Choi GH, Kim KS, Choi JS, et al: Prognostic value
of the 7th edition of the AJCC staging system as a clinical staging
system in patients with hepatocellular carcinoma. Eur J Cancer.
47:2568–2575. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang B, Zheng W, Fang L, Wu L, Zhou F,
Yin X, Yu X and Zou Z: Overexpressed targeting protein for Xklp2
(TPX2) serves as a promising prognostic marker and therapeutic
target for gastric cancer. Cancer Biol Ther. 17:824–832. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huitzil-Melendez FD, Capanu M, O'Reilly
EM, Duffy A, Gansukh B, Saltz LL and Abou-Alfa GK: Advanced
hepatocellular carcinoma: Which staging systems best predict
prognosis? J Clin Oncol. 28:2889–2895. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hao J, Yang Z, Wang L, Zhang Y, Shu Y,
Jiang L, Hu Y, Lv W, Dong P and Liu Y: Downregulation of BRD4
inhibits gallbladder cancer proliferation and metastasis and
induces apoptosis via PI3K/AKT pathway. Int J Oncol. 51:823–831.
2017. View Article : Google Scholar :
|
|
31
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fabregat I: Dysregulation of apoptosis in
hepatocellular carcinoma cells. World J Gastroenterol. 15:513–520.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin HL, Yang MH, Wu CW, Chen PM, Yang YP,
Chu YR, Kao CL, Ku HH, Lo JF, Liou JP, et al: 2-Methoxyestradiol
attenuates phosphatidylinositol 3-kinase/Akt pathway-mediated
metastasis of gastric cancer. Int J Cancer. 121:2547–2555. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rudmik LR and Magliocco AM: Molecular
mechanisms of hepatic metastasis in colorectal cancer. J Surg
Oncol. 92:347–359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kumar S and Weaver VM: Mechanics,
malignancy, and metastasis: The force journey of a tumor cell.
Cancer Metastasis Rev. 28:113–127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
El-Nahas AM: Plasticity of kidney cells:
Role in kidney remodeling and scarring. Kidney Int. 64:1553–1563.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|