Introduction
Cisplatin resistance can be provoked by drug
extrusion, multidrug resistance-associated protein activity and
upregulation of the DNA repair machinery. In addition, a series of
adaptive responses induced by cisplatin that maintain tumor cell
homeostasis and interfere with apoptotic signaling, including
metabolic reprogramming, autophagy and oxidative stress, can lead
to cisplatin resistance (1,2). In
the 1920s, Warburg et al first observed the metabolic shift
in energy in tumor cells, later termed the 'Warburg effect', in
which tumor cells shift towards using aerobic glycolysis to meet
energy requirements. Subsequent studies have linked this transition
with mitochondrial dysfunction (3–5).
Increasing evidence indicates that mitochondria, which integrate
metabolism and apoptotic activities, significantly influence cancer
cell survival, invasion, metastasis and drug resistance (6). Although mitochondria have their own
genome, the majority of mitochondrial proteins are encoded by the
nuclear genome (7,8). Through the mechanism known as the
nucleo-mitochondrial anterograde regulation system, these two
genomes coordinately regulate mitochondrial proteostasis and
maintain mitochondrial functional integrity to prevent tumor cells
from environmental stress-induced damage, including
chemotherapeutic stimulation (8–10).
Therefore, investigating the complex machinery of the
nucleo-mitochondrial anterograde regulation system is vital to
clarify the mechanism underlying cell apoptosis evasion and may
assist in identifying a novel anticancer therapy target to disturb
mitochondrial function.
Peroxisome proliferator-activated receptor
(PPAR)-coactivator 1α (PGC1α) is a member of the PGC1 family, which
functions as a transcriptional coactivator in the regulation of
multiple signal transduction pathways, and acts as a principal
regulator of cell metabolism and mitochondrial biogenesis (11). Accumulating evidence now suggests a
link between PGC1α-mediated mitochondrial biogenesis and tumor
apoptosis evasion (12,13). Permuth-Wey et al found that
inter-individual genetic variation in genes involved in
mitochondrial biogenesis, including PGC1α and nuclear respiratory
factor 1 (NRF1), was most markedly associated with epithelial
ovarian cancer (EOC) risk in a multi-center study of cases of EOC
in Caucasian individuals and controls, and the overexpression of
PGC1α was associated with poor prognosis in EOC (14). In addition, in BRAF V600E-positive
human melanoma, the expression of PGC1α resulted in the
upregulation of oxidative phosphorylation (OXPHOS) genes, energy
status and mitochondrial biogenesis, accompanied by acquired BRAF
inhibitor resistance (15). In
addition, PGC1α resisted mitogen-activated protein kinase
inhibitor-induced cell apoptosis in melanoma (16). Therefore, further elucidation of
the potential mechanism of the PGC1α-mediated upregulation of
mitochondrial bioenergy and biogenesis involved in apoptotic
evasion in chemotherapy-resistant tumor cell is important.
Previous studies in myocytes have found that the
transcriptional activity of NRF1 and NRF2 were significantly
increased following the overexpression of PGC1α, suggesting that
PGC1α may be a co-activator of NRF1 and NRF2 (17,18).
Other studies have shown that the PGC1α/NRF1/2-mitochondrial
transcription factor A (TFAM)/mitochondrial transcription factor B1
(TFB1M) axis may be involved in the upregulation of mitochondrial
mass, mitochondrial DNA (mtDNA) copy numbers and respiratory
complex protein expression to promote mitochondrial biogenesis
(7,17,19,20).
In the chemotherapeutic treatment of human colon cancer with
oxaliplatin, the genes involved in OXPHOS and mitochondrial
biogenesis, including PGC1α, were found to be markedly upregulated,
suggesting that chemotherapy induced a shift in tumor metabolism
from glycolysis to OXPHOS, eliciting tumor cell chemotherapeutic
resistance (21). Furthermore, the
sensitivity of estrogen receptor (ER)-positive breast cancer to
cisplatin was reported to increase when the PGC1α downstream gene
TFAM was knocked down, and cisplatin resistance was restored when
TFAM was reintroduced into ER-positive breast cancer cells
(22). These results confirmed
that the PGC1α-mediated mitochondrial biogenesis transcriptional
pattern may engage a tumor cell apoptosis evasion mechanism in
different stages of chemotherapeutic resistance.
As PGC1α interacts with different transcription
factors, the PGC1α-mediated multistep nuclear regulatory network
affects cancer cell survival and death from different aspects
(23–26). Therefore, to examine the action of
PGC1α in the evasion of apoptosis by upregulating mitochondrial
bioenergy and biogenesis in ovarian cancer, the mitochondrial
biogenesis-associated PGC1α signaling pathway and its changes under
external stimuli require examination. Our previous study
demonstrated differences in mitochondrial function between
cisplatin-sensitive SKOV3 ovarian cancer cells and their
cisplatin-resistant clones SKOV3/DDP cells (Xu et al,
unpublished data). The present study aimed to obtain further
insight into PGC1αG to examine the role of PGC1α-mediated
mitochondrial biogenesis in cisplatin resistance in ovarian cancer
cells, which may further substantiate the role of
nucleo-mitochondrial signaling communication in cell fate and
provide possibilities for promising mitochondria-targeted treatment
modalities for increasing cancer chemosensitivity.
Materials and methods
Reagents and antibodies
Cisplatin and
3-(4,5-dimetrylthi-azol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma-Aldrich; EMD Millipore (Billerica, MA,
USA). Enhanced chemiluminescence (ECL) reagents were from Thermo
Fisher Scientific, Inc. (Waltham, MA USA). The following antibodies
were used: Anti-PGC1α (cat. no. A12348), anti-NRF1 (cat. no.
A5246), anti-NRF2 (cat. no. A12306) and anti-TFAM (cat. no. A1926)
from ABclonal Biotech Co., Ltd. (Boston, MA, USA); anti-β actin
(cat. no. 60008-1-Ig), anti-B-cell lymphoma 2 (Bcl-2; cat. no.
12789-1-AP), anti-Bcl-2-associated X protein (Bax; cat. no.
50599-2-Ig), anti-cytochrome c oxidase subunit 5a (COX5A;
cat. no. 11448-1-AP), peroxidase-conjugated AffiniPure goat
anti-mouse IgG (H+L; cat. no. SA00001-1), and peroxidase-conjugated
AffiniPure goat anti-rabbit IgG (H+L; cat. no. SA00001-2) from
ProteinTech Group, Inc. (Chicago, IL, USA); anti-caspase-3 (cat.
no. ab32351), and anti-voltage-dependent anion-selective channel 1
(VDAC1; cat. no. ab14734) from Abcam (Cambridge, MA, USA);
anti-myeloid cell leukemia 1 (Mcl-1; cat. no. sc-12756),
anti-translocase of mitochondrial outer membrane complex 20 (TOM20;
cat. no. sc-17764), and anti-cytochrome c (cat. no.
sc-13561) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA).
Cell culture
The cisplatin-sensitive SKOV3 ovarian carcinoma
cells and their cisplatin-resistant clone SKOV3/DDP cells were
obtained from the Chinese Academy of Medical Sciences and Peking
Union Medical College (Beijing, China). The two cell lines were
maintained at 37°C in a 5% CO2 and 95% air atmosphere in
Roswell Park Memorial Institute-1640 culture medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(FBS; Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 U/ml streptomycin. The cisplatin-resistant
SKOV3/DDP cells were cultured in the presence of 1 µg/ml
cisplatin in RPMI-1640 culture medium at 37°C to maintain
resistance.
Cellular viability assays
Cellular viability was measured using MTT assays.
The cells were seeded in 96-well plates at a density of
1×104 cells/well. Following exposure of the cells to
cisplatin, 20 µl of MTT solution (5 mg/ml) was added to each
well and the cells were incubated for 4 h. DMSO (Beijing Chemical
Industry Co., Ltd., Beijing, China) was then added to the wells to
solubilize the formazan products following elimination of the
media. The absorbance was recorded at 570 nm using a CLARIOstar
microplate reader (BMG Labtech GmbH, Offenburg, Germany). The
growth inhibition rate was calculated as follows: Inhibition (%) =
[1 − (absorbance of experimental group / absorbance of control
group)] ×100.
