Introduction
Cisplatin (cis-diamminedichloroplatinum, CDDP) is a
front-line drug used in the clinical treatment of cancers of
various tissue types, including colorectal, lung, ovarian,
testicular, penile, cervical, head and neck, and bladder
carcinomas. CDDP was the first Food and Drug Administration
(FDA)-approved platinum (Pt)-based drug for cancer treatment in the
1970s, and continues to be one of the most widely used
chemotherapeutic drugs (1-4). CDDP is considered a cytotoxic agent,
as it binds and damages DNA, thus triggering various cytotoxic
events, such as the inhibition of DNA replication, transcription
arrest, DNA damage responses, cell cycle arrest, and apoptosis
(5). Such toxic effects limit the
clinical application of CDDP-based chemotherapy, which is
associated with nephrotoxicity, neurotoxicity and other significant
side-effects (6–8). CDDP-induced side-effects are
dose-dependent, limiting the administration of the effective dose,
thus compromising the therapeutic efficacy (9). The development of drug resistance is
another major challenge. Several factors may be responsible for
resistance to CDDP, such as reduced cellular uptake, increased
efflux and increased DNA repair (10,11).
Combining drugs is a potentially effective method for overcoming
resistance and minimizing toxic side-effects, as it allows for a
lower drug dose. Therefore, effective regimens that can potentiate
the therapeutic efficacy of CDDP are urgently required.
Pt-based drugs, including CDDP, carboplatin and
oxaliplatin, have been clinically evaluated in combination with
other chemotherapeutic agents, including etoposide, mitomycin C,
vinblastine, paclitaxel, docetaxel, vinorelbine, gemcitabine,
cyclophosphamide, doxorubicin, epirubicin, methotrexate, lonidamine
and 5-fluorouracil (12). Several
Pt-based drug combination regimens have achieved an improvement in
clinical response and have positively affected the overall survival
of patients (13,14). These drug combinations are largely
the results of empirical clinical trials rather than
mechanism-based design, as mechanistic studies and clinical
evaluation require vast resources (15). In recent years, the evaluation of
FDA-approved drugs for new disease indications (drug repurposing)
has emerged as an effective strategy for discovering novel
therapeutic measures that can be rapidly translated into clinical
applications due to the availability of prior knowledge regarding
the drug mechanisms of action, pharmacokinetics, formulation, and
toxicity/safety information (16–18).
In the present study, we used a cell-based
high-throughput screening (HTS) approach to screen the FDA-approved
drugs (1,280 compounds in total) in search of drugs that
potentially act synergistically with CDDP. We identified two
compounds, namely potassium antimony tartrate (PAT), and topotecan,
that were capable of significantly enhancing the anticancer
activity of CDDP. Topotecan was further investigated to gain
mechanistic insight into the synergic activity.
Materials and methods
Chemicals and reagents
The FDA-approved drug library of 1,280 compounds (10
mM concentration in DMSO, 80 compounds/96-well plate, 16 plates)
was purchased from MicroSource Discovery Systems (Gaylordsville,
CT, USA). CDDP and topotecan were purchased from Hospira (Lake
Forest, IL, USA) and GlaxoSmithKline (London, UK), respectively.
MTS, PAT and ethanol were purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany).
Cells and cell culture
All the cell lines used in this study were purchased
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). The human colorectal cancer cell line, DLD-1, and the
non-small-cell lung cancer (NSCLC) cell line, NCI-H460, were
cultured in RPMI-1640 (Corning Cellgro, Shanghai, China)
supplemented with 10% fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The colon cancer cell lines,
HT-29 and HCT-116, were maintained in McCoy's 5A (Sigma-Aldrich)
with 10% fetal bovine serum (FBS). The LS-174T and RKO human
colorectal cancer cell lines were cultured in DMEM and MEM (Corning
Cellgro) supplemented with 10% FBS, respectively. The cells were
cultured at 37°C in a humidified incubator (Thermo Fisher
Scientific, Inc.) containing 5% CO2, and were seeded and
incubated in culture flasks or plates overnight prior to each
treatment.
Cell viability assay and high-throughput
drug screening
Cell viability was measured using the MTS
colorimetric assay. Briefly, the cells were seeded in a 96-well
plate at a density of 2,000 cells per well, and treated with the
indicated drugs at the specified concentrations. Following a 72-h
incubation, 20 µl of MTS were added to each well and
incubated for a further 3 h. The absorbance of each well at 490 nm
was measured using a microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA) and each experiment was performed in triplicate
wells. The 1,280 FDA-approved drugs were stored as 10-mM solutions
in 100% DMSO and diluted to 2 mM with phosphate-buffered saline
(PBS) using a single-arm, multichannel workstation (Aurora Biomed,
Inc., Vancouver, BC, Canada). Using this workstation, the
high-throughput MTS viability assays were performed as described in
Table I.
 | Table IDrug combination screening protocol
used in this study. |
Table I
Drug combination screening protocol
used in this study.
