Introduction
Although the latest statistics suggest that the
overall incidence of prostate cancer (PCa) has rapidly declined in
the United States, accounting for approximately one-half of total
decline in male cancers (1), PCa
still remains a major health concern in developed countries. In
addition, the mortality rate associated with PCa is rising at a
rate of 5% per year in China (2).
Androgen deprivation therapy (ADT), such as abiraterone and
enzalutamide (mainly bicalutamide in China), is a mainstream
treatment strategy. The treatments are initially effective for
patients. However, the relief is temporary and castration-resistant
prostate cancer (CRPC) emergences within a few years (3). Chemotherapeutic agents, such as
docetaxel and cabazitaxel, are considered to be the preferred
treatment strategy following resistance to ADT (4,5).
However, sequential dual-resistance to androgen receptor (AR) axis
inhibitors and taxanes occurs with a lethal outcome within a few
months (6,7). However, there are few therapeutic
approaches available with which to combat the sequential
dual-resistant PCa. Thus, the development of novel therapeutic
strategies with which to combat refractory PCa is urgently
required.
Hepatocyte cell adhesion molecule (HepaCAM), a
member of the Ig superfamily, was first proven to be decreased or
undetectable in hepatocellular carcinoma (8). HepaCAM exerts a marked antitumor
effect by inhibiting proliferation, inducing apoptosis and
suppressing migration in multiple cancer types (9–16).
In our previous study, it was reported that HepaCAM downregulates
AR, leading to the suppression of the biological behavior of PCa
cell lines (17). However, the
role of HepaCAM remains unknown in CRPC. Moreover, as HepaCAM has
been identified to decrease AR amplification, which is responsible
for castration resistance, we wished to determine whether HepaCAM
can reverse the resistance of the resistant cells to the AR axis
inhibitor, enzalutamide.
The Notch signaling pathway has been proven to be
associated with cell differentiation, proliferation and apoptosis
(18). Furthermore, the
constitutive expression of the Notch intracellular domain (NICD)
has been shown to suppress the apoptosis of luminal epithelial
cells and stimulate luminal cell proliferation in the prostate
(18,19). Overactivated Notch signaling has
been found in PCa, including CRPC, which promotes PCa progression
(20,21). The downregulation of Notch has been
shown to significantly inhibit the proliferation, invasion and
migration of PCa cells in vitro (22–25).
PF-3084014, a γ-secretase inhibitor, suppresses Notch activity by
blocking NICD formation, and results in the inhibition of tumor
cells in diverse cancer types (26–28).
However, it is unclear as to whether PF-3084014 exerts an antitumor
effect on the resistant cells. A recent study demonstrated that
PF-3084014 restores the sensitivity of docetaxel-resistant PCa
cells to docetaxel through the downregulation of Notch signaling
in vitro and in vivo (22). However, it is unknown as to whether
PF-3084014 restores the sensitivity of enzalutamide-resistant
(Enza-R) cells to enzalutamide, and sequential dual-resistant
(E+D-R) cells to docetaxel.
In this study, we detected the expression of HepaCAM
in matched primary prostate cancer (PPC) and CRPC tissues, and
observed the differences in the expression of HepaCAM, Notch1 and
Hes1 between the matched PPC and CRPC specimens. We further
explored the correlations between the HepaCAM and Notch axis in
CRPC tissues and cell lines. Additionally, we evaluated the
sensitivities of Enza-R and E+D-R cells to enzalutamide and
docetaxel, respectively following the downregulation of Notch
activity by overexpressing HepaCAM and/or treatment with
PF-3084014. The findings of this study may provide a novel
treatment approach for patients with refractory PCa.
Materials and methods
Patients and tissue samples
Patients were included in this study by our
inclusion standard as follows: i) All patients met the EAU
guidelines for confirming CRPC (29). Serum testosterone levels at
castration levels (<1.7 nmol/l) plus either: a) Three
consecutive increases in serum prostate-specific antigen (PSA)
levels, 1 week apart, leading to two 50% increases over the nadir
with PSA levels >2.0 ng/ml; b) the appearance of new lesions and
the progression of the primary lesion: New bone lesions and a soft
tissue lesion (including prostate, bladder neck, seminal vesicle
and other viscera) using TRUS or/and MRI. ii) All patients had
available matched PPC and CRPC specimens. iii) All patients had
complete clinical data, including PPC and CRPC data. If patients
met the inclusion standard 'i', the tissues obtained from the
prostate lesions were regarded as CRPC specimens (30). According to the inclusion standard,
45 CRPC and 41 matched PPC samples (4 cases with clinical data of
PPC, but without PPC tissue specimens) were collected at the
Department of Urology at the First Affiliated Hospital of Chongqing
Medical University, Chongqing, China between April, 2008 and
September, 2016. CRPC specimens of prostate lesions were obtained
from the patients by transurethral resection of the prostate (TURP,
30 cases) or needle biopsy (15 cases). All samples were reviewed by
a pathologist for the confirmation of PCa. Gleason's score was
evaluated not only in the PPC tissues, but also in the CRPC tissues
with the help of a pathologist who was blinded to the clinical data
and assessed Gleason's scores in the tissue samples. This study was
approved by the Ethics Committee of Chongqing Medical University.
Informed consent was obtained from the patients or their family
members who agreed to the use of their samples in this study.
Immunohistochemistry assay
All the embedded samples, including the 41 PPC
specimens and 45 matched CRPC specimens (30 cases from TURP and 15
cases from needle biopsy), were cut into 5-μm-thick
sections. The immunoreactivities of HepaCAM, Notch1 and Hes1 were
detected using a standard immunoperoxidase staining procedure
(anti-HepaCAM, 1:200; cat. no. 18177-1-AP; ProteinTech, Wuhan,
China; anti-Notch1, 1:200, cat. no. ab52627; anti-Hes1, 1:200, cat.
no. ab108937; both from Abcam, Cambridge, UK). Staining scoring was
semi-quantitatively assessed using staining intensity and was
defined as 0, no staining; 1, weak staining; 2, light staining; 4,
moderate staining; and 6 and 8, strong staining. Staining scores of
≤1 were regarded as negative expression, while staining scores of
≥2 were regarded as positive expression.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from all cell lines using
TRIzol reagent, and reversed transcribed into cDNA using the Prime
Script™ RT reagent kit (both from Takara, Dalian, China). SYBR
PremixEx Taq™ II kit (Takara) was used for RT-qPCR with the CFX96™
Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The
sequences of the primers were as follows: HepaCAM sense,
5′-TACTGTAGATGTGCCCATTTCG-3′ and antisense,
5′-CTTCTGGTTTCAGGCGGTC-3′; Notch1 sense, 5′-GAAC
GGGGCUAACAAAGAUTT-3′ and antisense, 5′-AUCUUU GUUAGCCCCGUUCTT-3′;
Hes1 sense, 5′-GGACTAGTATGCCAGCTGATATAATGGAG-3′ and antisense,
5′-GAAGATCTAGGTGGGCTAGGGACTTTAC-3′; Jagged1 sense,
5′-GTGCCGCCATAGGTAGAGT-3′ and antisense, 5′-CCAGCCAACCACAGAAAC-3′;
and β-actin sense, 5′-TGACGTGGACATCCGCAAAG-3′ and antisense,
5′-CTGGAAGGTGGACAGCGAGG-3′. The thermocycling conditions of RT-qPCR
were as follows: Initial denaturation, 95°C for 3 min; 95°C for 10
sec, 60°C for 20 sec, 72°C for 20 sec, 40 cycles; final extension:
72 °C for 5 min. The mRNA expression levels were calculated using
the comparative 2−ΔΔCq method (31) and β-actin served as a calibrator.
All gene expression experiments were repeated at least 3 times.