Flow cytometric analysis
JC-1 (cat. no. C2005; Beyotime Institute of
Biotechnology, Haimen, China) was used to monitor the integrity of
mitochondria and fluorescent mitochondria dye MitoTracker™ Green FM
(cat. no. M7514; Invitrogen; Thermo Fisher Scientific, Inc.) was
used to detect the mass of mitochondria. Exponentially growing
SKOV3 and SKOV3/DDP cells were seeded in 6-well culture plates at a
density of 2×105 cells/well. Following exposure to
different experimental conditions, the cells were trypsinized and
resuspended in 1640 medium with 10% FBS at a concentration of
1×106 cells/ml. The cells were incubated with JC-1 (5
µg/ml) or MitoTracker™ Green (100 nm) in the dark at room
temperature for 20 min. The samples were examined using the BD
Accuri™ C6 Plus personal flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA).
Transmission electron microscopy
Electron microscopy and morphometric analysis were
performed as described previously (27). The cells were fixed for 30 min with
ice-cold 2.5% glutaraldehyde in 0.1 M cacodylate buffer, embedded
in Epon, and processed for transmission electron microscopy by
standard procedures. Representative areas were selected for
ultra-thin sectioning (70 nm) and examined on a transmission
electron microscope at ×20,000 magnification.
Immunofluorescence staining and confocal
laser microscopy
The cells were seeded onto coverslips in 24-well
plates (5×104 cells/well) overnight and exposed to
different experimental conditions. Following incubation with
MitoTracker™ Red CMXRos (cat. no. 7512; Invitrogen; Thermo Fisher
Scientific, Inc.) for 30 min, the cells were washed with cold PBS
three times, fixed with 4% (w/v) paraformaldehyde for 20 min and
washed with cold PBS three times. The cells were then stained with
Hoechst 33342 (1 µg/ml) for 5 min and then washed three
times with PBS. Following mounting, images were captured using an
Olympus FV1000 confocal laser microscope (Olympus Corporation,
Tokyo, Japan).
Western blot analysis
The cells subjected to the different treatments were
harvested, washed twice with cold PBS, and then gently scraped into
120 µl of RIPA buffer. The cell lysates were sonicated for
30 sec on ice and then lysed at 4°C for 45 min. The cell lysates
were centrifuged at 3,000 × g for 15 min at 4°C, and supernatant
protein concentrations were determined using the BCA Protein assay
kit (Pierce; Thermo Fisher Scientific, Inc.). For western blot
analysis, equivalent quantities of lysate proteins (30–50
µg) were separated by 12% w/v SDS-polyacrylamide gel
electrophoresis and transferred onto immobilon-P transfer membranes
(EMD Millipore). The membranes were blocked with 5% (w/v) skim milk
in buffer of PBST, containing 10 mM Tris-HCl (pH 7.6), 100 mM NaCl
and 0.1% (v/v) Tween-20, for 1 h at room temperature, then
incubated with the desired primary antibody (anti-PGC1α, 1:1,000
dilution; anti-NRF1, 1:1,000 dilution; anti-NRF2, 1:1,000 dilution;
anti-TFAM 1:1,000 dilution; anti-β-actin, 1:1,000 dilution;
anti-Bcl-2, 1:1,000 dilution; anti-Bax, 1:1,000 dilution;
anti-COX5A, 1:1,000 dilution; anti-caspase-3, 1:1,000 dilution;
anti-VDAC1, 1:1,000 dilution; anti-Mcl-1, 1:200 dilution;
anti-TOM20, 1:200 dilution and anti-cytochrome c, 1:200
dilution) overnight at 4°C. The following day, the membranes were
washed with PBST and incubated with the horseradish
peroxidase-conjugated secondary antibodies (1:2,000; ProteinTech
Group, Inc.) for 1 h at room temperature. Following washing the
membranes with PBST, immunodetection was performed using ECL
reagent (Thermo Fisher Scientific, Inc.) and visualized using a
Syngene Bio Imaging (Synoptics, Cambridge, UK). The protein levels
were quantified by densitometry using Quantity One version 4.6.2
software (Bio-Rad Laboratories, Inc.), normalized to β-actin.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total cellular RNA was extracted using TRIzol™
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), reverse
transcription was performed to generate cDNA, which was then
amplified by RT-qPCR. The sequences of the primers used are listed
in Table I. RT-qPCR analysis,
containing 0.2 µg cDNA, 0.2 µM forward primer, 0.2
µM reverse primer, 10 µl qPCR SuperMix and total
volume to 20 µl, was performed using TransStart Top Green
qPCR SuperMix (cat. no. AQ131, TransGen Biotech Co., Ltd., Beijing,
China) in the following conditions: 94.0°C for 30 sec, 40 cycles of
94.0°C for 5 sec and 60.0°C for 30 sec. A melting curve was
detected between 60 and 94°C to confirm the PCR product of
interest. Each sample was analyzed in triplicate in the CFX96
Touch™ Real-Time PCR detection system (Bio-Rad Laboratories, Inc.).
The relative expression was calculated using the 2−ΔΔCq
method (28) among the different
experimental groups normalized to the expression of GAPDH.
 | Table IPrimers for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primers for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene name | Primer
sequences |
|---|
| GAPDH | F:
5′-GGAGCGAGATCCCTCCAAAAT-3′ |
| R:
5′-GGCTGTTGTCATACTTCTCATGG-3′ |
| PPARGC1A | F:
5′-TGAAGACGGATTGCCCTCATT-3′ |
| R:
5′-GCTGGTGCCAGTAAGAGCTT-3′ |
| NRF1 | F:
5′-AGGAACACGGAGTGACCCAA-3′ |
| R:
5′-TGCATGTGCTTCTATGGTAGC-3′ |
| GABPA | F:
5′-TTAAACCTGCGGACACTGTTG-3′ |
| R:
5′-GTATCCCAAGGCGTTCTTGTT-3′ |
| TFAM | F:
5′-GCTCAGAACCCAGATGCAAAA-3′ |
| R:
5′-GCCACTCCGCCCTATAAGC-3′ |
| TFB1M | F:
5′-AGAGACTTGCAGCCAATACAGG-3′ |
| R:
5′-GTGTCGAACATTGCAGAGGTA-3′ |
| ATP5A1 | F:
5′-GTATTGCCCGCGTACATGG-3′ |
| R:
5′-AGGACATACCCTTTAAGCCTGA-3′ |
| VDAC1 | F:
5′-ACGTATGCCGATCTTGGCAAA-3′ |
| R:
5′-TCAGGCCGTACTCAGTCCATC-3′ |
| BCL2 | F:
5′-GGTGGGGTCATGTGTGTG-3′ |
| R:
5′-CGGTTCAGGTACTCAGTCATCC-3′ |
| 18s rRNA | F:
5′-TAGAGGGACAAGTGGCGTTC-3′ |
| R:
5′-CGCTGAGCCAGTCAGTGT-3′ |
| ND1 | F:
5′-CACCCAAGAACAGGGTTTGT-3′ |
| R:
5′-TGGCCATGGGATTGTTGTTAA-3′ |
PGC1α knockdown by short hairpin
(sh)RNA
shRNA sequences targeting human PGC1αG and a
non-target sequence were constructed by GeneChem Co., Ltd.
(Shanghai, China). The PGC1α shRNA sequences were
5′-GTT-ATA-CCT-GTG-ATG-CTT-T-3′ and 5′-CAG-CGA-AGA-TGA-AAG-TGA-T-3′
and the non-target shRNA (Scramble) sequence was
5′-TTC-TCC-GAA-CGT-GTC-ACG-T-3′. Transfections with the shRNAs were
performed using TurboFect™ transfection reagent (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, the SKOV3/DDP cells were plated in 6-well plates and
transfected the following day with 4 µg of PGC1α-shRNA or
shScramble using 6 µl of transfection reagent. The cells
were harvested 24 h following transfection, and whole cell lysates
were isolated for western blot and RT-qPCR analyses. For cellular
viability, the transfected cells were treated with cisplatin for 24
h and then analyzed by MTT assays.
Determination of the relative mtDNA copy
number
Total cellular DNA was extracted from
1×106 cells using the TIANamp Genomic DNA kit (Tiangen
Biotech Co., Ltd., Beijing, China). qPCR for mitochondrial DNA
content was determined using TransStart Top Green qPCR SuperMix
(TransGen Biotech Co. Ltd.) for measuring the mitochondrial-encoded
nicotinamide adenine dinucleotide dehydrogenase 1 (ND1) relative to
nuclear-encoded gene 18S rRNA (18S), as described previously
(29,30). The reactions, containing 0.2
µg DNA, 0.2 µM forward primer, 0.2 µM reverse
primer, 10 µl TransStart Top Green qPCR SuperMix and total
volume to 20 µl, were performed using the CFX96 Touch™
Real-Time PCR detection system (Bio-Rad Laboratories, Inc.) in the
following conditions: 94.0°C for 30 sec, 40 cycles of 94.0°C for 5
sec and 60.0°C for 30 sec. A melting curve was detected between 60
and 94°C to confirm the PCR product of interest. Each sample was
analyzed in triplicate and analyzed using Bio-Rad CFX Manager 3.0
software. The relative mtDNA copy number was calculated as the
ratio of the level of amplification obtained for ND1, vs. 18S
(primer sequences listed in Table
I) for each sample, and was normalized to the control
group.