| Step | Parameter | Description | Instrument |
|---|
| 1 Drug
dilution | 14 µl Drug
X | Dilute drug X | ABV1000 |
| 266 µl
DMSO | from 10 mM to 2
mM | ABV1000 |
| 2 Cell seeding | 100 µl | 2000
cells/well | |
| 3 Add reagent | 20 µl Drug
X | Add two
drugs/well | ABV1000 |
| 80 µl
CDDP | (final
concentration: 10 µM) | |
| 4 Incubate | 72 h | 5%
CO2/37°C incubation | Incubator |
| 5 Add reagents | 20 µl | MTS solution (5
mg/ml) | ABV1000 |
| 6 Incubate | 3 h | 5%
CO2/37°C incubation | Incubator |
| 7 Read | Fluorescence | 490 nm | Synergy MR |
Numerical characterization method of
synergy, additivity and antagonism
The drug combination index (CI) was calculated using
the CalcuSyn software developed by Chou (19) (CI <1, synergism; CI =1, additive
effect; CI >1, antagonism). The methods by which the normalized
cell survival rate was characterized were based on a previous
publication (20). Given the
viability of drugs A and B at the respective concentrations x and y
as VA and VB, the additive response viability
is predicted as VAV±B.
Colony formation assay
A total of 500 cells/well were plated in 6-well
plates and cultured with each drug for 10–14 days. Each of the
colonies was washed twice with PBS, fixed with methanol and stained
with crystal violet for 15 min at room temperature. After washing
with water, the colonies were photographed and counted using the
AlphaImager HP system (ProteinSimple, San Jose, CA, USA).
Cellular apoptosis assay
The cells were seeded in a 6-well plate as
2×105 cells/well, allowed to adhere overnight, and were
then treated with the indicated drugs as described in the figure
legends. The cells were harvested by trypsinization, washed with
PBS (4°C), and suspended in 500 µl buffer. The cells were
stained with Annexin V-FITC for 15 min and then stained with PI
(Apoptosis kit from KeyGen Biotech, Nanjing, China) for 5 min at
room temperature. The samples were analyzed by a FACSCalibur flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Comet assay (single-cell gel
electrophoresis assay)
A Comet assay was performed according to the method
previously described (21).
Briefly, following treatment with the indicated drugs for 6 h, the
cells were collected, washed and re-suspended in cold PBS. The cell
suspensions (2×106 cells/ml, 20 µl) were mixed
with 100 µl 0.5% low-melting-point agar (Sigma-Aldrich), and
were then placed onto a slide pre-coated with 1%
normal-melting-point agar (Sigma-Aldrich). When the agar was
solidified, the slides were submerged in fresh pre- chilled lysis
buffer (10 mM Tris-HCl, pH 10.0, 2.5 M NaCl, 100 mM
ethylenediaminetetraacetic acid and 1% Triton X-100) for at least 1
h at 4°C. After rinsing with a neutralization buffer (0.4 M
Tris-HCl, pH 7.5) for 5 min, the slides were soaked in the alkaline
electrophoresis buffer (300 mM NaOH, 1 mM
ethylenediaminetetraacetic acid, pH >13, 4°C) for 15 min and
then subjected to electrophoresis for a further 15–20 min (25 V,
300 mA). The slides were then stained with SYBR-Green I (Biotek,
Beijing, China) and photographed under a fluorescence microscope
(Nikon, Tokyo, Japan). The CASP software program, version 1.2.2,
provided by the CASPLab Comet Assay Project, was used to analyze
the percentage of tail DNA, which indicates damaged DNA.
Cellular Pt content analysis
The measurement of the total intracellular Pt
content was performed as previously described (22). In brief, following CDDP treatment,
the cells were collected, washed twice with cold PBS and counted.
Subsequently, 1 ml nitric acid was added to the cell pellets, mixed
and kept at 68°C for 1 h. The solution was diluted to 1:10 with
ddH2O and sent for a Pt content analysis using an
Agilent 7500ce inductively coupled plasma-mass spectrometer
(ICP-MS; Agilent Technologies, Inc., Santa Clara, CA, USA).
Western blot analysis and antibodies
The cells were washed twice with cold PBS and lysed
in radio immunoprecipitation assay (RIPA) lysis buffer containing
protease inhibitors and phosphatase inhibitors (Cell Signaling
Technology, Inc., Danvers, MA, USA) on ice for 20 min. Protein (30
µg) was separated by electrophoresis using 10% SDS-PAGE and
transferred onto polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). The membranes were incubated with mouse
anti-γH2AX (ab22551, dilution, 1:2,000) at 4°C overnight on a
shaking platform, washed, and then incubated with horseradish
peroxide-conjugated goat anti-mouse secondary antibody (ab6789;
dilution, 1:10,000) for 1 h at room temperature. The blots were
visualized using chemiluminescent reagents (KeyGen Biotech) and
revealed with X-ray film. α-tubulin was used as a loading control.
