Introduction
Acute promyelocytic leukemia (APL) is a type of
acute myeloid leukemia (AML), characterized by the expression of
retinoic acid receptor alpha (RARα) fusion genes, including
PML-RARα, NPM-RARα and PLZF-RARα, resulting in
blockage of myeloid differentiation and aberrant self-renewal of
promyelocytic cells (1,2). Fortunately, the majority of patients
with APL achieve complete remission upon treatment with
all-trans retinoic acid (ATRA) via degradation of RARα
fusion proteins (3,4). However, ATRA therapy has a number of
disadvantages, including drug resistance and a high recurrence rate
(5,6). The key molecular mechanism for
induced differentiation of leukemia cells has not been clarified.
Therefore, identifying the key molecular target of differentiation
disorders is vital for the diagnosis and treatment of leukemia.
The ubiquitin proteasome system (UPS), including
ubiquitination and deubiquitination, is accountable for the
majority of recycling and degradation of proteins within the cell.
Therefore, it regulates pathological changes, including
abnormalities of the immune system and tumor cells (7-11).
Ubiquitination, a common form of post-translational modification
(PTM), is the process by which ubiquitin attaches to lysine
residues on target proteins via a 3-enzyme cascade reaction that
involves the ubiquitin-activating enzyme (E1),
ubiquitin-conjugating enzyme (E2) and ubiquitin ligase (E3)
(12,13). This PTM is reversible by
deubiquitination by deubiquitinating enzymes (deubiquitinases or
DUBs), which can hydrolytically remove ubiquitin from protein
adducts (14-20). In humans, there are >100 types
of DUBs, which can be divided into 5 families: Ubiquitin-specific
proteases (USPs), ubiquitin carboxyl-terminal hydrolases, ovarian
tumor proteases, JAB1/MPN/MOV34 metalloenzymes, and
Machado-Josephin domain proteases (19). USP48 is a member of the USP family,
and there are 53 USP genes in the human genome (21). Previous studies have shown that
DUBs participate in cellular functions, including protein quality
control and degradation, DNA damage and repair, RNA transcription
and processing, and signal transduction (22).
In the present study, the role of USP48 in promoting
ATRA-induced differentiation of leukemia cells was examined. It was
determined that ATRA-induced differentiation was not attributable
to the regulation of p65, a substrate of USP48. In addition, it was
indicated that the expression of USP48 was regulated by
microRNA-301a-3p. These data suggest that dysregulation of USP48
may be an underlying mechanism for the abnormal differentiation of
APL, implicating USP48 as a potential therapeutic target for
APL.
Materials and methods
Cell culture and ATRA treatment
The human acute promy-elocytic leukemia cell lines,
NB4 and HL60, and the human acute monocytic leukemia cell line,
THP1, and 293T cells were purchased from the American Type Culture
Collection (Manassas, VA, USA). Leukemia cells were cultured in
RPMI-1640 medium and 293T cells cultured in DMEM medium
supplemented with 10% fetal bovine serum (FBS; HyClone; GE
Healthcare, Chicago, IL, USA), 100 U/ml penicillin and 100
µg/ml streptomycin, and incubated at 37°C in an atmosphere
with 5% CO2. Leukemia cells were treated with 1
µM ATRA or phorbol-12-myristate-13-acetate (PMA;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Wright-Giemsa staining and microarray
analysis
Leukemia cells were treated with 1 µM ATRA
for 72 h. Wright-Giemsa staining was performed using a
Wright-Giemsa staining kit (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China), according to the
manufacturer's protocol. For micro-array analysis, total RNA was
extracted from HL60 cells using TRIzol (Thermo Fisher Scientific,
Inc., Waltham, MA, USA), according to the manufacturer's protocol.
An Affymetrix Gene Chip Human Genome U133 Plus 2.0 Array analysis
was performed by Beijing CapitalBio Technology Co., Ltd. (Beijing,
China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. RNA was reverse-transcribed into cDNA using PrimeScript™
Reverse Transcriptase (Takara Bio, Inc., Otsu, Japan) for USP48 and
a miRcute Plus miRNA First-Strand cDNA Synthesis kit (Tiangen
Biotech Co., Ltd., Beijing, China), according to the manufacturer's
protocol, for microRNA. The cDNAs were used as templates in RT-qPCR
with the following primers: USP48, forward,
5′-TGGAGCCACTTGTTATGT-3′ and reverse, 5′-GGATCAATGTATCGCCTA-3′;
GAPDH, forward, 5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse,
5′-GCCATCACGCCACAGTTTC-3′; the microRNA primers were purchased from
Tiangen Biotech. RT-qPCR was performed using an Applied Biosystems
7500 real-time system using UltraSYBR Mixture (CWBiotech, Beijing,
China) for USP48, and miRcute Plus miRNA qPCR Detection kit (cat.
no. FP401-01; Tiangen Biotech) for microRNA. The following
conditions were used for PCR: 10 sec at 95°C, 40 cycles of 5 sec at
60°C, 10 sec at 72°C and 30 sec at 65°C. Relative quantity of
expression was calculated using the 2−ΔΔCq method
(23). USP48 and microRNA
expression were normalized to GAPDH and U6
expression, respectively.