Oxygen consumption rate (OCR)
The cellular OCR was measured using the
MitoXpress® Xtra-Oxygen Consumption assay (Luxcel
Biosciences, Cork, Ireland) using a CLARIOstar microplate reader
(BMG Labtech GmbH). Briefly, the SKOV3 cells were plated at
8×104 cells/well in 96-well plates and the SKOV3/DDP
cells were plated at 6×104 cells/well in 96-well plates
(clear-bottom, black-body plates) and allowed to adhere overnight.
The reaction was maintained at 37°C with a plate block heater. The
culture medium was removed from all wells and replaced with 160
µl of prewarmed mixed liquid comprising 10 µl
reconstituted medium MitoXpress® Xtra reagent, 10
µl cisplatin (final concentration of 6 or 30 µg/ml)
and 140 µl fresh culture media to each well. The wells were
sealed by adding two drops of pre-warmed HS mineral oil as
recommended by the manufacturer. Fluorescence decay was measured
kinetically immediately in the microplate reader at 37°C in
time-resolved fluorescence (TR-F) mode using a standard filter set
as follows, 380 nm excitation and 650 nm emission filters.
Mitochondrial isolation and protein
extraction
Mitochondrial isolation was performed using the
Minute™ Mitochondria Isolation kit (cat. no. MP-007; Invent
Biotechnologies, Inc., Plymouth, MN, USA) according to the
manufacturer's protocol. The isolated mitochondria was lysed in
0.5% (v/v) Triton X-100 in calcium-free PBS for western blot
analysis with antibodies against β-actin, VDAC1 and cytochrome
c to detect the release of cytochrome c.
Statistical analysis
Data are representative of three independent
experiments each performed in triplicate. Statistical analysis of
the data was performed using a one-way analysis of variance on IBM
SPSS version 22.0 (IBM SPSS, Armonk, NY, USA). Tukey's post hoc
test was used to determine the significance for all pairwise
comparisons of interest. P<0.05 was considered to indicate a
statistically significant difference.
Results
PGC1α-mediated mitochondrial biogenesis
is upregulated in cisplatin-resistant ovarian cancer cells
Previous studies have shown that SKOV3/DDP ovarian
cancer cells are resistant to cisplatin compared with SKOV3 cells
(31), and SKOV3/DDP cells are
more inclined to mitochondrial aerobic oxidation to sustain
cellular processes (Xu et al, unpublished data). As shown in
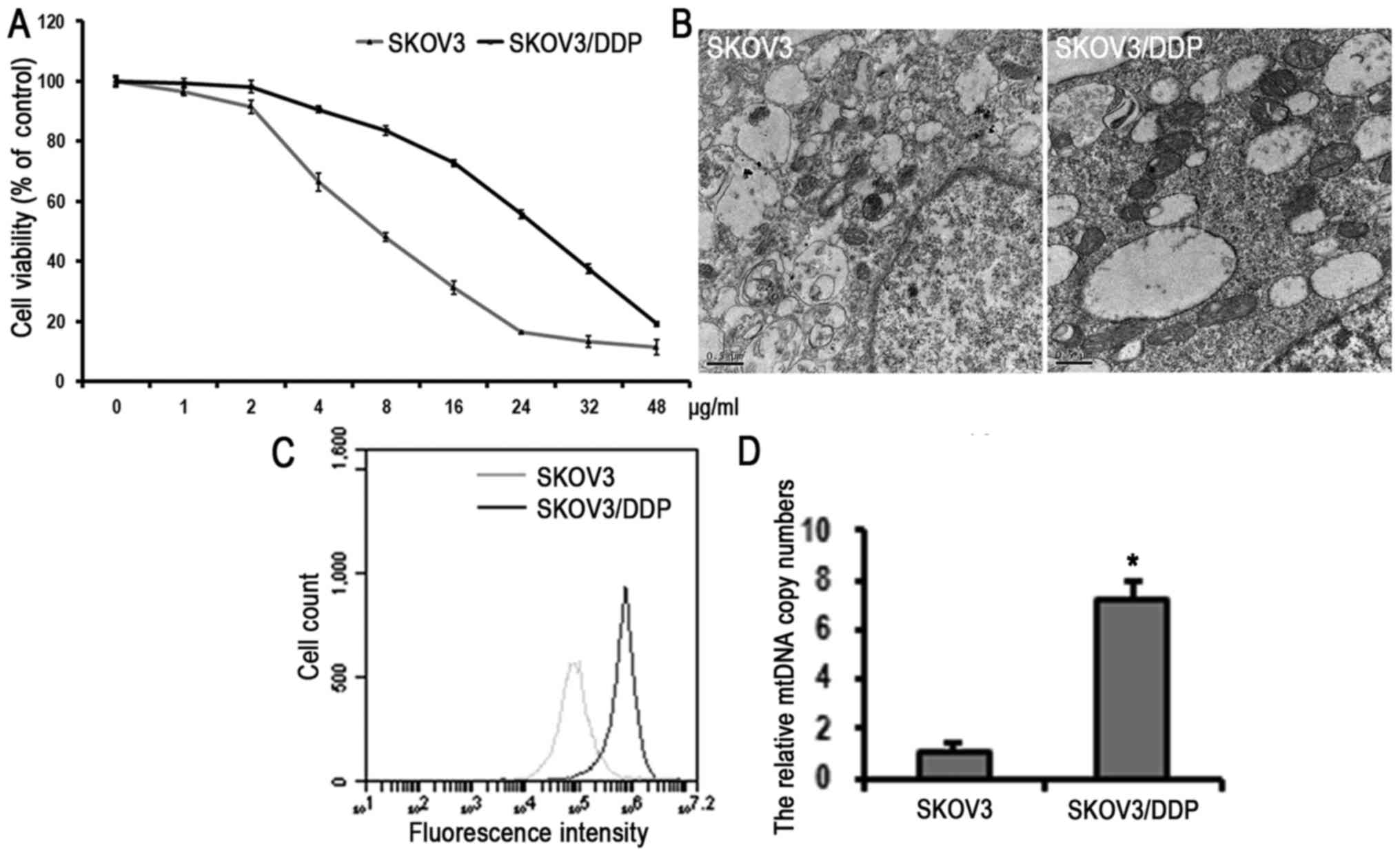
Fig. 1A, the IC50
values of SKOV3 and SKOV3/DDP cells were determined as 6 and 30
µg/ml, respectively. To further characterize the effect of
cisplatin resistance on mitochondria, the mitochondrial mass and
volume in cells were examined using transmission electron
microscopy. Higher numbers and a larger volume of mitochondria were
observed in the SKOV3/DDP cells compared with the SKOV3 cells
(Fig. 1B). In addition,
mitochondria were stained using the fluorescent mitochondria dye
MitoTracker™ Green to detect the mass of mitochondria using flow
cytometry. As shown in Fig. 1C,
the fluorescence intensity of the SKOV3/DDP cells was significantly
higher than that of the SKOV3 cells, indicating that the SKOV3/DDP
cells had higher mitochondria content than the SKOV3 cells. As
there is a positive correlation between mitochondrial mass and
mtDNA copy numbers (32), the
present study subsequently examined the mtDNA copy numbers in the
cell lines by qPCR. The relative mtDNA copy number of the SKOV3/DDP
cells was higher than that of the SKOV3 cells (Fig. 1D), suggesting that the SKOV3/DDP
cells had a higher level of mitochondrial biogenesis than the SKOV3
cells.