Western blot analysis
Total protein was extracted from the cell lines
(please see cell lines below) and tissue samples using RIPA buffer
containing the phosphatase inhibitors, NaF and
Na3VO4, and the protease inhibitor, PMSF
(Beyotime Institute of Biotechnology, Beijing, China). The protein
concentration was determined using the BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Protein samples (50
μg), stacked by 5% SDS-PAGE and separated by 10 or 12%
SDS-PAGE, were transferred to PVDF membranes (EMD Millipore,
Billerica, MA, USA). After blocking with 5% non-fat milk for 2 h at
room temperature, the membranes were incubated with the following
primary antibodies overnight at 4°C: Anti-E-Cadherin (1:1,000; cat.
no. 3195), anti-N-cadherin (1:1,000; cat. no. 4061), anti-Snail
(1:1,000; cat. no. 3895) were obtained from Cell Signaling
Technology (Danvers, MA, USA). Anti-Jagged1 (1:1,000; cat. no.
ab109536), anti-Notch1 (1:2,000; cat. no. ab52627), anti-NICD
(1:500; cat. no. ab83232), anti-Hes1 (1:1,000; cat. no. ab108937)
were from Abcam. Anti-HepaCAM (1:500; cat. no. 18177-1-AP) was
purchased from ProteinTech. Anti-GAPDH (1:1,000; cat. no. 5174;
Cell Signaling Technology) was used as loading control. The
membranes were then incubated with the following secondary
antibodies for 2 h at room temperature: Goat anti-mouse IgG
(1:3,000; cat. no. SA00001-1), goat anti-rabbit IgG (1:3,000; cat.
no. SA00001-2) (obtained from ProteinTech). The enhanced
chemiluminescent (ECL) kit was purchased from Merck Millipore
(Billerica, MA, USA). The intensity level of the protein expression
bands was evaluated using Image-Pro plus 6.0.
Cells cell culture, treatment and
transfection
Human prostate cell lines (RWPE-1, LNCaP and DU145)
were obtained from the American Type Culture Collection (ATCC,
Manassas, VA, USA). The 293A cell line was a gift from Professor
Wenli Luo, Key Laboratory of Laboratory Medical Diagnostics,
Ministry of Education, Department of Laboratory Medicine, Chongqing
Medical University, Chongqing, China. All the cell lines were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS) (both from Gibco-Life Technologies, Carlsbad, CA, USA) and 1%
penicillin/streptomycin (Beyotime Institute of Biotechnology). To
generate bicalutamide-resistant cells and enzalutamide-resistant
cells, the LNCaP cells, one of the androgen-dependent prostate
cancer cell strains, were treated with enzalutamide (10 μM)
(32) and bicalutamide (10
μM) (Selleck Chemicals, Houston, TX, USA), respectively for
at least 6 months. For the generation of bicalutamide-resistant
(Bica-R) cells, the cells were first cultured with 1 μM
(33), or 5, 10 or 25 μM
(34) bicalutamide, respectively.
We found that the concentration of 1 μM bicalutamide had
almost no effect on the LNCaP cells, and the concentration of 25
μM bicalutamide killed too many cells to induce the cells
continually (data not shown). Moreover, similar to treatment with
10 μM enzalutamide, treatment with 10 μM bicalutamide
inhibited cell growth by 60 to 70% (data not shown). After
screening, we selected the concentration of 10 μM of
bicalutamide by ourselves to generate Bica-R cells. The HepaCAM
plasmid was transfected into 293A cells using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's instructions. Adenoviruses carrying
HepaCAM (Ad-HepaCAM) were stored at −80°C and amplified in 293A
cells. The viral fluid was obtained after freezing and thawing the
293A cells repeatedly. The prostate cancer cell strains were
transfected with Ad-HepaCAM or Ad-GFP, respectively. After 72 h of
incubation, follow-up experiments were performed. The cells were
treated with the concentration of 5 μM PF-3084014 for 48 h
(Med Chem Express, Monmouth Junction, NJ, USA).
We also constructed docetaxel-resistant cells based
on the LNCaP cell line and sequential dual-resistant cells to
enzalutamide and docetaxel based on the Enza-R cells. The LNCaP and
Enza-R cells were respectively incubated with various
concentrations of docetaxel (0.1, 0.5, 1, 2 and 5 nM; Med Chem
Express) and the growth of the cells was observed. We found that
the concentrations of 0.1 and 0.5 nM docetaxel were not able to
inhibit cell growth effectively, and the concentrations of 2 and 5
nM docetaxel killed too many cells to culture continuously (data
not shown). Moreover, the concentration of 1 nM docetaxel inhibited
the growth of both the LNCaP and Enza-R cells by 60 to 70% (data
not shown). Therefore, we selected the concentration of 1 nM of
docetaxel as the initial concentration of administration. The LNCaP
and Enza-R cells were treated with 1 nM docetaxel every 24 h for 3
weeks. Moreover, when the morphology of the cells exhibited
alterations, such as cell membrane shrinkage and even disruption,
and acquired a thin and small, polygonal shape, or the cells
stopped growing, treatment was halted until the cells recovered.
The drug concentration was increased when the cells were able to
tolerate the current concentration. Each time the drug
concentration was increased, some aliquots of cells were stored.
When the cells were killed or contaminated, we resuscitated the
aliquots. The frozen cells were removed from liquid nitrogen and
placed in a 37°C water bath for 20–30 sec. The thawed cells were
added to RPMI-1640. Following centrifugation (1,000 rpm; 5 min),
the cells were incubated with a lower concentration of docetaxel.
By the stepwise exposure method, the cells were cultured until they
were able to tolerate 10 nM docetaxel (35) in 2 months. The cells were
maintained in 10 nM docetaxel for at least 4 months. The
docetaxel-resistant cells were termed Doce-R cells and sequential
dual-resistant cells (resistant to enzalutamide and docetaxel) were
termed E+D-R cells.
Cell counting kit-8 (CCK-8) assay
CCK-8 assay for cell viability, the cells were
plated in 96-well plates (2,000 cells/well), and incubated for 12
h. The cells were then cultured with the various treatment agents
in each 3 replicate wells. Each well was supplemented with 10
μl CCK-8 reagent (Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China). Following incubation for 1 h. Optical
density was detected at absorbance of 450 nm using a microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). CCK-8 assay
for the half maximal inhibitory concentration (IC50) of
enzalutamide or docetaxel to the cells, the resistant cells (4,000
cells/well) pretreated with various reagents, such as Ad-HepaCAM or
Ad-GFP, were seeded into 96-well plates and incubated for 12 h. The
cells were then treated with various concentrations of enzalutamide
or docetaxel in each 3 replicate wells for 24 h. DMSO
(Sigma-Aldrich; Thermo Fisher Scientific, Inc.) was used as the
control. For CCK-8 assay for the viability of the Enza-R, Doce-R,
E+D-R cells treated with PF-3084014, the cells (4,000 cells/well)
were treated with increasing concentrations of PF-3084014 for 48 h
(5, 10, 20, 30, 40, 60, 80 and 100 μM) and DMSO for 48 h was
used as the control.
Colony formation assay
The cells (400 cells/well) were plated in 6-well
plates, and were consecutively cultured until the numbers of each
clone reached 50 cells under a microscope (Nikon, Tokyo, Japan).
The clones were stained with 0.05% crystal violet solution
(Beyotime Institute of Biotechnology) for 20 min at room
temperature. The colony formation experiments were performed at
least 3 times.
Transwell and wound healing assay
For Transwell assay, 1.0×104 cells were
plated in the upper chamber of the insert with Matrigel (BD
Biosciences, San Jose, CA, USA). The cells were incubated with
serum-free medium for 48 h. The cells were then stained with 0.1%
crystal violet and 4% formaldehyde (Beyotime Institute of
Biotechnology). The number of cells, fixed on the bottom membrane
of the inserts was counted under a microscope (Nikon). For wound
healing assay, 5×104 cells/well were seeded into a
6-well plate. Following 24 h of incubation, the cells were wounded
with a yellow pipette tip. The cells were then cultured for 24 h
and the wound healing was observed under a microscope (Nikon) at
indicated time-points.
Immunofluorescence
A total of 1.0×105 cells/well were plated
into a 12-well plate inserted with glass coverslips. Following
incubation for 24 h, the cells were fixed in 4% paraformaldehyde
for 20 min, and incubated with primary antibody (anti-Notch1 1:50;
anti-Hes1, 1:100) (both from Abcam) overnight at 4°C. The cells
were then incubated with secondary antibody (Zhongshan Golden
Bridge Biotechnology, Beijing, China) for 50 min in a dark room at
room temperature. The cell nuclei were stained with DAPI (Zhongshan
Golden Bridge Biotechnology) for 10 min. Immunofluorescence images
were obtained using a fluorescence microscope (Nikon).