Western blotting
Western blot analyses were performed as previously
described (24). The following
primary antibodies were used: USP48 (dilution, 1:1,000; cat. no.
ab72226; Abcam, Cambridge, UK), p65 (dilution, 1:1,000; cat. no.
6956; Cell Signaling Technology, Inc., Danvers, MA, USA), tubulin
(dilution, 1:15,000; cat. no. RLM3030; Ruiyingbio, Suzhou, Jiangsu,
China), histone H3 (dilution, 1:20,000; cat. no. GB13102-1), and
GAPDH (dilution, 1:10,000; cat. no. GB13002-m-1) (both from
Servicebio, Wuhan, China).
Cell proliferation assay
A Cell Counting Kit-8 (CCK-8) assay (Solarbio,
Beijing, China) was used to detect cell proliferation, according to
the manufacturer's instructions. In brief, 1×104 cells
in 100 µl medium were plated per well of a 96-well plate in
triplicate. Following incubation for 0, 24 or 48 h, 10 µl
CCK-8 solution was added per well, and incubated for an additional
3 h at 37°C. Absorbance was measured at 450 nm using a microplate
reader (SynergyHTX; BioTek Instruments, Inc., Winooski, VT,
USA).
Flow cytometry (FCM)
A total of 1×106 cells treated with 1
µM ATRA were washed with serum-free RPMI-1640 medium and
then resuspended in serum-free RPMI-1640 medium. Cell cycle
analysis and apoptosis assessment were performed using Annexin
V/proprium iodide (PI) kit (cat. no. C1052; Beyotime Biotechnology,
Shanghai, China) and analyzed using a flow cytometer
(NovoCyteD1040; ACEA Biosciences, Inc., San Diego, CA, USA) with
NovoExpress1.2.1 software. To detect cluster of differentiation
(CD)11b expression, a FITC-conjugated anti-human CD11b antibody
(dilution, 1:50; cat. no. 555388; BD Pharmingen; BD Biosciences,
Franklin Lakes, NJ, USA) was added to the cells and incubated at
4°C for 30 min. Cells were washed with PBS and analyzed using a
flow cytometer (Epics XL4C) with a EXPO™32 ADC Analysis Software
(both from Beckman Coulter, Inc., Brea, CA, USA).
Immunofluorescence staining
A total of 2×105 cells were centrifuged
at 400 × g for 2 min at room temperature and fixed onto slides with
4% paraformaldehyde for 10 min. Then cells were permeabilized with
0.5% Triton X-100 for 20 min at room temperature. After blocking
with 3% BSA for 30 min at room temperature, USP48 (dilution, 1:100;
cat. no. ab72226; Abcam) and p65 (dilution, 1:100; cat. no. 6956;
Cell Signaling Technology, Inc.) primary antibodies were incubated
with the slides at 4°C overnight. Samples were then incubated with
a goat-anti-rabbit-CY3 (dilution, 1:300; cat. no. GB21303) or
goat-anti-mouse-488 nm (dilution, 1:400; cat. no. GB21301) (both
from Servicebio) fluorophore-conjugated IgG secondary antibodies. A
total of 100 µl DAPI (cat. no. G1012; Servicebio) was added
per slide and incubated at room temperature for 10 min to stain the
nuclei and. The images were acquired on a Nikon Eclipse C1
fluorescence microscope (Nikon Corporation, Tokyo, Japan).
Transfection
NB4 cells were transfected using a BTX ECM 830
electroporator (BTX Harvard Apparatus, San Diego, CA, USA) with one
125 V pulse for 15 msec. The cells were then transferred to
RPMI-1640 medium supplemented with 10% FBS and cultured for 24-48
h. For overexpression, USP48 was cloned into a pFlag-CMV-2 plasmid.