Based on the above results, the present study
subsequently examined the mitochondrial biogenesis regulatory
pathway in these two cell lines. The gene and protein expression
levels of mitochondrial biogenesis-related signaling molecules were
examined using RT-qPCR and western blot analyses. It was found that
the gene expression levels of NRF1, GA-binding protein
transcription factor-α (GABPA), TFAM and TFB1M were higher in the
SKOV3/DDP cells than in the SKOV3 cells (Fig. 2A). Consistent with these findings,
the protein expression levels of PGC1α, NRF1, NRF2, TFAM, VDAC1 and
COX5A were higher in the SKOV3/DDP cells than in the SKOV3 cells
(Fig. 2B). These results
demonstrated that the nuclear regulatory system of mitochondrial
biogenesis was upregulated in the cisplatin-resistant SKOV3/DDP
cells, suggesting the potential association between PGC1α-mediated
mitochondrial biogenesis and cisplatin resistance in ovarian
cancer.
 | Figure 2Discrepancies in the levels of PGC1α
downstream molecules between SKOV3 and SKOV3/DDP cells. (A) Reverse
transcription-quantitative polymerase chain reaction detection of
PGC1α downstream genes in SKOV3 and SKOV3/DDP cells (mean ±
standard deviation, n=3; *P<0.05, vs. SKOV3). (B)
Western blot detection of PGC1α, and PGC1α downstream proteins and
mitochondrial proteins in SKOV3 and SKOV3/DDP cells. (C)
Quantitation of PGC1α downstream and mitochondrial protein levels
(mean ± standard deviation, n=3; *P<0.05, vs. SKOV3).
SKOV3/DDP, cisplatin-resistant SKOV3 cells; PGC1α, peroxisome
proliferator-activated receptor-coactivator 1α; GABPA, GA-binding
protein transcription factor-αA; TFAM, mitochondrial transcription
factor A; TFB1M, mitochondrial transcription factor B1; TOM20,
translocase of mitochondrial outer membrane complex 20; VDAC1,
voltage-dependent anion-selective channel 1; COX5A, cytochrome
c oxidase subunit 5a. |
Cisplatin stimulates PGC1α-mediated
mitochondrial biogenesis in cisplatin-resistant ovarian cancer
cells
Previous results indicated that SKOV3/DDP cells
underwent reduced apoptosis, compared with SKOV3 cells treated with
cisplatin for 24 h (33). In the
present study, using confocal microscopy, apoptotic chromatin
condensation was examined in SKOV3 and SKOV3/DDP cells treated with
cisplatin (6 µg/ml) for 24 h with Hoechst 33342 staining.
Treatment with cisplatin induced apoptotic chromatin condensation
in the SKOV3 cells, whereas no significant changes were observed in
the SKOV3/DDP cells (Fig. 3A).
Changes in the mitochondrial membrane potential occur during the
early stages of apoptosis, therefore, the present study evaluated
the mitochondrial outer membrane integrity using JC-1 and flow
cytometry in the cell lines treated with cisplatin (6 µg/ml)
for various durations (0, 6, 12 and 24 h). The results showed that
cisplatin induced mitochondrial depolarization in the SKOV3 cells,
but not in the SKOV3/DDP cells (Fig.
3B–D), suggesting that ovarian cancer cisplatin-resistant cells
maintained mitochondrial structure stability under cisplatin
stress. Western blot analysis was also used to detect the release
of cytochrome c in SKOV3 and SKOV3/DDP cells treated with 6
µg/ml cisplatin for 24 h. Cisplatin induced the release of
cytochrome c in SKOV3 cells, but not in SKOV3/DDP cells
(Fig. 3E), indicating the
occurrence of apoptosis in the cisplatin-sensitive cells.
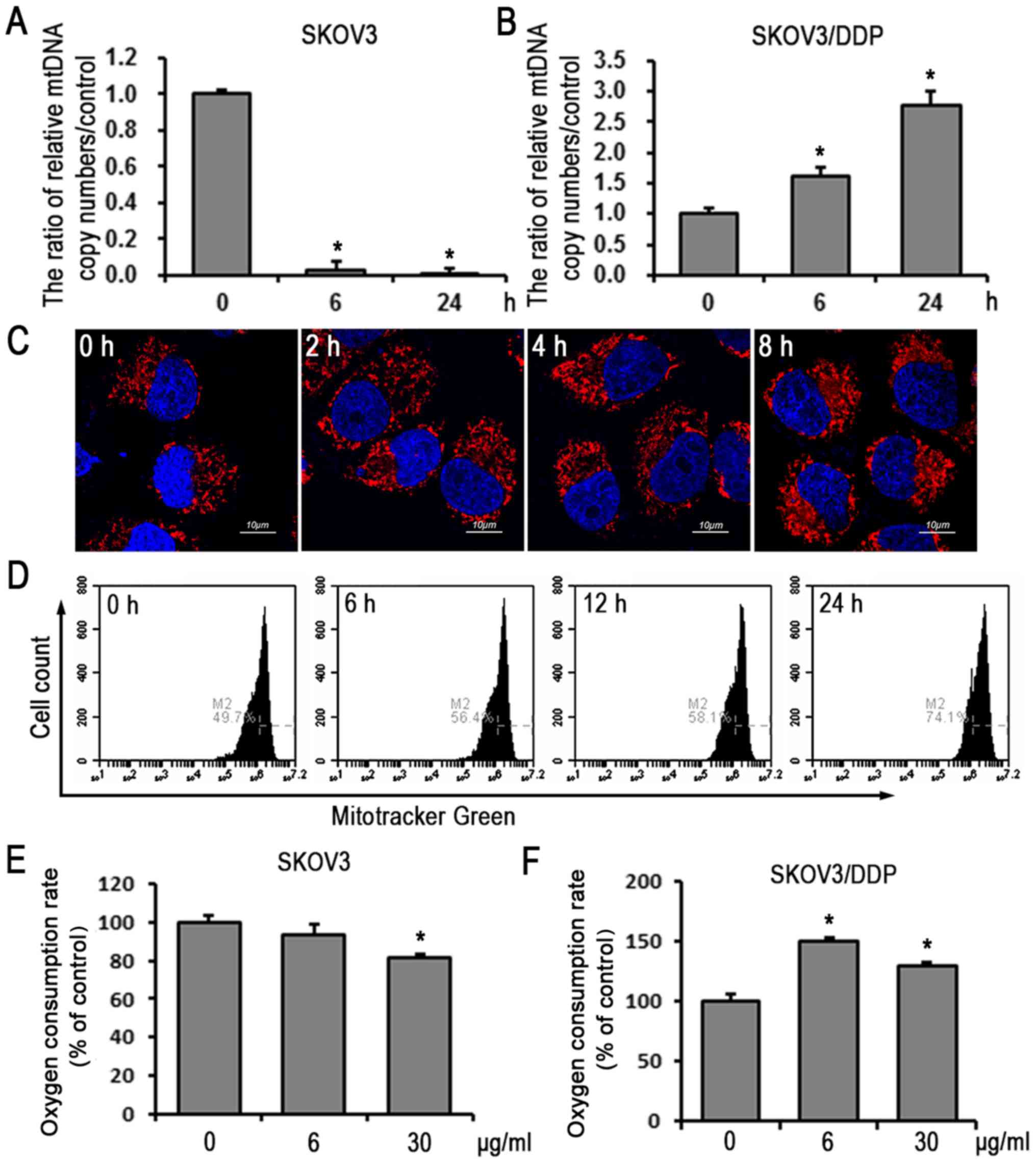
To further clarify the role of mitochondrial
biogenesis in the response to cisplatin-induced apoptosis, the
present study examined the mtDNA copy numbers in the two cell lines
treated with cisplatin (6 µg/ml) for 0, 3 and 24 h. The
mtDNA copy number was increased in the SKOV3/DDP cells in a
time-dependent manner, compared with that in the SKOV3 cells
(Fig. 4A and B). The mitochondrial
mass of the SKOV3/DDP cells treated with cisplatin (6 µg/ml)
for various 0, 2, 4 and 8 h was then examined using MitoTracker™
Red staining and confocal microscopy. In addition, MitoTracker™
Green staining and flow cytometry were used to examine SKOV3/DDP
cells treated with cisplatin (6 µg/ml) for 0, 6, 12 and 24
h. These results showed that the fluorescence intensity increased
in a time-dependent manner in SKOV3/DDP cells (Fig. 4C and D), demonstrating that
cisplatin-resistant ovarian cancer cells responded to cisplatin
stress by increasing intracellular mitochondrial mass.
Extracellular OCR was then evaluated. As shown in Fig. 4E and F, cisplatin induced an
increase of OCR in SKOV3/DDP cells and decreased OCR in SKOV3
cells, indicating that, in response to cisplatin,
cisplatin-resistant cells not only maintain mitochondrial mass and
integrity, but also enhance mitochondrial function to overcome
cisplatin-induced cytotoxic effects.