Statistical analysis
Statistical analyses were performed using SPSS 19.0
software (IBM SPSS Corp., Armonk, NY, USA). All the numerical data
are expressed as the means ± SD. Data were analyzed using
Kaplan-Meier survival analysis, one-way ANOVA, two-way ANOVA, the
Student's t-test, Pearson's correlation analysis, Spearman's
correlation analysis, the Mann-Whitney test, McNemer test, the
Chi-square test for trend and Pearson's Chi-square test where
appropriate. Values of P<0.05 were considered to indicate
statistically significant differences.
Results
HepaCAM negativity is associated with the
upregulation of Notch1 and Hes1 in CRPC samples
We collected 45 CRPC samples and 41 matched PPC
samples (PPC specimens of 4 patients were unavailable) (Table I). The expression of HepaCAM was
detected in the matched PPC and CRPC tissues by
immunohistochemistry assay. In total, 71% (32/45) of the CRPC
samples exhibited HepaCAM negative staining (staining scores ≤1),
whereas HepaCAM expression was negative in 58% (24/41) of the
matched PPC samples (Table I and
Fig. 1A).
 | Figure 1The expression levels of HepaCAM,
Notch1 and Hes1 in matched primary prostate cancer (PPC) and
castration-resistant prostate cancer (CRPC) samples. (A, panel 1)
Moderate staining of HepaCAM in sites where the gland structures
were presented (red arrows), and weak staining of HepaCAM in sites
where the gland structures disappeared (yellow arrows)
(magnification, ×200). (A, panel 2) The staining of HepaCAM was
undetectable in CRPC tissues (magnification, ×200). (B, panel 1)
Needle biopsy sample with moderate staining of Notch1 in PPC
tissues (magnification, ×200). (B, panel 2) Prostatectomy specimen
with strong staining of Notch1 in CRPC tissues (magnification,
×200). (C, panel 1) Hes1 protein was weakly expressed in sites
where the gland structure was presented (upper left-hand corner,
×400) and was moderately expressed in sites where the gland
structure was disorganized (upper right-hand corner; magnification,
400). (C, panel 2) Strong nuclear positivity of Hes1 in CRPC
tissues (magnification, ×400). (D–F) Average staining scores for
HepaCAM, Notch1 and Hes1 in matched PPC and CRPC tissues. (G and H)
The correlation curve analysis for HepaCAM staining scores versus
Notch1 and Hes1 staining scores in CRPC tissues. Values of
P<0.05 were considered to indicate statistically significant
differences. |
 | Table IDemographic and clinical
characteristics of the patients with castration-resistant prostate
cancer. |
Table I
Demographic and clinical
characteristics of the patients with castration-resistant prostate
cancer.
| Overall
n=45 | HepaCAM
| P-value |
|---|
Negative
32/45 (71%) | Positive
13/45 (29%) |
|---|
| Age, years | | | | P=0.93a |
| Median | 73 | 74 | 71 | |
| Quartiles
25–75 | 63–78 | 62–78 | 64–76 | |
| PSA of PPC
μg/l | n=45 | 24/41e | 17/41 | P=0.27a |
| Median | 92.49 | 134.61 | 71.19 | |
| Quartiles
25–75 | 28.34–239.4 | 63.06–195.00 | 19.83–186.15 | |
| PSA of CRPC
μg/l | n=45 | 32/45 | 13/45 | P=0.14a |
| Median | 39.98 | 37.85 | 41.47 | |
| Quartiles
25–75 | 19.32–152.9 | 17.79–99.50 | 23.55–198.50 | |
| Gleason score of
PPC | n=45 (%) | 24/41 (%) | 17/41 (%) | P=0.41b |
| ≤6 | 18/45 (40) | 8/24 (33) | 7/17 (41) | |
| 7 | 14/45 (31) | 7/24 (29) | 6/17 (35) | |
| ≥8 | 13/45 (29) | 9/24 (38) | 4/17 (24) | |
| Gleason score of
CRPCf | | 32/45 (%) | 13/45 (%) |
P=0.011b |
| ≤6 | 6/45 (13) | 2/32 (6) | 4/13 (31) | |
| 7 | 10/45 (22) | 6/32 (19) | 4/13 (31) | |
| ≥8 | 29/45 (65) | 24/32 (75) | 5/13 (38) | |
| Metastases sites of
PPC | | | | |
| Bone | 11/45 (24) | 7/11 (64) | 4/11 (36) | P=0.263c |
| Nodle | 10/45 (22) | 4/10 (40) | 6/10 (60) | P=0.388c |
| Visceral | 15/45 (33) | 6/15 (40) | 9/15 (60) | P=0.791c |
| Metastases sites of
CRPC | | | | |
| Bone | 32/45 (71) | 26/32 (81) | 6/32 (19) |
P=0.001c |
| Drugs for initial
treatment | n=45 (%) | 32/45 | 13/45 | P=0.607d |
| Bicalutamide | 35/45 (71) | 26/32 (81) | 9/13 (69) | |
| Other durgs | 10/45 (29) | 6/32 (19) | 4/13 (31) | |
We then determine whether there were any differences
in HepaCAM, Notch1 and Hes1 expression levels between the matched
PPC and CRPC tissues. In comparison to the matched PPC tissues, the
expression of HepaCAM was lost more frequently, (P=0.036; Fig. 1A, panels 1 and 2, and D), and the
expression of Hes1 was upregulated (P=0.0237; Fig. 1C, panels 1 and 2, and F) in the
CRPC tissues. We failed to observe any differences in the
expression of Notch1 between the matched PPC and CRPC tissues (P=
0.063; Fig. 1B, panels 1 and 2, and
E). We also evaluated whether the loss of HepaCAM correlated
with increased Notch1 and Hes1 expression levels in the CRPC
samples using Pearson's linear correlation. As shown in Fig. 1G and H, the loss of HepaCAM
negatively correlated with an increase in Notch1 expression
(r=−0.652, P<0.01), as well as an increase in Hes1 expression
(r=−0.442, P=0.02). The results of western blot analysis revealed a
similar result in 14 CRPC samples obtained by needle biopsy and
TURP (part of the 45 CRPC samples) (Fig. 2A–C).
To examine the association between the protein
expression of HepaCAM and dynamic alterations in gland morphology
in the matched tissues, Gleason's score, a system for assessing
gland morphology, was evaluated not only in the PPC tissues, but
also in CRPC tissues with the help of a pathologist. Our data
revealed that, compared to HepaCAM positivity in the CRPC tissues,
HepaCAM negativity was associated with higher Gleason scores
(P=0.011) (Table I), suggesting
that HepaCAM plays an important role in maintaining normal gland
morphology. Moreover, the loss of HepaCAM in the CRPC samples, but
not in the matched PPC samples was found to be associated with bone
metastases (P=0.001) (Table I)
suggesting that patients with CRPC with HepaCAM negativity are
prone to bone metastases. Kaplan-Meier survival analysis revealed
that the median PFS was 39 months (95% CI, 26–52 months) in the
patients with CRPC with HepaCAM positivity, while the median PFS
was 27 months (95% CI, 20–34 months) in the HepaCAM-negative
patients. HepaCAM negativity in the CRPC tissues was associated
with a shorter PFS in the patients with CRPC (P=0.039) (Fig. 2D).
Overexpression of HepaCAM suppresses the
proliferation, invasion and migration of the resistant cells
In China, the cost of the use of enzalutamide is
high, and thus the majority of patients cannot afford treatment
with this agent. Patients with PCa are willing to be treated with
bicalutamide. In this study, 78% of the patients (35/45) were
treated with bicalutamide (Table
I). Enzalutamide is widely used in the treatment of PCa in
developed countries. Therefore, in this study, we constructed both
Bica-R cells and Enza-R cells, as described in the Materials and
methods. Western blot analysis was performed to detect the
expression levels of HepaCAM in the RWPE-1, LNCaP, Bica-R cells and
Enza-R cells. As shown in Fig. 3A and
B, HepaCAM was highly expressed in the RWPE1 cells, whereas it
was almost undetectable in the other cell lines. To determine the
role of HepaCAM in the proliferative capacity of the resistant
cells, adenoviral vectors, carryuing the HepaCAM gene, were
transfected into the LNCap, Bica-R and Enza-R cells, respectively
(Fig. 3A and B). The results of
CCK-8 assay revealed that the overexpression of HepaCAM suppressed
the proliferation of the DU145, LNCaP, Bica-R and Enza-R cells
(Fig. 3C–F). The results of colony
formation assay revealed similar results (Fig. 4A). To explore the role of HepaCAM
in the invasion and migration of the resistant cells, Transwell
assay and wound healing assay were performed using the Bica-R and
Enza-R cells. The data revealed that the overexpression of HepaCAM
inhibited the invasion and migration of the resistant cells
(Fig. 4B and C).