Empty pFlag-CMV-2 plasmids were used as a negative control. A total
of 10 µg plasmid was used to transfect 5×106 NB4
cells/well for 48 h. Transient silencing was performed by
transfecting the following small interfering RNAs (siRNAs) into
2×106 NB4 cells/well: siUSP48-1,
5′-GCAGUUCUGUGGAGAAUAUTT AUAUUCUCCACAGAACUGCTT-3′; siUSP48-2,
5′-GCC CAACACUACUGUUCAATTUUGAACAGUAGUGUUGGG CTT-3′; siUSP48-3,
5′-GCUGGUAGAUCGGGAUAAUTTA UUAUCCCGAUCUACCAGCTT-3′, and negative
control, 5′-UUCUCCGAACGUGUCACGUTTACGUGACACGUUCG GAGAATT-3′. A total
of 20 µl siRNA was transfected per well, at a stock
concentration of was 1 OD/125 µl RNase-free H2O,
for 48 h. hsa-miR-301a-3p mimics and inhibitor (cat. nos.
miR10000688 and miR20000688, respectively) and negative control
miRNAs (cat. nos. miR01101 and miR02101) (both from Guangzhou
RiboBio Co., Ltd., Guangzhou, China) were transfected into
2×106 cells for 48 h, using 20 µl miRNA at a
stock concentration of 5 nmol/250 µl RNase-free
H2O. The transfection efficiency was confirmed by
detecting the mRNA or protein expression levels of USP48 using
RT-qPCR or western blotting, as aforementioned.
Luciferase assay
With wild-type 3′ untranslated region (UTR)
(USP48-WT) or mutant 3′UTR (USP48-Mut) sequence of USP48 was cloned
to a Dual-Luciferase Reporter vector (pmiR-RB-REPORT™; Guangzhou
RiboBio Co., Ltd.). 293T cells were co-transfected with negative
control or miR-301a-3p mimic and Dual-Luciferase Reporter plasmids
carrying USP48-WT or USP48-Mut of USP48 using Lipofectamine 2000
(Thermo Fisher Scientific, Inc.). After 48 h, luciferase activities
were measured with a Dual-Glo® Luciferase Assay system
(Promega, Madison, WI, USA), according to the manufacturer's
protocol. Renilla luciferase activity was used to normalize
the Firefly luciferase activity of the reporter construct.
miRNA prediction
To investigate the regulation of USP48 expression,
prediction of microRNAs targeting USP48 was performed using
TargetScanHuman 7.0 (http://www.targetscan.org/vert_70/).
Survival analysis
The USP48 gene was analyzed using the
cBioPortal database (http://www.cbioportal.org) (25,26).
All samples analyzed were derived from acute myeloid leukemia cases
(The Cancer Genome Atlas; dataset, NEJM2013) (27). A Z-score threshold of ±2.0 was used
for analysis of mRNA data (RNA Seq V2 RSEM; log; n=173).
Statistical analysis
Data are presented as the mean ± standard deviation
of ≥3 experiments. Statistical analysis was performed using SPSS
13.0 software (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 5.0
(GraphPad Software, Inc., La Jolla, CA, USA). Student's t-test was
used to analyze the difference between 2 groups, and one-way
analysis of variance was performed to analyze the difference
between ≥3 groups, followed by Dunnett's test. Survival was
analyzed using the Kaplan-Meier model. P<0.05 was considered to
indicate a statistically significant difference.