Taken together the results of the present study
indicated that, in response to cisplatin stress, SKOV3/DDP cells
maintained the mass, stability and function of mitochondria,
however, how the upstream regulatory pathway is involved in
mitochondrial biogenesis remained to be elucidated. The results of
RT-qPCR analysis revealed that the gene expression levels of PGC1α,
NRF1, GABPA and mitochondrial-related genes increased in SKOV3/DDP
cells treated with cisplatin (6 µg/ml) in a time-dependent
manner (Fig. 5A). The expression
of mitochondrial biogenesis-related signaling molecules were then
evaluated in SKOV3/DDP cells (Fig.
5B–E) treated with cisplatin for various times (0, 3, 6, 12 and
24 h). At a cisplatin concentration of 6 µg/ml, the protein
levels of PGC1αP NRF1 and NRF2 gradually increased with the
increase of exposure to cisplatin treatment; cisplatin also
enhanced the expression of mitochondrial proteins TFAM, TOM20 and
COX5A, but did not alter the levels of activated caspase-3
(Fig. 5B and D). Protein levels in
response to a high dose of cisplatin (30 µg/ml) in SKOV3/DDP
cells were also examined. Of note, the expression of PGC1α
gradually increased in a time-dependent manner (0–12 h), but then
decreased at 24 h compared with the controls (Fig. 5C and E), which may be due to
PGC1α-mediated mitochondrial biogenesis being unable to function
against the cisplatin-induced toxicity effect on mitochondria.
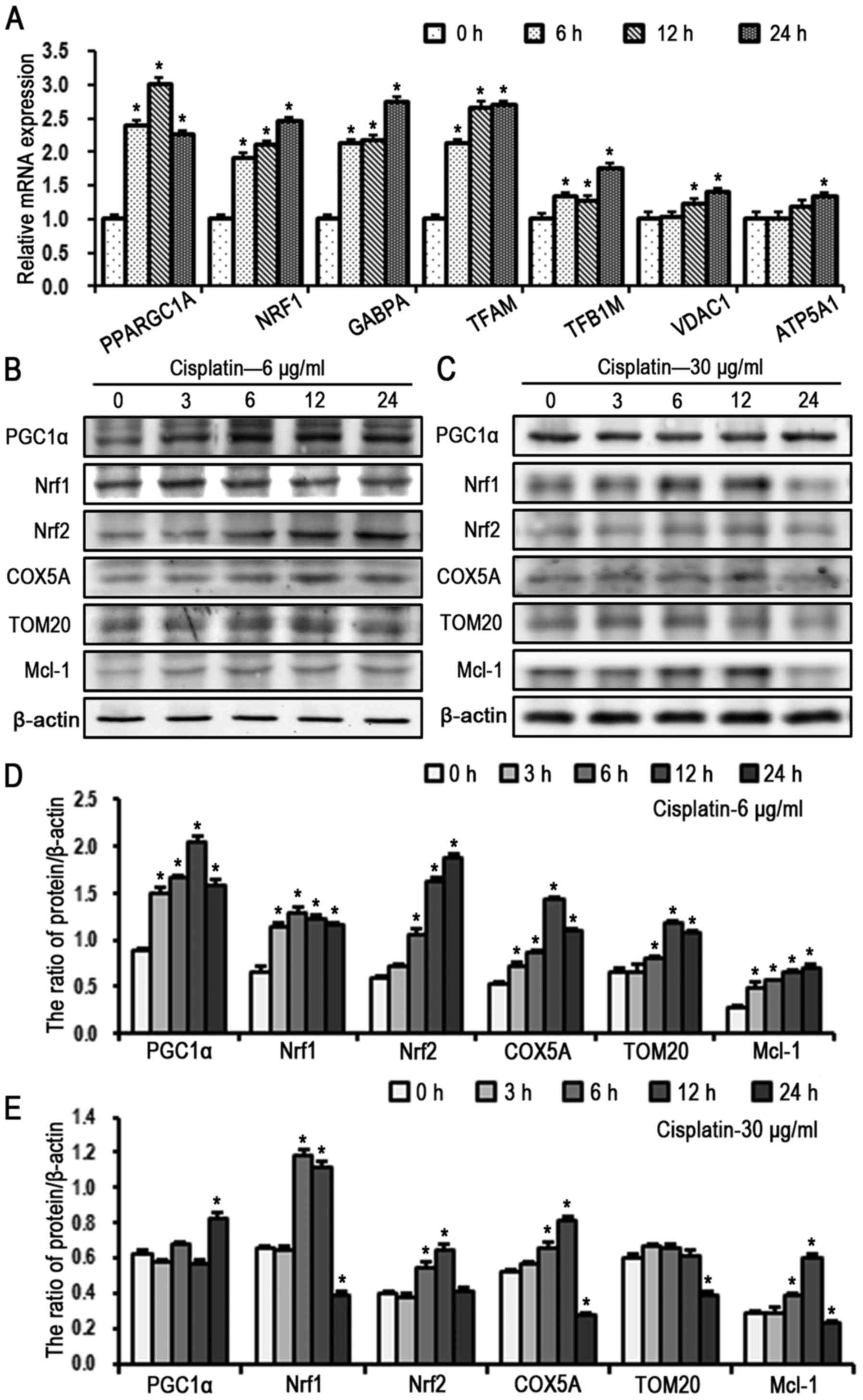 | Figure 5Cisplatin activates the PGC1α
signaling pathway in SKOV3/DDP cells. (A) mRNA levels of PGC1α
signal pathway components and mitochondrial proteins in SKOV3/DDP
cells treated with cisplatin (6 µg/ml) by reverse
transcription-quantitative polymerase chain reaction analysis (mean
± standard deviation, n=3; *P<0.05, vs. control).
Western blot detection of PGC1α signaling pathway and mitochondrial
proteins in SKOV3/DDP cells treated with (B) 6 µg/ml and (C)
30 µg/ml cisplatin. Quantitation of PGC1α signaling pathway
and mitochondrial protein levels in cells treated with (D) 6
µg/ml and (E) 30 µg/ml cisplatin (mean ± standard
deviation, n=3; *P<0.05, vs. control). SKOV3/DDP,
cisplatin-resistant SKOV3 cells; PGC1α, peroxisome
proliferator-activated receptor-coactivator 1α; NRF, nuclear
respiratory factor; GABPA, GA-binding protein transcription
factor-αA; TFAM, mitochondrial transcription factor A; TFB1M,
mitochondrial transcription factor B1; TOM20, translocase of
mitochondrial outer membrane complex 20; VDAC1, voltage-dependent
anion-selective channel 1; COX5A, cytochrome c oxidase
subunit 5a; Mcl-1, myeloid cell leukemia 1. |
These above results demonstrated that cisplatin
activated the PGC1α-mediated mitochondrial biogenesis signaling
pathway, and maintained the structure and function integrity of
mitochondria in SKOV3/DDP cells, with an upregulation of PGC1α.
This suggested that the PGC1α pathway may be involved in the
mechanism of cisplatin resistance in ovarian cancer
cisplatin-resistant cells.
Knockdown of PGC1α by shRNA decreases
mitochondrial biogenesis and increases sensitivity to cisplatin in
SKOV3/DDP cells
To further evaluate the action of PGC1α in cisplatin
resistance of ovarian cancer cells, PGC1α was knocked down in
SKOV3/DDP cells by transient transfection with shRNA, with the
protein expression of PGC1α shown to be downregulated (Fig. 6A). The mtDNA copy number was
significantly decreased in the PGC1α-shRNA knockdown group,
compared with that in the Scr-shRNA group (Fig. 6B). It was also observed that the
expression levels of PGC1α and PGC1α-related genes were decreased
in the PGC1α-shRNA knockdown group, compared with those in the
Scr-shRNA group (Fig. 6C).
Consistent with these results, the protein levels of PGC1α, NRF2,
TFAM, VDAC1 and TOM20 were decreased in the PGC1α-shRNA knockdown
group (Fig. 6D and E).
Furthermore, the level of apoptosis was increased following the
knockdown of PGC1α in SKOV3/DDP cells, as reflected in the increase
of activated caspase-3, the decreased ratios of anti-apoptotic
proteins Bcl-2 and Mcl-1 to pro-apoptotic protein Bax, and the
release of cytochrome c (Fig.