It is well known that AR amplification is
responsible for CRPC. Our previous study revealed that HepaCAM
decreased the expression of AR (17). Thus, we hypothesized that HepaCAM
may reverse the resistance of Enza-R cells to enzalutamide via the
downregulation of AR. The IC50 value of enzalutamide for
the Enza-R cells was determined by CCK-8 assay. Unexpectedly,
however, we failed to observe any significant changes in the
resistance of the Enza-R cells to enzlutamide when HepaCAM was
overexpressed (Fig. 4D).
HepaCAM suppresses the biological
behavior of the resistant cells through the downregulation of Notch
signalling
As mentioned above, the expression of HepaCAM
negatively correlated with Notch signaling in the CRPC samples
(Figs. 1G–H and 2A–C). Thus, we hypothesized that HepaCAM
may inhibit the proliferation and invasion of the resistant cells
through the downregulation of the Notch signaling pathway.
We also investigated members of Notch signaling by
western blot analysis and RT-qPCR in the LNCaP and CRPC cells. We
found that the levels of Jagged1, Notch1, NICD (protein level) and
Hes1 were enhanced in the resistant cells (Fig. 5A–C). To determine the role of Notch
signaling in the resistant cells, we treated the Enza-R cells with
5 μM PF-3084014 (a γ-secretase inhibitor) for 48 h, which
maintains Notch signaling in inactivation. As shown in Fig. 5D, PF-3084014 induced a decrease in
Enza-R cell viability, suggesting that Notch signaling plays an
important role in PCa cells which are resistant to treatment.
Moreover, when used in combination with Ad-HepaCAM and 5 μM
PF-3084014, the viability of the Enza-R cells was inhibited more
significantly than the use of either reagent alone (Fig. 5D). Furthermore, we found that the
combination of Ad-HepaCAM and PF-3084014 exerted a more potent
promoting effect on the protein expression of E-cadherin, and a
more potent suppressive effect on the protein expression of
N-cadherin and Snail (Fig. 5E and
F). Taken together, these findings indicated a synergistic
suppressive effect of HepaCAM and PF-3084014 on the proliferation
and migration of PCa cells which are resistant to treatment.
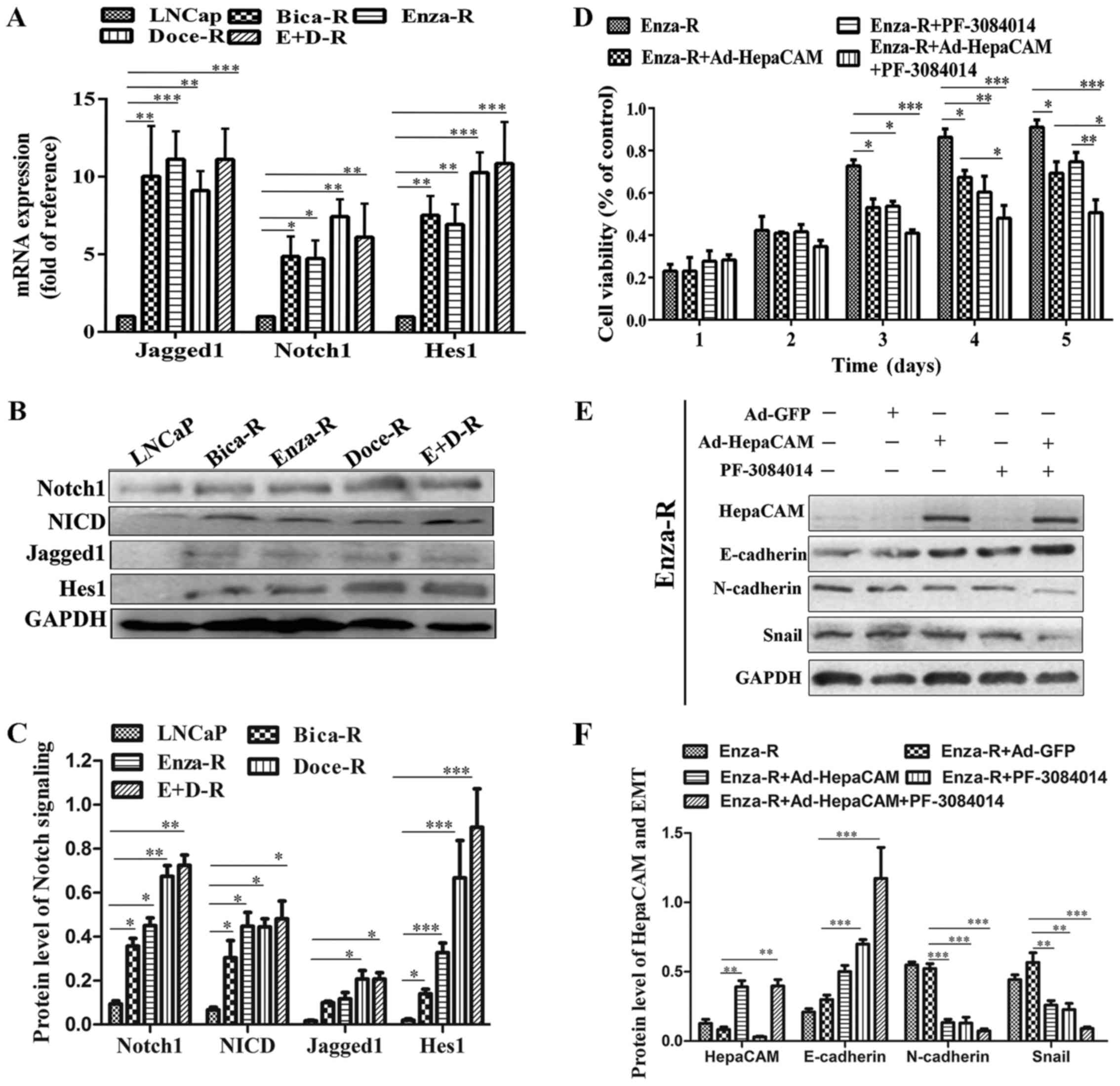 | Figure 5Notch signaling is upregulated in the
resistant cells. (A) The mRNA levels of Notch1, Jagged1 and Hes1 in
LNCap, Bica-R, Enza-R, Doce-R and E+D-R cells were detected by
RT-qPCR. (B and C) The protein levels of Jagged1, Notch1, NICD and
Hes1 in LNCap, Bica-R, Enza-R, Doce-R and E+D-R cells were
evaluated by western blot analysis. (D) Cell viability of Enza-R
cells was measured by CCK-8 assay following transfection with
Ad-GFP or Ad-HepaCAM for 72 h and/or 5 μM PF-3084014 for 48
h. (E and F) The expression of E-cadherin, N-cadherin and Snail in
Enza-R cells was examined by western blot analysis. The cells were
transfected with Ad-GFP or Ad-HepaCAM for 72 h and treated with 5
μM PF-3084014 for 48 h. GAPDH served as a loading control.
Bica-R, bicalutamide-resistant LNCaP cells; Enza-R,
enzalutamide-resistant LNCaP cells; Doce-R, docetaxel-resistant
LNCaP cells; E+D-R, sequential dual-resistant LNCap cells
(resistant to enzalutamide and docetaxel); *P<0.05,
**P<0.01 and ***P<0.001. |
We then determined the possible mechanisms
responsible for the suppressive effects of HepaCAM overexpression
on the survival of PCa cells which are resistant to treatment. The
results of immunofluorescence assay revealed that the
overexpression of HepaCAM decreased Notch1 and Hes1 expression in
the Enza-R cells, which was similar to the effects of PF-3084014.