The α-tubulin antibody (GTX628802; dilution, 1:5,000) was purchased
from GeneTex (Irvine, CA, USA), and all other antibodies were
purchased from Abcam (Cambridge, MA, USA).
Statistical analysis
The cytotoxic combined effect of PAT/topotecan and
CDDP was calculated using CalcuSyn software (Biosoft, Cambridge,
UK). The data were analyzed with GraphPad Prism 5 (GraphPad
Software, Inc., La Jolla, CA, USA). A Student's t-test was used to
analyze the statistical difference between two groups, while a
two-way ANOVA with Bonferroni's correction were used to determine
the statistical difference among multiple groups, as indicated in
the figure legends. The data are reported as the means ± SD of 3
independent experiments. The results were deemed statistically
significant at a value of P<0.05.
Results
Screening of drugs that potentiate the
anticancer activity of CDDP
To identify drugs that may enhance the effects of
CDDP, a quantitative cell-based drug screening approach was used to
assess the combinational activity of CDDP and one of the 1,280
FDA-approved drugs. The sensitivity to CDDP was first evaluated in
several colorectal cancer cell lines to select the optimal drug
concentrations and cell lines for the screening. Two cell lines
with relatively low sensitivities to CDDP, HT-29 and DLD-1, were
selected. As shown in Fig. 1A, the
IC50 value for CDDP was ~10 µM for both cell
lines, which was selected for further combination analyses. Various
experimental conditions, including the number of cells per well,
the length of the drug incubation period, and the volume of the
reagents, were tested for optimization of the assay. The protocol
for the screening study is summarized in Table I. The screening procedures included
three main steps, as shown in Fig.
1B. During the primary screening, we compared the viability of
the two cell lines exposed to CDDP (10 µM) alone or to CDDP
in combination with 10 µM drug X (one of the 1,280 drugs in
the FDA-approved drug library). A drug that enhanced the CDDP
inhibition of cell growth to <20% viable cells was considered as
a potential hit in the primary screening (Fig. 1C). Using this criterion, 40 drugs
capable of enhancing the cytotoxicity of CDDP were identified.
Subsequently, the 40 compounds identified from the primary
screening were further evaluated at a dose-range of 0.1–30
µM to further evaluate the potential synergistic effects in
combination with CDDP. The top 10 compounds that were validated in
the second test step are listed in Table II. In the third step, the
dose-effect analysis described by Chou and Talalay (19) was used to determine the drug CI
values of the top hits for the two colon cancer cell lines. Through
these 3 steps, we identified two drugs, namely PAT and topotecan,
that consistently showed synergy with CDDP.
| Table IITop 10 drugs that potentiate the
anticancer activity of cisplatin. |
Table II
Top 10 drugs that potentiate the
anticancer activity of cisplatin.
| Rank | Drug name | Classification |
|---|
| 1 | Antimony potassium
tartrate trihydrate |
Antischistosomal |
| 2 | Topotecan
hydrochloride | Antineoplastic;
topoisomerase I inhibitor |
| 3 | Thioridazine
hydrochloride | Antipsychotic |
| 4 |
Oxyphenbutazone |
Anti-inflammatory |
| 5 | Emetine
dihydrochloride | Inhibits RNA, DNA
and protein synthesis |
| 6 | Oxyquinoline
sulfate | Anti-infective,
complexing agent |
| 7 | Monensin sodium
(monensin A) | Antibacterial |
| 8 | Piroctone
olamine | Antiseborrheic |
| 9 | Amsacrinea | Antineoplastic,
immune suppressive |
| 10 | Phenylmercuric
acetate | Antifungal,
antimicrobial |
Synergistic effects of CDDP and
topotecan
Since CDDP is often used in the treatment of colon
cancer and NSCLC (23,24), we selected NSCLC (NCI-H460) and
colon cancer (DLD-1) cell lines to further investigate the
chemosensitization effect of topotecan on CDDP. MTS cell growth
inhibition, colony formation and apoptosis assays were used to
evaluate the drug combination effects. The results of MTS assay
demonstrated that the combination of CDDP and topotecan resulted in
a concentration-dependent increase in cell growth inhibition. To
examine whether the drug combination was more than additive, cell
growth inhibition curves were normalized to the growth inhibition
induced by the corresponding concentration (5–10 µM) of CDDP
alone, according to a previously described method (20). The normalized curves exhibited a
significant low-left shift with the drug combination in DLD-1
(Fig. 2A) and NCI-H460 cells
(Fig. 2B) respectively, indicating
more than an additive effect. The colony formation assay also
confirmed a significant loss of more cell colonies when CDDP and
topotecan were used in combination in DLD-1 cells (Fig. 2C) and NCI-H460 cells (Fig. 2D). In addition, an apoptosis assay
was performed to evaluate the synergistic killing effect of CDDP
and topotecan after the DLD-1 cells or NCI-H460 cells were
double-stained with Annexin V/PI (Fig.