7A–E). No significant differences were observed between the
Scr-shRNA group and the controls. The MTT assay demonstrated that
the knockdown of PGC1α increased in the SKOV3/DDP cell sensitivity
to cisplatin (IC50 of 14.6 µg/ml) compared with
that in the control and Scr-shRNA groups (Fig. 7F).
 | Figure 6Knockdown of PGC1α decreases
mitochondrial biogenesis in SKOV3/DDP cells. (A) Western blot
analysis of the knockdown efficiency of PGC1α. (B) Relative mtDNA
copy numbers were detected using qPCR analysis in SKOV3/DDP cells
transfected with shRNA for 24 h (mean ± standard deviation, n=3;
#P<0.05, vs. Scr-shRNA). (C) RT-qPCR analysis of mRNA
levels of PGC1α signaling pathway and mitochondrial genes in
SKOV3/DDP cells transfected with shRNA for 24 h (mean ± standard
deviation, n=3; #P<0.05, vs. Scr-shRNA). (D) Western
blot detection of mitochondrial biogenesis proteins in SKOV3/DDP
cells. (E) Quantitation of mitochondrial biogenesis protein levels
(mean ± standard deviation, n=3; *P<0.05, vs.
control). SKOV/DDP, cisplatin-resistant SKOV cells; PGC1α,
peroxisome proliferator-activated receptor-coactivator 1α; NRF,
nuclear respiratory factor; GABPA, GA-binding protein transcription
factor-αA TFAM, mitochondrial transcription factor A; TFB1M,
mitochondrial transcription factor B1; VDAC1, voltage-dependent
anion-selective channel 1; BCL2, B-cell lymphoma 2; shRNA, short
hairpin RNA; Scr, scrambled; mtDNA, mitochondrial DNA; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction. |
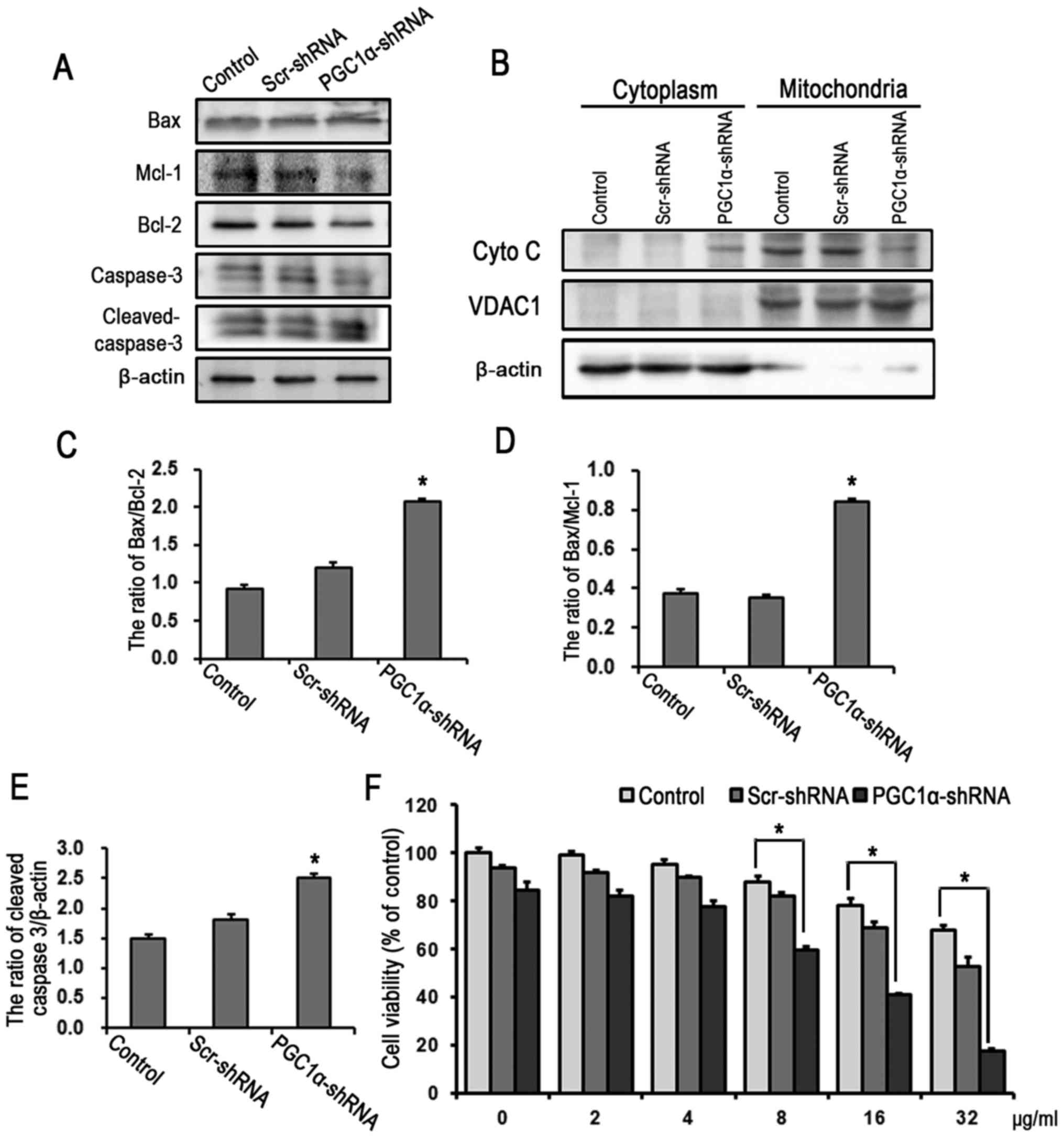 | Figure 7Knockdown of PGC1α increases the
level of apoptosis and decreases the cisplatin resistance in
SKOV3/DDP cells. (A) Western blot detection of mitochondrial
apoptosis-related proteins in SKOV3/DDP cells transfected with
shRNA for 24 h. (B) Western blot detection of the release of Cyto C
in SKOV3 and SKOV3/DDP cells treated with 6 µg cisplatin for
24 h. Quantitation of protein levels of (C) Bax/Bcl-2, (D)
Bax/Mcl-1 and (E) cleaved caspase-3 (mean ± standard deviation,
n=3; *P<0.05, vs. control). (F) SKOV3/DDP cells were
treated with varying doses of cisplatin for 24 h following PGC1α
knockdown by shRNA. Cell viability was determined using a
3-(4,5-dimetrylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
(mean ± standard deviation, n=3; *P<0.05, vs.
control). SKOV/DDP, cisplatin-resistant SKOV cells; PGC1α,
peroxisome proliferator-activated receptor-coactivator 1α; shRNA,
short hairpin RNA; Scr, scrambled; Mcl-1, myeloid cell leukemia 1;
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; Cyto C,
cytochrome c; VDAC1, voltage-dependent anion-selective
channel 1. |
The above results indicated that the deficiency of
PGC1α in SKOV3/DDP cells resulted in downregulation of the
mitochondrial biogenesis signaling pathway and reductions in the
mass and function of mitochondria, resulting in increased
sensitivity to cisplatin. These results further verified the
hypothesis that, in response to cisplatin stress, ovarian cancer
cisplatin-resistant cells show upregulated expression of PGC1α and
activation of the mitochondrial biogenesis signaling pathway, which
contributes to cisplatin resistance by maintaining the abundance
and functional integrity of mitochondria.
Discussion
Cisplatin resistance remains a major obstacle in the
clinical treatment of ovarian cancer (34). Increasing evidence indicates that,
in addition to DNA lesions, complex signal pathways in the nucleus
and cytoplasm can be involved in the cisplatin resistance mechanism
(1,2). Vellinga et al showed that
chemotherapy induced the upregulation of PGC1α signaling
pathway-dependent respiratory complex protein and enhanced the OCR
in colon cancer and, under this condition, the cancer cells
survived cisplatin cytotoxicity through transforming tumor
metabolism from aerobic glycolysis to OXPHOS (21). Similar to these findings, our
previous study revealed that cisplatin-resistant SKOV3/DDP ovarian
cancer cells were more dependent on mitochondrial aerobic oxidation
to support their growth (Xu et al, unpublished data), and
the expression of PGC1α, a crucial regulator of mitochondrial
biogenesis, was significantly higher in SKOV3/DDP cells than SKOV3
cells (Fig. 1E). Transmission
electron microscopy and flow cytometry showed that the mass and
volume of mitochondria were larger in SKOV3/DDP cells than in SKOV3
cells (Fig. 1B and C). It was also
found that the mtDNA copy number was higher in SKOV3/DDP cells than
in SKOV3 cells (Fig. 1D).
Collectively, these results demonstrate a higher level of
mitochondrial biogenesis in cisplatin-resistant ovarian cancer
cells and suggested an association between the high level of
mitochondrial energy metabolism and the cisplatin resistance
mechanism.