Importantly, when the cells were treated with a combination of
Ad-HepaCAM and PF-3084014, the expression levels of Notch1 and Hes1
were downregulated more significantly. Note that in order for
Fig. 6 to be more concise, the
panels for DAPI staining alone are not shown (Fig. 6A). Western blot analysis and
RT-qPCR were performed to further determine the mechanisms
responsible for the suppressive effects of HepaCAM overexpression
on the viability of the resistant cells. As shown in Fig. B–E, both the mRNA and protein levels
of Jagged1, Notch1, NICD (only the protein level) and Hes1 were
downregulated when the resistant cells were treated with Ad-HepaCAM
or/and PF3084014. Taken together, our data indicated that HepaCAM
inhibited the biological behavior of the PCa cells which are
resistant to treatment via the downregulation of Notch
signaling.
 | Figure 6Overexpression of HepaCAM
downregulates Notch signaling. (A) Notch1 and Hes1 were detected by
immunofluorescence assay; cell nuclei were stained with DAPI
(magnification, ×200); in order for the figure to be more concise,
the panels for DAPI staining alone are not shown. (B) RT-qPCR
analysis of the mRNA expression levels of Notch1, Jagged1 and Hes1
in the Enza-R cells cultured alone or transfected with Ad-HepaCAM
(72 h) and/or treated with 5 μM PF-3084014 (48 h). (C–E) The
protein levels of Jagged1, Notch1, NICD and Hes1 in Bica-R and
Enza-R cells were detected by western blot analysis; GAPDH served
as a loading control. Bica-R, bicalutamide-resistant LNCaP cells;
Enza-R, enzalutamide-resistant LNCaP cells; Doce-R,
docetaxel-resistant LNCaP cells; E+D-R, sequential dual-resistant
LNCap cells (resistant to enzalutamide and docetaxel);
*P<0.05, **P<0.01 and
***P<0.001. |
Construction of docetaxel-resistant cells
and sequential dual-resistant cells (resistant to enzalutamide and
docetaxel)
A recent study indicated that overactivated Notch
signaling plays an important role in the resistant of PCa to
docetaxel, and that the downregulation of Notch reverses docetaxel
resistance (22). The results of
this study revealed that the overexpression of HepaCAM
downregulated Notch signaling in the resistant cells. Thus, we
hypothesized that the downregulation of Notch signaling induced by
the overexpression of HepaCAM possibly re-sensitizes the
docetaxel-resistant cells to docataxel, instead of re-sensitizing
the Enza-R cells to enzalutamide.
To confirm our hypothesis, we constructed Doce-R
cells and sequential dual-resistant cells (resistant to
enzalutamide and docetaxel) (E+D-R cells) as described in the
Materials and methods. The IC50 values of docetaxel for
the Doce-R, E+D-R and their parental cells were evaluated by CCK-8
assay. As shown in Fig. 7A and B,
compared to their parental cells, the LNCaP, Doce-R cells exhibited
an 81-fold increase in their resistance to docetaxel, whereas the
E+D-R cells displayed a 56-fold increase in their resistance to
docetaxel, compared to the Enza-R cells. Moreover, our data
indicated that docetaxel inhibited the viability of both the LNCaP
and Enza-R cells within the same range of concentrations,
indicating that there was no cross-resistance between enzalutamide
and docetaxel (Fig. 7A and B).
HepaCAM overexpression fails to
re-sensitize the Doce-R and E+D-R cells to docetaxel, and
PF-3084014 partly restores the sensitivity of the Enza-R, E+D-R
cells to enzalutamide and docetaxel, respectively in vitro
Our data indicated that the overexpression of
HepaCAM suppressed the proliferation of both the Doce-R and E+D-R
cells (Fig. 7C–E). When the cells
were treated with PF-3084014 followed by Ad-HepaCAM, the inhibitory
effects were enhanced in the E+D-R cells (Fig. 7E). However, we failed to observe
that the overexpression of HepaCAM restores the sensitivity of the
Doce-R and E+D-R cells to docetaxel (Figs. 7A and B, and 8B and C). After the Enza-R and E+D-R
cells were treated with 5 μM PF-3084014 for 48 h, the
IC50 values of enzalutamide and docetaxel were
determined by CCK-8 assay, respectively. Surprisingly, we found
that PF-3084014 restored the sensitivity of the Enza-R cells to
enzalutamide by 4-fold, and that of the E+D-R cells to docetaxel by
7-fold (Fig. 8A and C) indicating
that PF-3084014 may be regarded as a sensitizer of docetaxel and
enzalutamide in the treatment of refractory PCa.
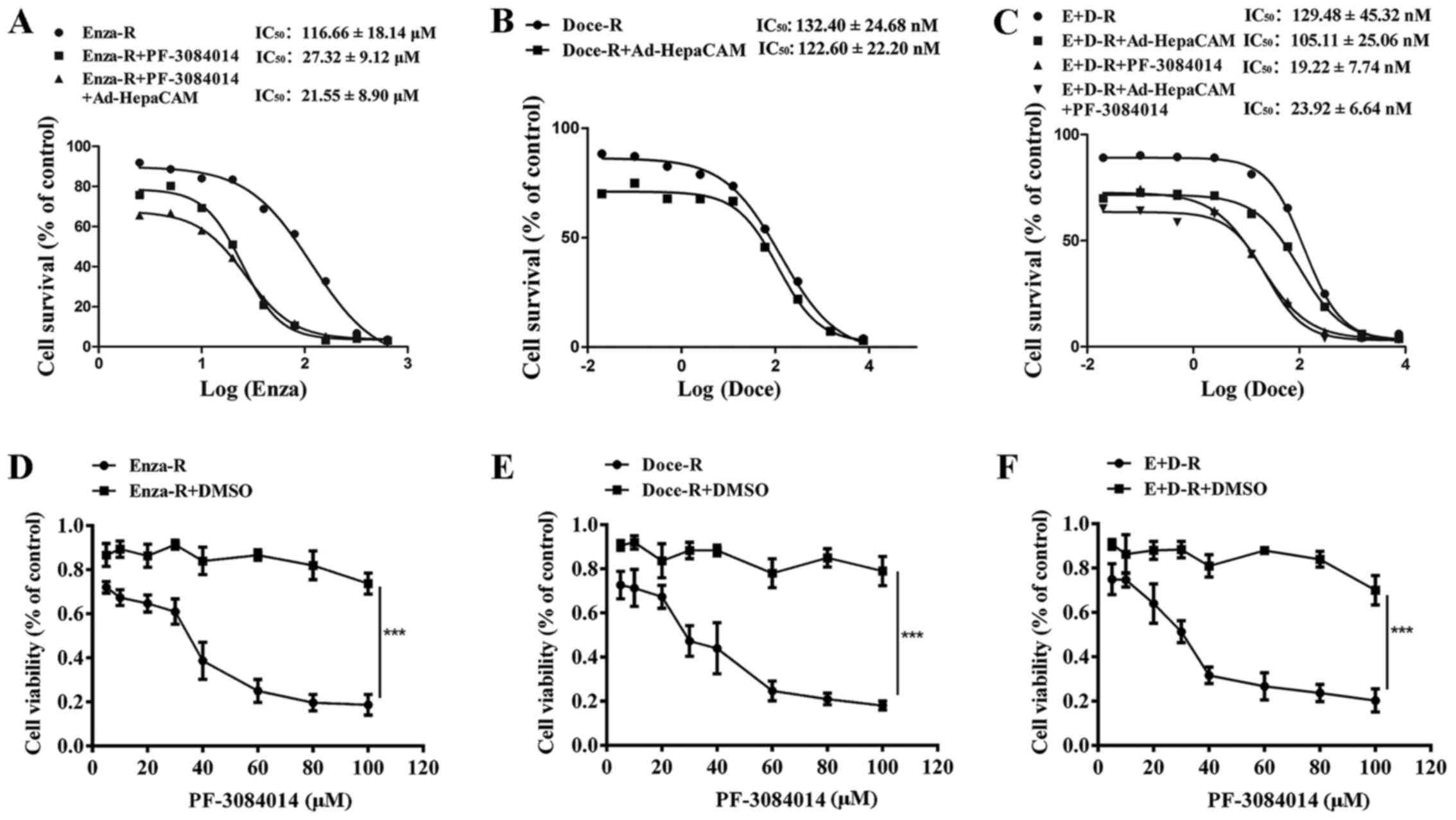 | Figure 8PF-3084014 re-sensitizes the
resistant cells to the corresponding drugs, and exerts an antitumor
effect on the resistant cells. (A–C) Enza-R, Doce-R and E+D-R cells
were exposed to increasing concentrations of enzalutamide and
docetaxel for 48 h, respectively, and the half maximal inhibitory
concentration (IC50) was determined by CCK-8 assay. The
cells were transfected with Ad-HepaCAM for 72 h and/or treated with
PF-3084014 for 48 h. (D–F) Enza-R, Doce-R and E+D-R cells were
treated with increasing concentrations of PF-3084014 (5, 10, 20,
30, 40, 60, 80 and 100 μM) or DMSO for 48 h. Cell viability
was detected by CCK-8 assay. ***P<0.001. Enza-R,
enzalutamide-resistant LNCaP cells; Doce-R, docetaxel-resistant
LNCaP cells; E+D-R, sequential dual- resistant LNCaP cells
(resistant to enzalutamide and docetaxel). |
To determine whether PF-3084014 exerts antitumor
effect on refractory PCa, CCK-8 assay was performed to evaluate the
viability of the Enza-R, Doce-R and E+D-R cells following treatment
with various concentrations of PF-3084014 (5, 10, 20, 30, 40, 60,
80 and 100 μM) for 48 h. The results revealed that, at an
increasing concentration, PF-3084014 exerted a gradual but potent
antitumor effect on the Enza-R, Doce-R and E+D-R cells (Fig. 8D–F), indicating that PF-3084014 may
be considered as a novel therapy for refractory PCa.