2E and F). Quantitative analysis of triplicate experiments
demonstrated that the observed cell survival rate of 54.5% was
significantly lower than the calculated additive effect (73.6%;
P<0.05, Fig. 2G) in the DLD1
cells. A similar synergistic drug combination effect was also
observed in the NCI-H460 cells (Fig.
2F and G). Quantitative analysis of the drug combination index
revealed that the CI values were predominately <1.0 (Fig. 2H), confirming the synergy between
CDDP and topotecan.
 | Figure 2Synergistic effect of cisplatin
(CDDP) in combination with topotecan in colon and lung cancer
cells. (A) Normalized cell survival curves in DLD-1 cells treated
with the the indicated concentrations of topotecan and CDDP
(square, 5 µM; triangle, 10 µM). Graph shows means ±
SD, n=3 experiments; the P-values for 5 µM CDDP + TPT and 10
µM CDDP + TPT compared to TPT alone are 2.45×10−4
and 1.06×10−11, respectively (two-way ANOVA). (B)
Normalized viability of NCI-H460 cells treated with the respective
concentration of CDDP alone or combined with topotecan. Graph shows
the means ± SD, n=3 experiments; the P-values for 1 µM CDDP
+ TPT and 5 µM CDDP + TPT compared to TPT alone are
1.33×10−10 and 6.77×10−14, respectively
(two-way ANOVA). (C and D) The colony formation assay of (C) DLD-1
and (D) NCI-H460 cells treated with the indicated concentrations of
CDDP and topotecan for 2 weeks. (E and F) Apoptosis induced by CDDP
(10 µM) and combination treatment with topotecan (2.5
µM) for 48 h was detected by Annexin V/PI double staining
followed by flow cytometric analysis in (E) DLD-1 and (F) NCI-H460
cells. (G) Quantitative data of the apoptosis assay in DLD-1 cells
and NCI-H460 cells. Additive, indicates the estimated additive
effect based on the effect of each drug alone. Data are shown as
the means ± SD. n=3 experiments, *P<0.05. (H) Drug
combination index (CI) between CDDP and topotecan in DLD-1 and
NCI-H460 cells based on colony formation assay. (CI <1,
synergism; CI =1, additive; CI >1, antagonism). |
Enhancement of the CDDP anticancer
activity by PAT
Similar to the synergistic effects between CDDP and
topotecan described above, the ability of PAT to enhance the
anticancer activity of CDDP was validated using multiple assays. As
is shown in Fig. 3A and B, PAT
alone did not exert a significant effect on cell survival within
the range of concentrations tested; however, the combination of PAT
and CDDP led to a significant decrease in the survival of both the
DLD-1 and NCI-H460 cells. The evident low-left shift of the
normalized cell survival curves with the drug combination indicated
more than an additive effect. Colony formation assay in the DLD-1
(Fig. 3C) and NCI-H460 cells
(Fig. 3D) and flow cytometric
apoptosis assay (Fig. 3E and F)
also confirmed the synergy between CDDP and PAT in both cell lines.
All CI values calculated from the colony formation assay with
different concentrations of the drug combinations were <1.0
(Fig. 3H), indicating a
synergistic effect of the two drugs in both cell lines.
 | Figure 3Effect of cisplatin (CDDP) and
potassium antimony tartrate (PAT) alone or in combination on the
viability of colon and lung cancer cell lines. (A) Normalized cell
survival curves of DLD-1 colon cancer cells treated with the
indicated concentrations of CDDP and PAT. Data are shown as the
means ± SD, n=3 experiments; The P-values for 5 µM CDDP +
PAT and 10 µM CDDP + PAT compared to PAT alone group are
3.21×10−9 and 1.67×10−11, respectively
(two-way ANOVA). (B) Normalized cell survival curves of NCI-H460
lung cancer cells treated with the indicated concentrations of CDDP
and PAT. Data are shown as the means ± SD, n=3 experiments; The
P-values for 5 µM CDDP + PAT and 10 µM CDDP + PAT
compared to PAT alone group are 3.82×10−4 and
3.68×10−6, respectively (two-way ANOVA). (C and D) The
colony formation assay of (C) DLD-1 and (D) NCI-H460 cells treated
with the indicated concentration of CDDP, PAT or their combination.
(E and F) Induction of apoptosis of (E) DLD-1 and (F) NCI-H460
cells by CDDP, PAT, or their combination for 48 h. (G) Quantitative
data of the apoptosis assay in DLD-1 cells. Additive, indicates the
estimated additive effect based on the effect of each drug alone.
Columns, mean (n=3 experiments); bars, SD; *P<0.05.