Results
USP48 expression decreases during
ATRA-induced granulocytic differentiation of APL cells
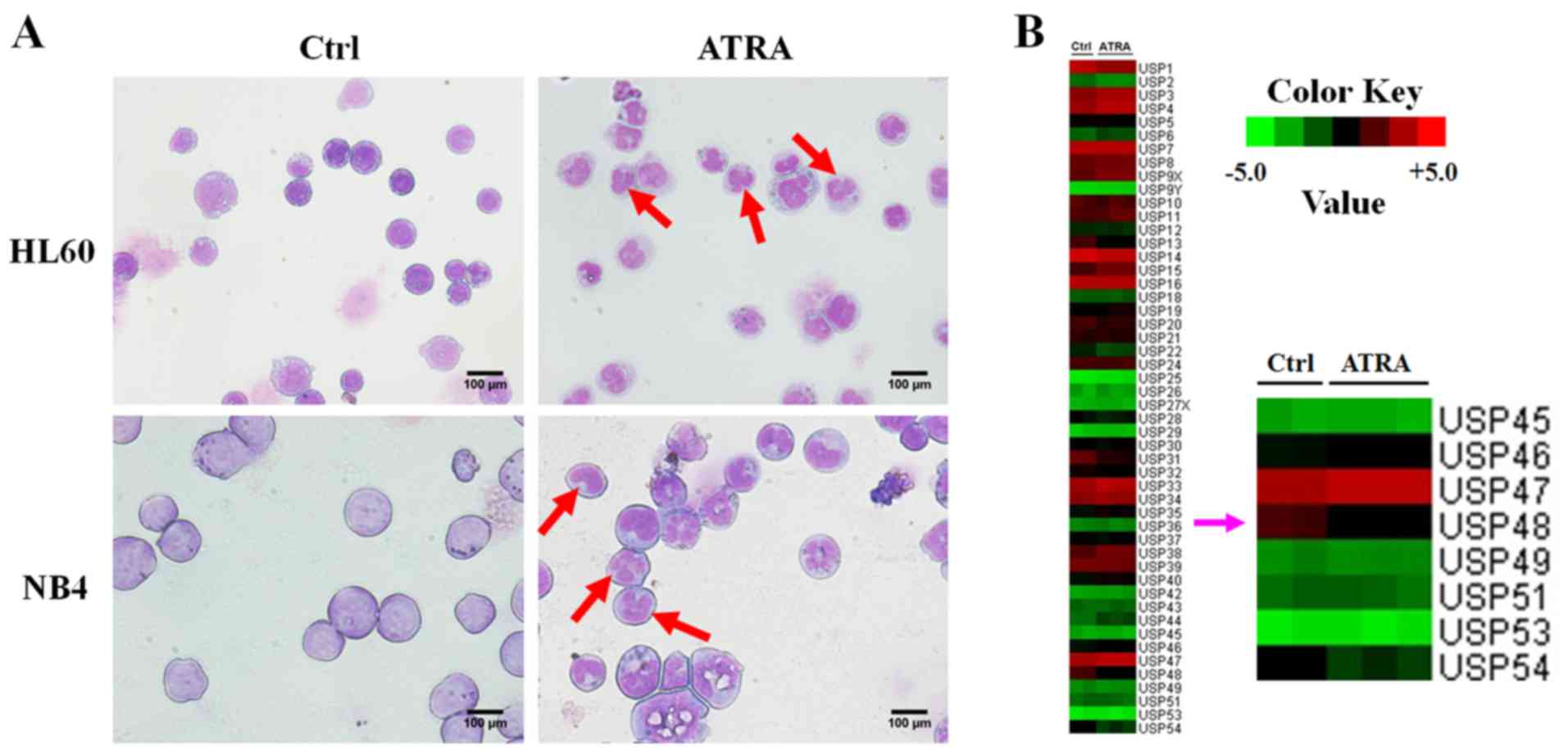
To systematically explore the underlying mechanism
of abnormal granulocytic differentiation in APL, leukemia cells
were treated with or without 1 µM ATRA for 72 h.
Morphological analysis, the emergence of nuclear lobulation and
pyknosis (Fig. 1A) confirmed the
differentiation of APL cells induced by ATRA. In addition, gene
microarray analysis was performed and the gene expression profiles
of HL-60 cells were changed following ATRA treatment. Comparison of
the expression variation of USP family members revealed that the
expression of USP48 was decreased in HL60 cells treated with ATRA
(Fig. 1B). RT-qPCR and western
blotting revealed that ATRA stimulation significantly downregulated
USP48 expression at the mRNA and protein levels in NB4 and HL-60
cells after 24 h treatment compared with untreated controls
(Fig. 2A and B). In contrast,
decreased expression of USP48 was not stimulated by PMA treatment
in THP1 cells (Fig. 2C). These
results indicate that USP48 expression was decreased during
ATRA-induced granulocytic differentiation of APL cells.
USP48 inhibits the proliferation and
promotes the ATRA-induced differentiation of NB4 cells
To investigate the role of USP48 in cell
proliferation, a CCK-8 assay was performed, which indicated that
NB4-cell proliferation was inhibited by overexpression of USP48
(Fig. 3A). The inhibition of cell
proliferation by USP48 was further confirmed by cell cycle analysis
(Fig. 3B). However, overexpression
of USP48 did not affect the rate of apoptosis (Fig. 3C). To examine whether USP48
contributed to the ATRA-induced differentiation of APL cells, USP48
was overexpressed or silenced in NB4 cells. siUSP48-3 was the most
effective oligonucleotide for silencing USP48 (Fig. 3D), and was selected for use in
subsequent experiments. FCM analysis demonstrated that the
expression of CD11b was increased following ATRA treatment of NB4
cells compared with control. Furthermore, CD11b expression was
promoted by electroporation-mediated overexpression of USP48 in NB4
cells in response to ATRA treatment compared with untreated cells
(Fig. 3E). In contrast, silencing
of USP48 inhibited the expression of CD11b induced by ATRA in NB4
cells compared with untreated cells (Fig. 3E). The results demonstrated that
USP48 inhibited the proliferation and promoted the ATRA-induced
granulocytic differentiation of NB4 cells.