Discussion
Cell adhesion molecules have been studied for many
years in various types of cancer. Some studies have indicated that
some adhesion molecules, such as CEACAM5 and CEACAM6 play important
roles in tumor initiation and progression (36,37).
Other adhesion molecules, such as CEACAM1 and CEACAM1-4S have been
shown to exert anti-proliferative effects on some cancer types
(38–40). For example, a high expression of
CEACAM1-4S has been detected in normal breast epithelial cells;
however, its expression is lost in breast cancer cells (MCF7). With
the enforced expression of CEACAM1-4S, MCF7 cells have been shown
to return to a morphological phenotype in Matrigel, which is
similar to normal breast acini (38). In this study, HepaCAM, one of the
cell adhesion molecules, was found to be expressed in PPC tissues
where gland structures were presented. However, the expression of
HepaCAM was downregulated in sites where gland structures were
disorganized. Moreover, when gland structures disappeared, it was
undetectable (Fig. 1A, panels 1 and
2). In addition, HepaCAM negativity in the CRPC tissues was
associated with more severe Gleason scores. Therefore, we
hypothesized that HepaCAM, similar to CEACAM1-4S, may be associated
with maintaining the normal morphological phenotype of prostate
epithelial cells. We aim to confirm this hypothesis in follow-up
experiments.
In the present study, we also found that the loss of
HepaCAM was more frequent in CRPC tissues than that in matched PPC
tissues (Table I and Fig. 1A and D). This finding suggested
that, along with tumor progression and the emergence of castration
resistance, the downregulation and loss of HepaCAM gradually and
continuously occurs over a few years. Importantly, our data
demonstrated that HepaCAM negativity was associated with a shorter
PFS in patients with CRPC (Fig.
2D). The results suggested that the loss of HepaCAM was
associated with the poor prognosis of patients with CRPC. In the
future, we aim to analyze the overall survival (OS) when the death
endpoint occurs in patients with CRPC.
The activities of Notch signaling have been proven
to be elevated in PCa (20,
21). More interestingly, Notch
activities are more intensive in specimens of metastatic PCa than
in specimens of PPC (41,42). The findings of this study yielded a
similar result in that Notch signaling was markedly increased in
CRPC samples in compared to matched PPC tissues, indicating that
Notch signaling plays an important role in the emergence and
progression of CRPC. A recent study revealed that Notch signaling
was upregulated in patients with docetaxel-resistant PCa, and
inhibiting Notch signaling eliminates subpopulation of the cells
which are responsible for docetaxel resistance and delays the
initiation of the resistance (43). In present study, Notch signaling
was upregulated in the Bica-R, Enza-R, Doce-R and E+D-R cells
(Fig. 5A and B). When HepaCAM was
overexpressed, mRNA and protein levels of Notch were decreased. The
viability and growth of the cells was decreased, suggesting that
HepaCAM exerted antitumor effects through the downregulation of
Notch activity in refractory PCa.
HepaCAM, an upstream cellular regulator, is involved
in the regulation of many cell signaling pathways. For example, the
knockdown of interleukin-6 (IL-6) upregulates HepaCAM expression
via the STAT3/DNMTs axis, and reduces the proliferation of renal
cell carcinoma cells (15).
HepaCAM also increases the proportion of c-Myc phosphorylation in
human renal carcinoma cells (44).
The overexpression of HepaCAM downregulates p-AKT and p-FoxO
expression, and inhibits the proliferation and viability of bladder
cancer cells (45). Moreover, in
glioblastoma cells, HepaCAM is able to keep stabilizing connexin 43
protein, a well-established tumor suppressor, and enhances its
localization to the plasma membrane at cellular junctions (46).
AR axis inhibitors remain the major therapeutic
strategies for patients with PCa (47,48).
However, the inevitable transition from hormone-sensitive PCa
(HSPC) to CRPC remains an ever-present challenge in the treatment
of PCa. When ADT fails and CRPC develops, docetaxel has been proven
to prolong the OS of patients with CRPC (49). However, docetaxel resistance occurs
within a few months. However,, there are no effective approaches
for dual-resistant PCa. In the present study, in order to observe
the sequential dual resistance to AR axis inhibitors and taxanes,
we constructed sequential dual-resistant cells (E+D-R) for the
first time, at least to the best of our knowledge. As shown by our
data (Fig. 7D and E), the
overexpression of HepaCAM suppressed the growth of E+D-R cells,
indicating that HepaCAM possibly represents a novel therapeutic
target for patients with refractory PCa.
PF-3084014, a γ-secretase inhibitor, has displayed
antitumor activity in several types of cancer, such as breast
cancer (50) and acute
lymphoblastic leukemia (26). It
has entered clinical trials for the treatment of multiple tumors
(26,51,52).
More surprisingly, in a recent study, PF-3084014 was used in a
phase II clinical trial for patients with advanced desmoid tumors,
and clinical benefits with no instances of progressive disease and
measurable regression in tumor volume were observed in 11 of 17
patients (53). A recent study
also revealed that PF-3084014 sensitized docetaxel-resistant cells
to docetaxel both in vitro and in vivo (22). In present study, we revealed that
PF-3084014 also partly restored sensitivity of the E+D-R, Enza-R
cells to docetaxel and to enzalutamide in vitro, suggesting
that PF-3084014, as sensitizer of both enzalutamide and docetaxel,
may be a novel adjuvant drug for use in the treatment of refractory
PCa.
Unexpectedly, we failed to prove that the
overexpression of HepaCAM restored the sensitivity of the Enza-R,
Doce-R and E+D-R cells to corresponding drugs. A previous study
demonstrated that Notch4 activation, but not Notch1 and Notch2,
rendered MCF7 cells unresponsive to tamoxifen (54). Another study demonstrated that the
upregulation of Notch4, but not Notch1, was responsible for
tamoxifen resistance in specific breast cancer. The downregulation
of Notch4 by MRK-003 (another γ-secretase inhibitor) has also been
shown to reverse tamoxifen resistance and the hormone-dependent
phenotype (55). In our opinion,
Notch4, not Notch1, may also be responsible for the resistance of
Enza-R and E+D-R cells. PF-3084014 partly reverses resistance by
decreasing Notch4. However, HepaCAM may only affect Notch1, but not
Notch4, resulting in failing to restore sensitivity of Doce-R,
E+D-R cells to docetaxel.
Importantly, we further revealed that the use of
PF-3084014 alone exerted an antitumor effect in vitro,
suggesting that PF-3084014 may be not only function as a
sensitizer, but may also be a promising reagent for use in the
treatment of refractory PCa. Our results were not consistent with a
those of a previous study (22),
in which the use of PF-3084014 alone did not exert an antitumor
effect on docetaxel-resistant cells. This may be explained by the
fact that these authors treated the cells with a constant
concentration of PF-3084014, 5 μM. However, when the cells
were treated with 20 μM PF-3084014, as in this study, its
antitumor effect was highlighted (Fig.
8D–F).
Taken together, the present study demonstrates the
following: HepaCAM expression was lost and Notch signaling was
excessively activated in the majority of CRPC tissues. HepaCAM
negativity was associated with a worse PFS of patients with CRPC.