(H) Drug combination index (CI) of CDDP and PAT in DLD-1 and
NCI-H460 cells (CI <1, synergism; CI =1, additive; CI >1,
antagonism). |
Topotecan impairs the ability of cells to
repair CDDP-induced DNA damage
The cytotoxicity of CDDP is mainly induced by DNA
damage from the formation of drug-DNA adducts, leading to
interstrand and intrastrand crosslinks, and double-strand breaks
(DSBs) (5). Considering the
important role of topoisomerase I, a therapeutic target of
topotecan, in chromatin remodeling and DNA repair (25–27),
we hypothesized that the CDDP-induced DNA damage may be enhanced by
topotecan due to its inhibition of topoisomerase I. Using the
alkaline comet assay, we observed that DNA strand breaks induced by
CDDP were significantly increased by topotecan, as evidenced by the
appearance of increased comet tails after single-cell gel
electrophoresis (Fig. 4A and B).
In the cells treated with CDDP alone, the drug-induced DNA damage
was largely repaired at 4 h after the removal of CDDP, as evidenced
by the disappearance or reduction in the number of DNA tails. The
addition of topotecan rendered the cells unable to repair the DNA
damage, resulting in the persistence of strand breaks (comet tails)
at 4 h after drug removal. The quantitative data for the DNA strand
breaks are presented in Fig. 4C and
D.
 | Figure 4Effect of topotecan on DNA damage
induced by cisplatin (CDDP). (A) Comet assay of DNA damage in DLD-1
cells treated with CDDP (10 µM), topotecan (1 µM), or
their combination. Cells were treated with the indicated
concentrations of the drugs for 6 h, and the samples were then
either subjected to comet assay or incubated in drug-free medium
for another 4 h to allow potential DNA repair. The bright green
dots represent the cellular nuclei area; the 'tail' length and
intensity on the left side of each nucleus represent the degree of
DNA fragmentations eluted out from the cell during electrophoresis.
(B) Comet assay of DNA damage in NCI-H460 cells treated with the
indicated concentrations of CDDP, topotecan (0.5 µM), or
their combination. (C) Quantitative data of DNA damage in DLD-1
cells treated with CDDP (10 µM), topotecan (1 µM), or
their combination. The comparison between the 6-h drug exposure
group and the 4-h drug removal group within each independent
treatment (CDDP, TPT or C + T) was analyzed by a Student's t-test.
The comparison of (C + T) vs. the CDDP group or (C + T) vs. the TPT
group was analyzed by the Student's t-test with Bonferroni
correction. (D) Quantitative data of DNA damage in NCI-H460 cells
treated with CDDP (10 µM), topotecan (0.5 µM), or
their combination. The comparison between the 6-h drug exposure
group and the 4-h drug removal group within each independent
treatment (CDDP, TPT, or C + T) was analyzed by the Student's
t-test. The comparison of (C + T) vs. the CDDP group or (C + T) vs.
the TPT group was analyzed by the Student's t-test with Bonferroni
correction. Data are shown as the means ± SD, n=4 experiments.
*P<0.05; **P< 0.01;
***P<0.001 following Bonferroni correction. |
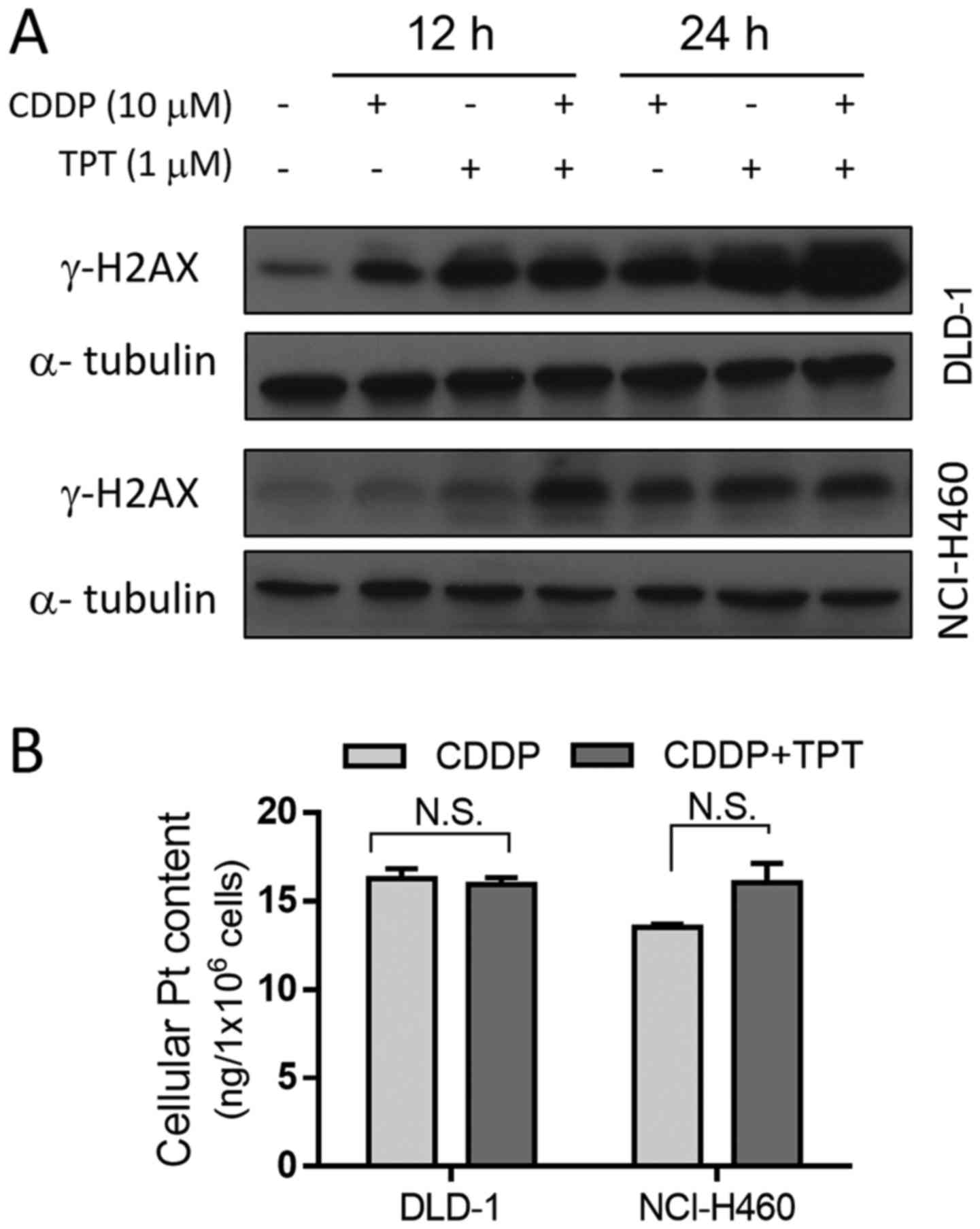
Topotecan enhances the formation of
CDDP-induced DNA DSBs without affecting cellular Pt content
The replication DSBs produce several
well-characterized molecular responses, including the