The function of USP48 is not dependent on
the regulation of p65
In further study of the underlying mechanisms of the
role of USP48 in ATRA-induced granulocytic differentiation, western
blotting demonstrated that the expression of USP48 increased in NB4
cells following ATRA treatment up to 24 h, and decreased after 24 h
(Fig. 4A). Furthermore, USP48
expression in the nucleus was decreased in the cytoplasm of NB4
cells treated with ATRA overall; however, this was preceded by an
initial increase (Fig. 4B).
Immunofluorescence staining also indicated that the localization of
USP48 was predominantly in the nucleus of NB4 cells following ATRA
treatment (Fig. 4C). These results
implied that USP48 may function primarily in the nucleus during
differentiation. The primary target of USP48 during ATRA-mediated
differentiation was then investigated. The expression of p65, a
reported target of USP48 in the nucleus (28), was suppressed by siRNA-mediated
USP48 silencing in NB4 cells (Fig.
4D). This data indicated that the pathway mediated by nuclear
factor-κB (NF-κB) in ATRA-induced differentiation may contribute to
the function of USP48. However, immunofluorescence staining
revealed inconsistent findings, as p65 and USP48 were not
co-localized in NB4 cells following ATRA treatment (Fig. 4E). These results suggested that the
function of USP48 in ATRA-induced differentiation may not depend on
the regulation of p65.
USP48 is regulated by miR-301a-3p
A total of 3 candidate microRNAs, miR-148a-3p,
miR-301a-3p and miR-454a-3p, were selected based on chip results of
microRNA expression profiles (data not shown). The expression of
the candidate microRNAs in NB4 cells was confirmed by RT-qPCR: The
expression of miR-148a-3p was decreased and that of miR-301a-3p was
significantly increased by ATRA compared with untreated cells.
However, no significant change in the expression of miR-454a-3p was
observed (Fig. 5A). Therefore,
miR-301a-3p was selected for further evaluation of the regulation
of USP48 expression. Western blot analysis demonstrated that the
expression of USP48 was decreased by transfection with miR-301a-3p
mimics, and increased by transfection with miR-301a-3p inhibitor
(Fig. 5B). To determine whether
USP48 was an miR-301a-3p target gene, the 3′UTR of the cDNA
transcript was examined using TargetScanHuman 7.0. A 7mer-m8
binding site for miR-301a-3p was identified, located at position
174-180 of the 3′UTR (Fig. 5C). To
deter-mine whether USP48 was a direct target of miR-301a-3p,
luciferase reporter vectors harboring USP48-WT and USP48-Mut were
constructed. Co-transfection of miR-301a-3p mimics and the
luciferase-USP48-WT fusion construct resulted in decreased
luciferase activity compared with NC mimics-luciferase-USP48-WT
fusion. However, this effect was not evident with transfection of
the luciferase-USP48-Mut fusion construct (Fig. 5D). These observations suggested
that miR-301a-3p may regulate USP48 via the binding site in the
3′UTR.
Upregulation of USP48 expression is a
potential positive prognostic indicator in AML
cBioPortal results indicated that USP48 was
upregulated in the majority of the considered patients with AML
(Fig. 6A). Furthermore,
upregulation of USP48 in AML may be associated with increased
overall survival time (median, 25.8 months in patients exhibiting
overexpressed USP48 vs. 17.4 months in the remaining patients;
log-rank test, P=0.356; Fig. 6B).
Furthermore, upregulation of USP48 expression may be associated
with increased disease-free survival in AML (log-rank test,
P=0.153; Fig. 6C). However, these
associations were not statistically significant, which may be due
to the small samples sizes. These analyses should be repeated using
large cohorts, to confirm whether upregulation of USP48 expression
is predictive of good prognosis in AML.
Discussion
Although the biological functions and substrates of
the majority of DUBs remain unclear, DUBs have been suggested to be
important targets for the treatment of human diseases, including
cancer. For example, USP9X has been demonstrated regulate the
destruction of Ets-1, which serves important functions in the
tumorigenic program of metastatic melanoma (29). Additionally, USP15 has been
demonstrated to bind to and stabilize p53, which serves a critical
role in cancer progression through deubiquitination (30). A previous study reported that USP49
deubiquitinates and stabilizes FKBP51 to negatively regulate
tumorigenesis and chemoresistance through AKT signaling (31). In addition, USP4-mediated
deubiquitination and stabilization of PRL-3 is required for
potentiating colorectal oncogenesis (32). In the present study, deubiquitinase
USP48 was identified by microarray analysis, which was performed to
comparatively evaluate the expression profile of HL60 cells
following ATRA-induced differentiation. USP48 expression was
inhibited significantly following ATRA expo-sure. Thus, the present
study highlights USP48 as a potential therapeutic target for
APL.