Treatment strategies targeting mitochondria
metabolic function in cancer cells have gained wide acceptance
(35,36), and earlier studies have shown that
the overexpression of PGC1α restores the intracellular oxidative
metabolism and ATP content, which prevents cell death by enhancing
bioenergy homeostasis and the antioxidant defense system. Even if
PGC1α is overexpressed, the OCR cannot be restored with knockout of
NRF1 and NRF2 (37). Of note,
there are discrepancies in PGC1α in tumor cells of different
histologic origin, and the overexpression of PGC1αis not beneficial
in all cases. LaGory et al found that, under hypoxia, the
downregulation of the PGC1α/estrogen-related receptor α axis
induced tumor chemotherapy resistance in renal cell carcinoma
(38), suggesting the specific
effect of PGC1α depends on the cellular environment and the
interaction of PGC1α with different molecules. Therefore, the
present study focused on the PGC1α/NRF1/2-axis-regulated
mitochondrial biogenesis pathway. NRF1 and NRF2 transcriptionally
regulate the majority of aerobic respiratory complex proteins
(39) and mtDNA transcription
factors (40,41), and control mitochondrial biogenesis
through their interactions with PGC1α (42–44),
which ensures the integration between the nuclear and mitochondrial
genomes for mitochondrial function in a direct and indirect manner.
In the present study, the gene expression levels of NRF1, GABPA
TFAM and TFB1M were higher in SKOV3/DDP cells than in SKOV3 cells
(Fig. 2A), and similar results
were shown for protein expression (Fig. 2B). These results demonstrated
differences in the mitochondrial biogenesis status between the two
cell lines, and provided preliminary data supporting a role for
PGC1α in cisplatin resistance via regulating mitochondrial
biogenesis in ovarian cancer.
Several reports have shown that an increase of mtDNA
copy number, an indicator of mitochondrial biogenesis, is
associated with poor prognosis in colorectal cancer, gastric cancer
and lymphoblastic leukemia glioma (45–47).
The present study found that the mtDNA copy number in SKOV3/DDP
cells gradually increased with cisplatin exposure duration, whereas
the copy number in SKOV3 cells decreased (Fig. 4A and B). With the increase of
mitochondrial mass (Fig. 4C and
D), the OCR of SKOV3/DDP cells also increased under cisplatin
treatment, and that of SKOV3 cells decreased (Fig. 4E and F). At the molecular level,
cisplatin promoted the gene and protein expression of PGC1α, and
upregulated the PGC1α-mediated mitochondrial biogenesis pathway
(Fig. 5). The JC-1 staining and
flow cytometry revealed that the SKOV3/DDP cells maintained
mitochondrial integrity on exposure to cisplatin, as reflected in
the high mitochondrial membrane potential, but not in SKOV3 cells
(Fig. 3B and C), leading to the
release of cytochrome c and chromatin condensation in SKOV3
cells. These results demonstrated that cisplatin-resistant and
non-resistant ovarian cancer cells show distinct responses to
cisplatin stimulation, with differences in the mass and integrity
of mitochondria in the two cells, eventually leading to differences
in mitochondrial function and cell fate.
To further clarify the role of PGC1α in
cisplatin-resistant ovarian cancer cells, knockdown assays were
performed through transient transfection with shRNA. The mtDNA copy
number and PGC1α-mediated mitochondrial biogenesis pathway were
decreased upon PGC1α knockdown (Fig.
6). The majority of Bcl-2 family proteins are located in the
mitochondrial outer membrane through the carboxy-terminal
transmembrane domain, whereas the localization and function of
Bcl-2 family anti-apoptotic proteins are regulated by the protein
and lipids in the mitochondrial outer membrane (48). In addition to the Bcl-2 family
proteins involved in regulation of the mitochondrial localization
of Bax, several other receptors with components involved in
mitochondrial transport mechanisms, including the TOM complex, and
mitochondrial carrier homolog 2, facilitate Bcl-2 family protein
localization in the mitochondrial outer membrane (49). The results of the present study
showed that, following knockdown of PGC1α, the ratio of
anti-apoptotic proteins (Bcl-2 and Mcl-1) and pro-apoptotic protein
(Bax) in the SKOV3/DDP cells were significantly reduced (Fig. 7A and C–E), as was the release of
cytochrome c, which supported the hypothesis that PGC1α
affects the localization and function of Bcl-2 family proteins and
the level of mitochondrial apoptosis through mitochondrial
biogenesis. Finally, consistent with this hypothesis, the cell
viability decreased in response to cisplatin following the
knockdown of PGC1α (Fig. 7F),
suggesting that the cisplatin-resistant ovarian cancer cells
upregulate mitochondrial bioenergy and biogenesis against cisplatin
toxicity depending on the action of PGC1α and its signaling
pathway.
However, there numerous questions remain unanswered.
The present study observed that the expression of PGC1α was
increased, however, the expression of its downstream protein were
decreased following a high dose of cisplatin for 24 h in the
SKOV3/DDP cells (Fig. 5C). There
are two possible reasons for these observations, one of which is
that cisplatin-induced mitochondrial toxicity exceeded the
supplementation of PGC1α-mediated mitochondrial biogenesis or,
under the toxic effect of cisplatin, PGC1α may initiate other
unknown signal pathways triggering cell death. As discussed above,
PGC1α may be involved in different cellular process depending upon
its interaction with different molecules under various cellular
environments.
Mitochondrial targeting offers additional
possibilities for cancer therapy, including the inhibition of
mitochondrial metabolism, interruption of mitochondria protein
location or destruction of mitochondrial membrane structure.
However, overcoming biologic barriers and accurate location are
major challenges for targeting mitochondrial therapy (50). Although research has suggested the
potential application of nanocarriers to deliver agents, further
investigation is required in the clinical setting.
Collectively, the results of the present study
provided evidence that cisplatin activated PGC1α-mediated
mitochondrial biogenesis in cisplatin-resistant ovarian cancer
cells, which led to the maintenance of mitochondrial mass and
functional stability, and enhanced the mitochondrial anti-apoptotic
capacity. Upon depletion of PGC1α in the cisplatin-resistant
ovarian cancer cells, mitochondrial biogenesis decreased, followed
by a decrease in mitochondrial stability and function; in these
conditions, the anti-apoptotic capacity was unable to respond to
cisplatin toxicity and enhanced the sensitivity of the cell to
cisplatin. These data showed that PGC1α-mediated mitochondrial
biogenesis in cisplatin-resistant cells ovarian cancer may provide
a specific targeting approach for mitochondrial
respiration-dependent chemotherapy-resistant tumors and alter the
clinical efficacy of cisplatin via the nuclear transcription system
in these tumors.
Acknowledgments
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81772794, 81472419,
81672948, 81501982 and 81572927), the Jilin Provincial Research
Foundation for the Development of Science and Technology Projects
(grant nos. 20170623021TC and 20160414005GH) and the Jilin
University Bethune Plan B Projects (grant no. 2015222).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LS conceived and designed the experiments, performed
the experiments, analyzed the data and wrote the manuscript. BS and
JS performed the experiments, analyzed the data and were the major
contributors in writing the manuscript. SY, YL and HX analyzed the
data, prepared figures and/or tables. JS and LS conceived and
designed the experiments, reviewed and edited the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warburg O: Iron, the oxygen-carrier of
respiration-ferment. Science. 61:575–582. 1925. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
6
|
Zong WX, Rabinowitz JD and White E:
Mitochondria and cancer. Mol Cell. 61:667–676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scarpulla RC: Transcriptional paradigms in
mammalian mitochondrial biogenesis and function. Physiol Rev.