HepaCAM exerted antitumor effects on CRPC cells through the
downregulation of Notch signaling. More importantly, PF-3084014
partly restored the sensitivity of the Enza-R and E+D-R cells to
enzalutamide and docetaxel, respectively.
Acknowledgments
The authors would like to thank Dr Dan Yang, Key
Laboratory of Laboratory Medical Diagnostics, Ministry of
Education, Department of Laboratory Medicine, Chongqing Medical
University, Chongqing, China, for providing technical assistance
with the immunohistochemistry and immunofluorescence assays.
Funding
This study was supported by a grant from the Natural
Science Foundation of China (no. 81272572).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZD and XW designed the experiments. ZD, LL, YZ and
WS collected the specimens and analyzed the clinical data. ZD, LL,
MY, ZQ, YH, TL and JW carried out the experiments. ZD and LL
co-wrote the manuscript. CL provided technical support of this
research project and supervised the progress of the experiments.
ZD, ZC, WS and NL analyzed statistical data. ZD, LL and XW
assembled and installed the figures. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Chongqing Medical University. Informed consent was obtained from
the patients or their family members who agreed to the use of their
samples in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Egan A, Dong Y, Zhang H, Qi Y, Balk SP and
Sartor O: Castration-resistant prostate cancer: Adaptive responses
in the androgen axis. Cancer Treat Rev. 40:426–433. 2014.
View Article : Google Scholar
|
|
4
|
van Soest RJ, Nieuweboer AJ, de Morrée ES,
Chitu D, Bergman AM, Goey SH, Bos MM, van der Meer N, Hamberg P, de
Wit R, et al: Dutch Uro-Oncology Studygroup (DUOS): The influence
of prior novel androgen receptor targeted therapy on the efficacy
of cabazitaxel in men with metastatic castration- resistant
prostate cancer. Eur J Cancer. 51:2562–2569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Al Nakouzi N, Le Moulec S, Albigès L, Wang
C, Beuzeboc P, Gross-Goupil M, de La Motte Rouge T, Guillot A,
Gajda D, Massard C, et al: Cabazitaxel remains active in patients
progressing after docetaxel followed by novel androgen receptor
pathway targeted therapies. Eur Urol. 68:228–235. 2015. View Article : Google Scholar
|
|
6
|
Mezynski J, Pezaro C, Bianchini D, Zivi A,
Sandhu S, Thompson E, Hunt J, Sheridan E, Baikady B, Sarvadikar A,
et al: Antitumour activity of docetaxel following treatment with
the CYP17A1 inhibitor abiraterone: Clinical evidence for
cross-resistance? Ann Oncol. 23:2943–2947. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Soest RJ, van Royen ME, de Morrée ES,
Moll JM, Teubel W, Wiemer EA, Mathijssen RH, de Wit R and van
Weerden WM: Cross-resistance between taxanes and new hormonal
agents abiraterone and enzalutamide may affect drug sequence
choices in metastatic castration-resistant prostate cancer. Eur J
Cancer. 49:3821–3830. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chung Moh M, Hoon Lee L and Shen S:
Cloning and characterization of hepaCAM, a novel Ig-like cell
adhesion molecule suppressed in human hepatocellular carcinoma. J
Hepatol. 42:833–841. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu B, He Y, Wu X, Luo C, Liu A and Zhang
J: Exploration of the correlations between interferon-γ in patient
serum and HEPACAM in bladder transitional cell carcinoma, and the
interferon-γ mechanism inhibiting BIU-87 proliferation. J Urol.
188:1346–1353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xun C, Luo C, Wu X, Zhang Q, Yan L and
Shen S: Expression of hepaCAM and its effect on proliferation of
tumor cells in renal cell carcinoma. Urology. 75:828–834. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang XL, Zhang Y, Tan B, Luo CL and Wu
XH: Renal tumor-derived exosomes inhibit hepaCAM expression of
renal carcinoma cells in a p-AKT-dependent manner. Neoplasma.
61:416–423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tao J, Liu Q, Wu X, Xu X, Zhang Y, Wang Q
and Luo C: Identification of hypermethylation in hepatocyte cell
adhesion molecule gene promoter region in bladder carcinoma. Int J
Med Sci. 10:1860–1867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du HF, Ou LP, Lv CK, Yang X, Song XD, Fan
YR, Wu XH and Luo CL: Expression of hepaCAM inhibits bladder cancer
cell proliferation via a Wnt/β-catenin-dependent pathway in vitro
and in vivo. Cancer Biol Ther. 16:1502–1513. 2015. View Article : Google Scholar
|
|
14
|
Wang Q, Luo C, Wu X, Du H, Song X and Fan
Y: hepaCAM and p-mTOR closely correlate in bladder transitional
cell carcinoma and hepaCAM expression inhibits proliferation via an
AMPK/mTOR dependent pathway in human bladder cancer cells. J Urol.
190:1912–1918. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quan Z, He Y, Luo C, Xia Y, Zhao Y, Liu N
and Wu X: Interleukin 6 induces cell proliferation of clear cell
renal cell carcinoma by suppressing hepaCAM via the STAT3-dependent
up-regulation of DNMT1 or DNMT3b. Cell Signal. 32:48–58. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang T, Moh MC, Lee LH and Shen S: The
immunoglobulin-like cell adhesion molecule hepaCAM is cleaved in
the human breast carcinoma MCF7 cells. Int J Oncol. 37:155–165.
2010.PubMed/NCBI
|
|
17
|
Song X, Wang Y, Du H, Fan Y, Yang X, Wang
X, Wu X and Luo C: Overexpression of HepaCAM inhibits cell
viability and motility through suppressing nucleus translocation of
androgen receptor and ERK signaling in prostate cancer. Prostate.
74:1023–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guruharsha KG, Kankel MW and
Artavanis-Tsakonas S: The Notch signalling system: Recent insights
into the complexity of a conserved pathway. Nat Rev Genet.
13:654–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kwon OJ, Valdez JM, Zhang L, Zhang B, Wei
X, Su Q, Ittmann MM, Creighton CJ and Xin L: Increased Notch
signalling inhibits anoikis and stimulates proliferation of
prostate luminal epithelial cells. Nat Commun. 5:44162014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bin Hafeez B, Adhami VM, Asim M, Siddiqui
IA, Bhat KM, Zhong W, Saleem M, Din M, Setaluri V and Mukhtar H:
Targeted knockdown of Notch1 inhibits invasion of human prostate
cancer cells concomitant with inhibition of matrix
metalloproteinase-9 and urokinase plasminogen activator. Clin
Cancer Res. 15:452–459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santagata S, Demichelis F, Riva A,
Varambally S, Hofer MD, Kutok JL, Kim R, Tang J, Montie JE,
Chinnaiyan AM, et al: JAGGED1 expression is associated with
prostate cancer metastasis and recurrence. Cancer Res.
64:6854–6857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cui D, Dai J, Keller JM, Mizokami A, Xia S
and Keller ET: Notch pathway inhibition using PF-03084014, a
γ-secretase inhibitor (GSI), enhances the antitumor effect of
docetaxel in prostate cancer. Clin Cancer Res. 21:4619–4629. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo Y, Zhang K, Cheng C, Ji Z, Wang X,
Wang M, Chu M, Tang DG, Zhu HH and Gao WQ: Numb−/low
enriches a castration-resistant prostate cancer cell subpopulation
associated with enhanced notch and hedgehog signaling. Clin Cancer
Res. 23:6744–6756. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stoyanova T, Riedinger M, Lin S,
Faltermeier CM, Smith BA, Zhang KX, Going CC, Goldstein AS, Lee JK,
Drake JM, et al: Activation of Notch1 synergizes with multiple
pathways in promoting castration-resistant prostate cancer. Proc
Natl Acad Sci USA. 113:E6457–E6466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Wu X, Ou L, Yang X, Wang X, Tang
M, Chen E and Luo C: PLCε knockdown inhibits prostate cancer cell
proliferation via suppression of Notch signalling and nuclear
translocation of the androgen receptor. Cancer Lett. 362:61–69.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wei P, Walls M, Qiu M, Ding R, Denlinger
RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, et al:
Evaluation of selective gamma-secretase inhibitor PF-03084014 for
its antitumor efficacy and gastrointestinal safety to guide optimal
clinical trial design. Mol Cancer Ther. 9:1618–1628. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yabuuchi S, Pai SG, Campbell NR, de Wilde
RF, De Oliveira E, Korangath P, Streppel MM, Rasheed ZA, Hidalgo M,
Maitra A, et al: Notch signaling pathway targeted therapy
suppresses tumor progression and metastatic spread in pancreatic
cancer. Cancer Lett. 335:41–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arcaroli JJ, Quackenbush KS, Purkey A,
Powell RW, Pitts TM, Bagby S, Tan AC, Cross B, McPhillips K, Song
EK, et al: Tumours with elevated levels of the Notch and Wnt
pathways exhibit efficacy to PF-03084014, a γ-secretase inhibitor,
in a preclinical colorectal explant model. Br J Cancer.