phosphorylation of the H2AX histone variant, which occurs within
minutes after the formation of DSBs (28). The phosphorylated form of H2AX,
termed γ-H2AX, can be detected by immunofluorescence or
immunostaining as it accumulates and forms nuclear foci at DSBs
(29). Thus, we used western blot
analysis of γ-H2AX to detect the DSBs induced by CDDP in the
presence and absence of topotecan. The γ-H2AX level increased after
each of the drug treatments alone, and was further enhanced when
the two drugs were combined (Fig.
5A). The results revealed that DNA damage at 24 h was
significantly more severe when the DLD-1 cells were treated with
both CDDP and topotecan compared with when they were treated with
either drug alone. A similar combination result was observed in the
NCI-H460 cells following treatment with the drugs for 12 h.
As nucleotide excision repair (NER) is the primary
mechanism for the removal of CDDP from the cell nucleus (30), and Pt efflux is a critical
mechanism of resistance to CDDP (31), the impact of topotecan on cellular
Pt content was measured to examine the possibility that topotecan
may enhance the anticancer activity of CDDP by increasing cellular
Pt content. Quantitative analysis of Pt revealed that, following a
24-h incubation of the DLD-1 cells with 10 µM CDDP in the
presence or absence of topotecan, there was no difference in
intracellular Pt content between the two groups (Fig. 5B). Similar results were also
observed in the NCI-H460 cells (Fig.
5B). These results indicate that the increase in DNA DSBs in
cells treated with both CDDP and topotecan was not due to an
increase in the cellular accumulation of Pt, but was likely due to
the inhibition of DNA repair.
Discussion
CDDP-based chemotherapy is one of the conventional
regimens used in the clinical treatment of various cancers of
different tissue types. However, the development of drug resistance
and toxic side-effects pose major challenges to the clinical use of
CDDP (1–3,6,7).
Even with newer-generation Pt drugs, such as oxaliplatin and
carboplatin, cross-resistance and toxic effects still develop
(32,33). Thus, an effective regimen combining
CDDP with another clinical drug to overcome resistance to treatment
and reduce the incidence and severity of toxic side-effects is
urgently required. Various high-throughput screening technologies
for the discovery of potential drug combination regimens have been
reported (34), although there
remain challenges in establishing effective combination
screening.
In the present study, we were able to identify
clinical drugs that enhance the anticancer activity of CDDP by
using a cell-based assay to screen a collection of FDA-approved
drugs. Among the library of 1,280 FDA-approved compounds screened,
10 drugs exhibited potential ability to enhance the cytotoxicity of
CDDP. One of the compounds identified, topotecan, has previously
been demonstrated to improve the therapeutic efficacy when combined
with CDDP in the clinical treatment of cancers (35). Thus, it appears that this
cell-based screening strategy is robust and may yield clinically
relevant hits. Therefore, it is possible to use this method to
identify FDA-approved drugs that may improve the therapeutic
efficacy of other chemotherapeutic agents.