88:611–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mercer TR, Neph S, Dinger ME, Crawford J,
Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O,
Stamatoyannopoulos JA, et al: The human mitochondrial
transcriptome. Cell. 146:645–658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quirós PM, Mottis A and Auwerx J:
Mitonuclear communication in homeostasis and stress. Nat Rev Mol
Cell Biol. 17:213–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marchetti P, Guerreschi P, Mortier L and
Kluza J: Integration of mitochondrial targeting for molecular
cancer therapeutics. Int J Cell Biol. 2015:2831452015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y and Xu H: Translational regulation
of mitochondrial biogenesis. Biochem Soc Trans. 44:1717–1724. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fernandez-Marcos PJ and Auwerx J:
Regulation of PGC-1α, a nodal regulator of mitochondrial
biogenesis. Am J Clin Nutr. 93:884S–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan Z, Luo X, Xiao L, Tang M, Bode AM,
Dong Z and Cao Y: The role of PGC1α in cancer metabolism and its
therapeutic implications. Mol Cancer Ther. 15:774–782. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Permuth-Wey J, Chen YA, Tsai YY, Chen Z,
Qu X, Lancaster JM, Stockwell H, Dagne G, Iversen E, Risch H, et
al: Inherited variants in mitochondrial biogenesis genes may
influence epithelial ovarian cancer risk. Cancer Epidemiol
Biomarkers Prev. 20:1131–1145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haq R, Shoag J, Andreu-Perez P, Yokoyama
S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL,
et al: Oncogenic BRAF regulates oxidative metabolism via PGC1α and
MITF. Cancer Cell. 23:302–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gopal YN, Rizos H, Chen G, Deng W,
Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V,
et al: Inhibition of mTORC1/2 overcomes resistance to MAPK pathway
inhibitors mediated by PGC1α and oxidative phosphorylation in
melanoma. Cancer Res. 74:7037–7047. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Z, Puigserver P, Andersson U, Zhang C,
Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et
al: Mechanisms controlling mitochondrial biogenesis and respiration
through the thermogenic coactivator PGC-1. Cell. 98:115–124. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Puigserver P and Spiegelman BM: Peroxisome
proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1
alpha): Transcriptional coactivator and metabolic regulator. Endocr
Rev. 24:78–90. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gleyzer N, Vercauteren K and Scarpulla RC:
Control of mitochondrial transcription specificity factors (TFB1M
and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and
PGC-1 family coactivators. Mol Cell Biol. 25:1354–1366. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scarpulla RC: Nuclear control of
respiratory chain expression by nuclear respiratory factors and
PGC-1-related coactivator. Ann N Y Acad Sci. 1147:321–334. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vellinga TT, Borovski T, de Boer VC,
Fatrai S, van Schelven S, Trumpi K, Verheem A, Snoeren N, Emmink
BL, Koster J, et al: SIRT1/PGC1α-dependent increase in oxidative
phosphorylation supports chemotherapy resistance of colon cancer.
Clin Cancer Res. 21:2870–2879. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gao W, Wu MH, Wang N, Ying MZ, Zhang YY,
Hua J, Chuan L and Wang YJ: Mitochondrial transcription factor A
contributes to cisplatin resistance in patients with estrogen
receptor-positive breast cancer. Mol Med Rep. 14:5304–5310. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhalla K, Hwang BJ, Dewi RE, Ou L, Twaddel
W, Fang HB, Vafai SB, Vazquez F, Puigserver P, Boros L, et al:
PGC1α promotes tumor growth by inducing gene expression programs
supporting lipogenesis. Cancer Res. 71:6888–6898. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vazquez F, Lim JH, Chim H, Bhalla K,
Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman
BM, et al: PGC1α expression defines a subset of human melanoma
tumors with increased mitochondrial capacity and resistance to
oxidative stress. Cancer Cell. 23:287–301. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Do MT, Kim HG, Choi JH and Jeong HG:
Metformin induces microRNA-34a to downregulate the
Sirt1/Pgc-1α/Nrf2 pathway, leading to increased susceptibility of
wild-type p53 cancer cells to oxidative stress and therapeutic
agents. Free Radic Biol Med. 74:21–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jones AW, Yao Z, Vicencio JM,
Karkucinska-Wieckowska A and Szabadkai G: PGC-1 family coactivators
and cell fate: Roles in cancer, neurodegeneration, cardiovascular
disease and retrograde mitochondria-nucleus signalling.
Mitochondrion. 12:86–99. 2012. View Article : Google Scholar
|
|
27
|
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo
J and Hu X: Shikonin circumvents cancer drug resistance by
induction of a necroptotic death. Mol Cancer Ther. 6:1641–1649.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Eaton JS, Lin ZP, Sartorelli AC, Bonawitz
ND and Shadel GS: Ataxia-telangiectasia mutated kinase regulates
ribonucleotide reductase and mitochondrial homeostasis. J Clin
Invest. 117:2723–2734. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lebedeva MA, Eaton JS and Shadel GS: Loss
of p53 causes mitochondrial DNA depletion and altered mitochondrial
reactive oxygen species homeostasis. Biochim Biophys Acta.
1787:328–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu H, Su J, Xu Y, Kang J, Li H, Zhang L,
Yi H, Xiang X, Liu F and Sun L: p62/SQSTM1 involved in cisplatin
resistance in human ovarian cancer cells by clearing ubiquitinated
proteins. Eur J Cancer. 47:1585–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Medeiros DM: Assessing mitochondria
biogenesis. Methods. 46:288–294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma L, Xu Y, Su J, Yu H, Kang J, Li H, Li
X, Xie Q, Yu C, Sun L, et al: Autophagic flux promotes cisplatin
resistance in human ovarian carcinoma cells through ATP-mediated
lysosomal function. Int J Oncol. 47:1890–1900. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Agarwal R and Kaye SB: Ovarian cancer:
Strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schulze A and Harris AL: How cancer
metabolism is tuned for proliferation and vulnerable to disruption.
Nature. 491:364–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan Y, Cao M, Liu J, Yang Q, Miao X, Go
VLW, Lee PWN and Xiao GG: Metabolic regulation in mitochondria and
drug resistance. Adv Exp Med Biol. 1038:149–171. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shao D, Liu Y, Liu X, Zhu L, Cui Y, Cui A,
Qiao A, Kong X, Liu Y, Chen Q, et al: PGC-1 beta-regulated
mitochondrial biogenesis and function in myotubes is mediated by
NRF-1 and ERR alpha. Mitochondrion. 10:516–527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
LaGory EL, Wu C, Taniguchi CM, Ding CC,
Chi JT, von Eyben R, Scott DA, Richardson AD and Giaccia AJ:
Suppression of PGC-1α is critical for reprogramming oxidative
metabolism in renal cell carcinoma. Cell Reports. 12:116–127. 2015.
View Article : Google Scholar
|
|
39
|
Cam H, Balciunaite E, Blais A, Spektor A,
Scarpulla RC, Young R, Kluger Y and Dynlacht BD: A common set of
gene regulatory networks links metabolism and growth inhibition.
Mol Cell. 16:399–411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kelly DP and Scarpulla RC: Transcriptional
regulatory circuits controlling mitochondrial biogenesis and
function. Genes Dev. 18:357–368. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Scarpulla RC: Nuclear activators and
coactivators in mammalian mitochondrial biogenesis. Biochim Biophys
Acta. 1576:1–14. 2002. View Article : Google Scholar
|
|
42
|
Scarpulla RC: Metabolic control of
mitochondrial biogenesis through the PGC-1 family regulatory
network. Biochim Biophys Acta. 1813:1269–1278. 2011. View Article : Google Scholar :
|
|
43
|
Mootha VK, Handschin C, Arlow D, Xie X, St
Pierre J , Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N,
et al: Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative
phosphorylation gene expression that is altered in diabetic muscle.
Proc Natl Acad Sci USA. 101:6570–6575. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Girnun GD: The diverse role of the PPARγ
coactivator 1 family of transcriptional coactivators in cancer.
Semin Cell Dev Biol. 23:381–388. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jain A, Bakhshi S, Thakkar H, Gerards M
and Singh A: Elevated mitochondrial DNA copy numbers in pediatric
acute lymphoblastic leukemia: A potential biomarker for predicting
inferior survival. Pediatr Blood Cancer. Nov 14–2017.Epub ahead of
print. PubMed/NCBI
|
|
46
|
Wang Y, He S, Zhu X, Qiao W and Zhang J:
High copy number of mitochondrial DNA predicts poor prognosis in
patients with advanced stage colon cancer. Int J Biol Markers.
31:e382–e388. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang G, Qu Y, Dang S, Yang Q, Shi B and
Hou P: Variable copy number of mitochondrial DNA (mtDNA) predicts
worse prognosis in advanced gastric cancer patients. Diagn Pathol.
8:1732013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hatok J and Racay P: Bcl-2 family
proteins: Master regulators of cell survival. Biomol Concepts.
7:259–270. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zaltsman Y, Shachnai L, Yivgi-Ohana N,
Schwarz M, Maryanovich M, Houtkooper RH, Vaz FM, De Leonardis F,
Fiermonte G, Palmieri F, et al: MTCH2/MIMP is a major facilitator
of tBID recruitment to mitochondria. Nat Cell Biol. 12:553–562.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Milane L, Trivedi M, Singh A, Talekar M
and Amiji M: Mitochondrial biology, targets, and drug delivery. J
Control Release. 207:40–58. 2015. View Article : Google Scholar : PubMed/NCBI
|