Recently, it has been reported that USP48 can
regulate the NF-κB signaling pathway by regulating the stability of
the RelA (p65) in the nucleus (28). Furthermore, previous study
indicated that USP48 regulates the stability of Mdm2 protein and
thus the p53 signaling pathway, which is not dependent on its
activity as a ubiquitination enzyme (33). In addition, USP48 has been
demonstrated to reduce E-cadherin-mediated adhe-rens junctions
through increasing TNF receptor associated factor 2 stability
(34). Furthermore, high
expression of USP48 has been correlated with glioma malignancy, and
USP48 has been demonstrated to activate Gli-dependent transcription
and stabilize Gli1 protein through direct cleavage of its
ubiquitin, which is critical for glioma cell proliferation and
glioblastoma tumorigenesis (35).
A previous study predicted that an imbalance in the BRCA1-BRCA1
associated RING domain 1-USP48 circuit has deleterious consequences
for genome stability and that it may have significance in the
prevention and progression of cancer (36). Therefore, numerous studies indicate
that USP48 is involved in immunoregulation and cancer pathogenesis,
which further prompted the clarification of its function in myeloid
differentiation in the present study.
To investigate whether USP48 contributes to the
degradation of the is promyelocytic leukemia-retinoic acid receptor
α (PML-RARα) fusion protein in response to ATRA exposure, a
molecular mechanism that can induce myeloid differentiation, NB4
(PML-RARα positive) and HL60 (PML-RARα negative) cells were used
(37). The results revealed that
USP48 expression was inhibited in NB4 and HL60 cells treated with
ATRA compared with untreated cells. This data indicated that the
expression of USP48 may not be dependent on the degradation of
PML-RARα. To elucidate the mechanism of USP48 function in
ATRA-mediated differentiation, the co-localization of p65 and USP48
after ATRA exposure using immunofluorescence staining. p65 protein
expression was not co-localized with that of USP48, suggesting that
USP48 may regulate ATRA-induced differentiation of APL cells via
other signaling pathways. Furthermore, the possible regulation of
USP48 by miR-301a-3p was investigated. Reduced USP48-promoter
activity was observed upon miR-301a-3p treatment in the luciferase
assay; however, the effect was not significant. Thus, USP48 may be
partially regulated by miR-301a-3p. It is believed that USP48
expression is induced by the Sonic Hedgehog pathway through
Gli1-mediated transcriptional activation (35) and that its ubiquitin chain trimming
activity is regulated by casein-kinase-2-mediated phosphorylation
in response to cytokine-stimulation (28). Therefore, USP48 may serve an
important role in leukemia-cell differentiation, however, the
underlying mechanism requires further investigation. In addition to
USP48, other DUBs have been associated with leukemia. For example,
HAUSP (USP7) aberrantly regulates the nuclear exclusion of the
tumor suppressor phosphate and tensin homolog in APL via
deubiquitinase activity (38).
Other DUBs, including USP9X (39),
CYLD (40) and A20 (41), are associated with the occurrence
and ATRA-induced differentiation of leukemia cells. In consistence
with previous studies, the present study suggests that DUBs
function in ATRA-induced differentiation of leukemia cells.
In summary, it was demonstrated that USP48 inhibits
the proliferation of leukemia cells and promotes ATRA-induced
differentiation of leukemia cells. In addition, the expression of
USP48 was demonstrated to be partially regulated by miR-301a-3p.
Therefore, the present study eludes to a previously unknown
miR-301a-3p-USP48 molecular network, which regulates the
differentiation of leukemia cells (Fig. 7). This implies that dysregulation
of USP48 may underlie the abnormal differentiation in APL, and that
USP48 is a potential therapeutic target for APL.
Acknowledgments
Not applicable.
Funding
The present study was supported by the grants from
the Shandong Medical and Health Science and Technology Plan Project
(grant nos. 2015WS0193 and 2014WS0067), the Foundation for
Outstanding Young Scientists in Shandong Province (grant no.
2014BSC03013) and the Innovation Project of the Shandong Academy of
Medical Sciences.