PAT was the top hit from our screening assay. This
compound, also known as tartar emetic, was previously used as an
anti-parasitic agent for the treatment of leishmaniosis and
schistosomiasis (36,37). PAT, similar to other
metal-containing compounds, such as arsenic trioxide and CDDP,
possesses anticancer properties and has been proposed as a
potential novel therapy for acute promyelocytic leukemia and other
malignancies (38). It has been
reported that PAT may induce the caspase- and reactive oxygen
species (ROS)-dependent apoptosis of human myeloid leukemia HL-60
cells and lymphoid tumor cells (39,40).
Of note, a recent study reported that PAT exerted anti-angiogenic
effects on lung cancer (41). The
present study revealed the significant ability of PAT to enhance
the anticancer activity of CDDP in a synergistic manner, suggesting
that this drug may potentially be used in combination with CDDP to
improve the therapeutic efficacy. It should be noted, however, that
PAT is a strong emetic agent that may induce vomiting (42,43),
and such an effect may limit its potential use as an anticancer
agent in combination with CDDP, which is also known to cause nausea
and vomiting. The mechanisms through which PAT enhanced the
anticancer activity of CDDP are currently unclear. One possibility
is that PAT-induced ROS generation (39,40)
may increase the DNA damage induced by CDDP. Evidently, further
investigation is required to gain further insight into the
underlying mechanisms, which may serve as a basis for developing
more effective CDDP sensitizers.
Topotecan
[10-hydroxy-9-dimethylaminomethyl-(S)-camptothecin] is an inhibitor
of topoisomerase I, and has been used in the clinical treatment of
ovarian, cervical and lung cancer. This compound exerts its
cytotoxic effects by inhibiting the enzymatic complex
DNA-topoisomerase I in the nucleus, thus blocking the normal DNA
replication process (44,45). Previous studies have revealed that
topotecan may potentiate the cytotoxic activity of CDDP, etoposide
and paclitaxel in cervical cancer cell lines (46,47).
The combination of CDDP and topotecan as a clinical regimen is
based on the hypothesis that the concomitant administration of
these two agents is advantageous compared with either agent alone
(48). This drug combination was
approved by the FDA in 2006 for the treatment of patients with
advanced-stage cervical carcinoma who were unsuitable for surgery
or radiation therapy (49). Our
unbiased cell-based screening identified topotecan as one of the
top hits that synergistically enhanced the anticancer activity of
CDDP in both lung and colon cancer cells. Further mechanistic
analyses demonstrated that topotecan was able to enhance the number
of DNA DSBs induced by CDDP, and inhibited the repair of DNA stand
breaks after drug removal without affecting the intracellular Pt
content.
In conclusion, the findings of this study suggest
that the cell-based high-throughput screening of the FDA-approved
drugs in combination with a major chemotherapeutic agent represents
an effective strategy for the identification of synergistic drug
combination regimens with feasibility to be translated into the
clinical treatment of cancer patients. Further mechanistic studies
of the identified drug combinations may provide valuable new
information to serve as a basis for developing more effective
chemosensitizers and improve the clinical outcome for cancer
patients.
Abbreviations:
|
CDDP
|
cis-diamminedichloroplatinum/cisplatin
|
|
PAT
|
potassium antimony tartrate
|
|
HTS
|
high-throughput screening
|
Acknowledgments
The authors would like to acknowledge Dr Huaiqiang
Ju and Ms. Alice Qiu for providing technical assistance, as well as
Ms. Ting Li and Dr Yongqiao He for their advice on statistical
analysis.
References
|
1
|
Rosenberg B, VanCamp L, Trosko JE and
Mansour VH: Platinum compounds: A new class of potent antitumour
agents. Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jamieson ER and Lippard SJ: Structure,
recognition, and processing of cisplatin-DNA adducts. Chem Rev.
99:2467–2498. 1999. View Article : Google Scholar
|
|
3
|
Giacchetti S, Perpoint B, Zidani R, Le
Bail N, Faggiuolo R, Focan C, Chollet P, Llory JF, Letourneau Y,
Coudert B, et al: Phase III multicenter randomized trial of
oxaliplatin added to chronomodulated fluorouracil-leucovorin as
first-line treatment of metastatic colorectal cancer. J Clin Oncol.
18:136–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vasey PA, Paul J, Birt A, Junor EJ, Reed
NS, Symonds RP, Atkinson R, Graham J, Crawford SM, Coleman R, et al
Scottish Gynaecological Cancer Trials Group: Docetaxel and
cisplatin in combination as first-line chemotherapy for advanced
epithelial ovarian cancer. J Clin Oncol. 17:2069–2080. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang D and Lippard SJ: Cellular processing
of platinum anticancer drugs. Nat Rev Drug Discov. 4:307–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Santos NA, Catão CS, Martins NM, Curti C,
Bianchi ML and Santos AC: Cisplatin-induced nephrotoxicity is
associated with oxidative stress, redox state unbalance, impairment
of energetic metabolism and apoptosis in rat kidney mitochondria.