109:667–675. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heidenreich A, Bastian PJ, Bellmunt J,
Bolla M, Joniau S, van der Kwast T, Mason M, Matveev V, Wiegel T,
Zattoni F, et al: European Association of Urology: EAU guidelines
on prostate cancer. Part II: Treatment of advanced, relapsing, and
castration-resistant prostate cancer. Eur Urol. 65:467–479. 2014.
View Article : Google Scholar
|
|
30
|
Matei DV, Renne G, Pimentel M, Sandri MT,
Zorzino L, Botteri E, De Cicco C, Musi G, Brescia A, Mazzoleni F,
et al: Neuroendocrine differentiation in castration-resistant
prostate cancer: A systematic diagnostic attempt. Clin Genitourin
Cancer. 10:164–173. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
32
|
Kuruma H, Matsumoto H, Shiota M, Bishop J,
Lamoureux F, Thomas C, Briere D, Los G, Gleave M, Fanjul A, et al:
A novel antiandrogen, compound 30, suppresses castration-resistant
and MDV3100-resistant prostate cancer growth in vitro and in vivo.
Mol Cancer Ther. 12:567–576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kawata H, Ishikura N, Watanabe M,
Nishimoto A, Tsunenari T and Aoki Y: Prolonged treatment with
bicalutamide induces androgen receptor overexpression and androgen
hypersensitivity. Prostate. 70:745–754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kawabata R, Oie S, Oka T, Takahashi M,
Kanayama H and Itoh K: Hydroxyflutamide enhances cellular
sensitivity to 5-fluorouracil by suppressing thymidylate synthase
expression in bicalutamide-resistant human prostate cancer cells.
Int J Oncol. 38:665–676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takeda M, Mizokami A, Mamiya K, Li YQ,
Zhang J, Keller ET and Namiki M: The establishment of two
paclitaxel-resistant prostate cancer cell lines and the mechanisms
of paclitaxel resistance with two cell lines. Prostate. 67:955–967.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Govindan SV, Cardillo TM, Moon SJ, Hansen
HJ and Goldenberg DM: CEACAM5-targeted therapy of human colonic and
pancreatic cancer xenografts with potent labetuzumab-SN-38
immunoconjugates. Clin Cancer Res. 15:6052–6061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han SU, Kwak TH, Her KH, Cho YH, Choi C,
Lee HJ, Hong S, Park YS, Kim YS, Kim TA, et al: CEACAM5 and CEACAM6
are major target genes for Smad3-mediated TGF-beta signaling.
Oncogene. 27:675–683. 2008. View Article : Google Scholar
|
|
38
|
Kirshner J, Chen CJ, Liu P, Huang J and
Shively JE: CEACAM1-4S, a cell-cell adhesion molecule, mediates
apoptosis and reverts mammary carcinoma cells to a normal
morphogenic phenotype in a 3D culture. Proc Natl Acad Sci USA.
100:521–526. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weigelt B, Ghajar CM and Bissell MJ: The
need for complex 3D culture models to unravel novel pathways and
identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev.
69–70:42–51. 2014. View Article : Google Scholar
|
|
40
|
Stubblefield K, Chean J, Nguyen T, Chen CJ
and Shively JE: The adaptor SASH1 acts through NOTCH1 and its
inhibitor DLK1 in a 3D model of lumenogenesis involving CEACAM1.
Exp Cell Res. 359:384–393. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Danza G, Di Serio C, Ambrosio MR, Sturli
N, Lonetto G, Rosati F, Rocca BJ, Ventimiglia G, del Vecchio MT,
Prudovsky I, et al: Notch3 is activated by chronic hypoxia and
contributes to the progression of human prostate cancer. Int J
Cancer. 133:2577–2586. 2013.PubMed/NCBI
|
|
42
|
Zhu H, Zhou X, Redfield S, Lewin J and
Miele L: Elevated Jagged-1 and Notch-1 expression in high grade and
metastatic prostate cancers. Am J Transl Res. 5:368–378.
2013.PubMed/NCBI
|
|
43
|
Domingo-Domenech J, Vidal SJ,
Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco
R, Bonal DM, Charytonowicz E, Gladoun N, de la Iglesia-Vicente J,
et al: Suppression of acquired docetaxel resistance in prostate
cancer through depletion of notch- and hedgehog-dependent
tumor-initiating cells. Cancer Cell. 22:373–388. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang QL, Luo CL, Wu XH, Wang CY, Xu X,
Zhang YY, Liu Q and Shen SL: HepaCAM induces G1 phase arrest and
promotes c-Myc degradation in human renal cell carcinoma. J Cell
Biochem. 112:2910–2919. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tang M, Zhao Y, Liu N, Chen E, Quan Z, Wu
X and Luo C: Overexpression of HepaCAM inhibits bladder cancer cell
proliferation and viability through the AKT/FoxO pathway. J Cancer
Res Clin Oncol. 143:793–805. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu M, Moh MC and Schwarz H: HepaCAM
associates with connexin 43 and enhances its localization in
cellular junctions. Sci Rep. 6:362182016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yap TA, Smith AD, Ferraldeschi R,
Al-Lazikani B, Workman P and de Bono JS: Drug discovery in advanced
prostate cancer: Translating biology into therapy. Nat Rev Drug
Discov. 15:699–718. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sharp A, Welti J, Blagg J and de Bono JS:
Targeting androgen receptor aberrations in castration-resistant
prostate cancer. Clin Cancer Res. 22:4280–4282. 2016. View Article : Google Scholar
|
|
49
|
Tannock IF, de Wit R, Berry WR, Horti J,
Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, et
al: TAX 327 Investigators: Docetaxel plus prednisone or
mitoxantrone plus prednisone for advanced prostate cancer. N Engl J
Med. 351:1502–1512. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang CC, Pavlicek A, Zhang Q, Lira ME,
Painter CL, Yan Z, Zheng X, Lee NV, Ozeck M, Qiu M, et al:
Biomarker and pharmacologic evaluation of the γ-secretase inhibitor
PF-03084014 in breast cancer models. Clin Cancer Res. 18:5008–5019.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Papayannidis C, DeAngelo DJ, Stock W,
Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA,
Ozeck M, et al: A Phase 1 study of the novel gamma-secretase
inhibitor PF-03084014 in patients with T-cell acute lymphoblastic
leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J.
5:e3502015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Carol H, Maris JM, Kang MH, Reynolds CP,
Kolb EA, Gorlick R, Keir ST, Wu J, Kurmasheva RT, Houghton PJ, et
al: Initial testing (stage 1) of the notch inhibitor PF-03084014,
by the pediatric preclinical testing program. Pediatr Blood Cancer.
61:1493–1496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kummar S, O'Sullivan Coyne G, Do KT,
Turkbey B, Meltzer PS, Polley E, Choyke PL, Meehan R, Vilimas R,
Horneffer Y, et al: Clinical activity of the γ-secretase inhibitor
PF-03084014 in adults with desmoid tumors (aggressive
fibromatosis). J Clin Oncol. 35:1561–1569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lombardo Y, Faronato M, Filipovic A,
Vircillo V, Magnani L and Coombes RC: Nicastrin and Notch4 drive
endocrine therapy resistance and epithelial to mesenchymal
transition in MCF7 breast cancer cells. Breast Cancer Res.
16:R622014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yun J, Pannuti A, Espinoza I, Zhu H, Hicks
C, Zhu X, Caskey M, Rizzo P, D'Souza G, Backus K, et al: Crosstalk
between PKCα and Notch-4 in endocrine-resistant breast cancer
cells. Oncogenesis. 2:e602013. View Article : Google Scholar
|


