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LLL wrote the manuscript. LLL, HYL and GSJ designed
the experiments, LLL, YW, XYZ, GHS, QG, ZYZ, YTD and HPY performed
the experiments and analyzed the data. The final version of the
manuscript has been read and approved by all authors.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fialkow PJ, Janssen JW and Bartram CR:
Clonal remissions in acute nonlymphocytic leukemia: Evidence for a
multistep pathogenesis of the malignancy. Blood. 77:1415–1417.
1991.PubMed/NCBI
|
|
2
|
Mistry AR, Pedersen EW, Solomon E and
Grimwade D: The molecular pathogenesis of acute promyelocytic
leukaemia: Implications for the clinical management of the disease.
Blood Rev. 17:71–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nasr R, Guillemin MC, Ferhi O, Soilihi H,
Peres L, Berthier C, Rousselot P, Robledo-Sarmiento M,
Lallemand-Breitenbach V, Gourmel B, et al: Eradication of acute
promyelocytic leukemia-initiating cells through PML-RARA
degradation. Nat Med. 14:1333–1342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dos Santos GA, Kats L and Pandolfi PP:
Synergy against PML-RARa: Targeting transcription, proteolysis,
differentiation, and self-renewal in acute promyelocytic leukemia.
J Exp Med. 210:2793–2802. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dombret H, Castaigne S, Fenaux P,
Chomienne C and Degos L: Induction treatment of acute promyelocytic
leukemia using all-trans retinoic acid. Controversies about dosage,
advantages and side-effect management. Leukemia. 8(Suppl 3):
S73–S75. 1994.PubMed/NCBI
|
|
6
|
Tomita A, Kiyoi H and Naoe T: Mechanisms
of action and resistance to all-trans retinoic acid (ATRA) and
arsenic trioxide (As2O3) in acute
promyelocytic leukemia. Int J Hematol. 97:717–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilkinson KD: Ubiquitination and
deubiquitination: Targeting of proteins for degradation by the
proteasome. Semin Cell Dev Biol. 11:141–148. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goldberg AL: Protein degradation and
protection against misfolded or damaged proteins. Nature.
426:895–899. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ciechanover A: Proteolysis: From the
lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol.
6:79–87. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ciechanover A: Intracellular protein
degradation: From a vague idea thru the lysosome and the
ubiquitin-proteasome system and onto human diseases and drug
targeting. Cell Death Differ. 12:1178–1190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ciechanover A and Schwartz AL:
Ubiquitin-mediated degradation of cellular proteins in health and
disease. Hepatology. 35:3–6. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pickart CM: Mechanisms underlying
ubiquitination. Annu Rev Biochem. 70:503–533. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schulman BA: Twists and turns in
ubiquitin-like protein conjugation cascades. Protein Sci.
20:1941–1954. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Komander D, Clague MJ and Urbé S: Breaking
the chains: Structure and function of the deubiquitinases. Nat Rev
Mol Cell Biol. 10:550–563. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Komander D: Mechanism, specificity and
structure of the deubiquitinases. Subcell Biochem. 54:69–87. 2010.
View Article : Google Scholar
|
|
16
|
Amerik AY and Hochstrasser M: Mechanism
and function of deubiquitinating enzymes. Biochim Biophys Acta.
1695:189–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eletr ZM and Wilkinson KD: Regulation of
proteolysis by human deubiquitinating enzymes. Biochim Biophys
Acta. 1843:114–128. 2014. View Article : Google Scholar
|
|
18
|
Wilkinson KD: DUBs at a glance. J Cell
Sci. 122:2325–2329. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nijman SM, Luna-Vargas MP, Velds A,
Brummelkamp TR, Dirac AM, Sixma TK and Bernards R: A genomic and
functional inventory of deubiquitinating enzymes. Cell.
123:773–786. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Love KR, Catic A, Schlieker C and Ploegh
HL: Mechanisms, biology and inhibitors of deubiquitinating enzymes.
Nat Chem Biol. 3:697–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quesada V, Díaz-Perales A,
Gutiérrez-Fernández A, Garabaya C, Cal S and López-Otín C: Cloning
and enzymatic analysis of 22 novel human ubiquitin-specific
proteases. Biochem Biophys Res Commun. 314:54–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sowa ME, Bennett EJ, Gygi SP and Harper
JW: Defining the human deubiquitinating enzyme interaction
landscape. Cell. 138:389–403. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Shi M, Ren X, Wang X, Wang H, Liu G, Yuan
X, Zheng S, Yu L, Pan S, Song G, et al: A novel combination of
oridonin and valproic acid in enhancement of apoptosis induction of
HL-60 leukemia cells. Int J Oncol. 48:734–746. 2016. View Article : Google Scholar
|
|
25
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multi-dimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ley TJ, Miller C, Ding L, Raphael BJ,
Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty
JD, et al Cancer Genome Atlas Research Network: Genomic and
epigenomic landscapes of adult de novo acute myeloid leukemia. N
Engl J Med. 368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schweitzer K and Naumann M: CSN-associated
USP48 confers stability to nuclear NF-κB/RelA by trimming
K48-linked Ub-chains. Biochim Biophys Acta. 1853:453–469. 2015.
View Article : Google Scholar
|
|
29
|
Potu H, Peterson LF, Kandarpa M, Pal A,
Sun H, Durham A, Harms PW, Hollenhorst PC, Eskiocak U, Talpaz M, et
al: Usp9x regulates Ets-1 ubiquitination and stability to control
NRAS expression and tumorigenicity in melanoma. Nat Commun.
8:144492017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu WT, Huang KY, Lu MC, Huang HL, Chen
CY, Cheng YL, Yu HC, Liu SQ, Lai NS and Huang HB: TGF-β upregulates
the translation of USP15 via the PI3K/AKT pathway to promote p53
stability. Oncogene. 36:2715–2723. 2017. View Article : Google Scholar
|
|
31
|
Luo K, Li Y, Yin Y, Li L, Wu C, Chen Y,
Nowsheen S, Hu Q, Zhang L, Lou Z, et al: USP49 negatively regulates
tumori-genesis and chemoresistance through FKBP51-AKT signaling.
EMBO J. 36:1434–1446. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xing C, Lu XX, Guo PD, Shen T, Zhang S, He
XS, Gan WJ, Li XM, Wang JR, Zhao YY, et al: Ubiquitin-specific
protease 4-mediated deubiquitination and stabilization of PRL-3 is
required for potentiating colorectal oncogenesis. Cancer Res.
76:83–95. 2016. View Article : Google Scholar
|
|
33
|
Cetkovská K, Šustová H and Uldrijan S:
Ubiquitin-specific peptidase 48 regulates Mdm2 protein levels
independent of its deubiquitinase activity. Sci Rep. 7:431802017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li S, Wang D, Zhao J, Weathington NM,
Shang D and Zhao Y: The deubiquitinating enzyme USP48 stabilizes
TRAF2 and reduces E-cadherin-mediated adherens junctions. FASEB J.
32:230–242. 2018. View Article : Google Scholar
|
|
35
|
Zhou A, Lin K, Zhang S, Ma L, Xue J,
Morris SA, Aldape KD and Huang S: Gli1-induced deubiquitinase USP48
aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep.
18:1318–1330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Uckelmann M, Densham RM, Baas R,
Winterwerp HH, Fish A, Sixma TK and Morris JR: USP48 restrains
resection by site-specific cleavage of the BRCA1 ubiquitin mark
from H2A. Nat Commun. 9:2292018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song G, Shi L, Guo Y, Yu L, Wang L, Zhang
X, Li L, Han Y, Ren X, Guo Q, et al: A novel PAD4/SOX4/PU.1
signaling pathway is involved in the committed differentiation of
acute promyelocytic leukemia cells into granulocytic cells.
Oncotarget. 7:3144–3157. 2016.
|
|
38
|
Song MS, Salmena L, Carracedo A, Egia A,
Lo-Coco F, Teruya-Feldstein J and Pandolfi PP: The
deubiquitinylation and localization of PTEN are regulated by a
HAUSP-PML network. Nature. 455:813–817. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schwickart M, Huang X, Lill JR, Liu J,
Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F,
Eastham-Anderson J, et al: Deubiquitinase USP9X stabilizes MCL1 and
promotes tumour cell survival. Nature. 463:103–107. 2010.
View Article : Google Scholar
|
|
40
|
Espinosa L, Cathelin S, D'Altri T,
Trimarchi T, Statnikov A, Guiu J, Rodilla V, Inglés-Esteve J,
Nomdedeu J, Bellosillo B, et al: The Notch/Hes1 pathway sustains
NF-κB activation through CYLD repression in T cell leukemia. Cancer
Cell. 18:268–281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Novak U, Rinaldi A, Kwee I, Nandula SV,
Rancoita PM, Compagno M, Cerri M, Rossi D, Murty VV, Zucca E, et
al: The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated
by somatic mutations and genomic deletions in marginal zone
lymphomas. Blood. 113:4918–4921. 2009. View Article : Google Scholar : PubMed/NCBI
|