Arch Toxicol. 81:495–504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Macciò A and Madeddu C: Cisplatin : An old
drug with a newfound efficacy - from mechanisms of action to
cytotoxicity. Expert Opin Pharmacother. 14:1839–1857. 2013.
View Article : Google Scholar
|
|
10
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kelland LR, Sharp SY, O'Neill CF, Raynaud
FI, Beale PJ and Judson IR: Mini-review: Discovery and development
of platinum complexes designed to circumvent cisplatin resistance.
J Inorg Biochem. 77:111–115. 1999. View Article : Google Scholar
|
|
12
|
Wong E and Giandomenico CM: Current status
of platinum-based antitumor drugs. Chem Rev. 99:2451–2466. 1999.
View Article : Google Scholar
|
|
13
|
Martín M: Platinum compounds in the
treatment of advanced breast cancer. Clin Breast Cancer. 2:190–209.
2001. View Article : Google Scholar
|
|
14
|
Cosaert J and Quoix E: Platinum drugs in
the treatment of non-small-cell lung cancer. Br J Cancer.
87:825–833. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al-Lazikani B, Banerji U and Workman P:
Combinatorial drug therapy for cancer in the post-genomic era. Nat
Biotechnol. 30:679–692. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chong CR and Sullivan DJ Jr: New uses for
old drugs. Nature. 448:645–646. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weir SJ, DeGennaro LJ and Austin CP:
Repurposing approved and abandoned drugs for the treatment and
prevention of cancer through public-private partnership. Cancer
Res. 72:1055–1058. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pessetto ZY, Weir SJ, Sethi G, Broward MA
and Godwin AK: Drug repurposing for gastrointestinal stromal tumor.
Mol Cancer Ther. 12:1299–1309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mathews Griner LA, Guha R, Shinn P, Young
RM, Keller JM, Liu D, Goldlust IS, Yasgar A, McKnight C, Boxer MB,
et al: High-throughput combinatorial screening identifies drugs
that cooperate with ibrutinib to kill activated B-cell-like diffuse
large B-cell lymphoma cells. Proc Natl Acad Sci USA. 111:2349–2354.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan S, Wang F, Chen G, Zhang H, Feng L,
Wang L, Colman H, Keating MJ, Li X, Xu RH, et al: Effective
elimination of cancer stem cells by a novel drug combination
strategy. Stem Cells. 31:23–34. 2013. View Article : Google Scholar :
|
|
22
|
Di Pasqua AJ, Hong C, Wu MY, McCracken E,
Wang X, Mi L and Chung FL: Sensitization of non-small cell lung
cancer cells to cisplatin by naturally occurring isothiocyanates.
Chem Res Toxicol. 23:1307–1309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Spigel DR and Greco FA: Chemotherapy in
metastatic and locally advanced non-small cell lung cancer. Semin
Surg Oncol. 21:98–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hata F, Sasaki K, Hirata K, Yamamitsu S
and Shirasaka T: Efficacy of a continuous venous infusion of
fluorouracil and daily divided dose cisplatin as adjuvant therapy
in resectable colorectal cancer: A prospective randomized trial.
Surg Today. 38:623–632. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pommier Y: Topoisomerase I inhibitors:
Camptothecins and beyond. Nat Rev Cancer. 6:789–802. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang JC: Cellular roles of DNA
topoisomerases: A molecular perspective. Nat Rev Mol Cell Biol.
3:430–440. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Staker BL, Hjerrild K, Feese MD, Behnke
CA, Burgin AB Jr and Stewart L: The mechanism of topoisomerase I
poisoning by a camptothecin analog. Proc Natl Acad Sci USA.
99:15387–15392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Redon C, Pilch D, Rogakou E, Sedelnikova
O, Newrock K and Bonner W: Histone H2A variants H2AX and H2AZ. Curr
Opin Genet Dev. 12:162–169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Furuta T, Takemura H, Liao ZY, Aune GJ,
Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT,
et al: Phosphorylation of histone H2AX and activation of Mre11,
Rad50, and Nbs1 in response to replication-dependent DNA
double-strand breaks induced by mammalian DNA topoisomerase I
cleavage complexes. J Biol Chem. 278:20303–20312. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Costa RM, Chiganças V, Galhardo RS,
Carvalho H and Menck CF: The eukaryotic nucleotide excision repair
pathway. Biochimie. 85:1083–1099. 2003. View Article : Google Scholar
|
|
31
|
Komuro Y, Udagawa Y, Susumu N, Aoki D,
Kubota T and Nozawa S: Paclitaxel and SN-38 overcome cisplatin
resistance of ovarian cancer cell lines by down-regulating the
influx and efflux system of cisplatin. Jpn J Cancer Res.
92:1242–1250. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stordal B, Pavlakis N and Davey R:
Oxaliplatin for the treatment of cisplatin-resistant cancer: A
systematic review. Cancer Treat Rev. 33:347–357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Terakawa T, Miyake H, Yokoyama N, Miyazaki
A, Tanaka H, Inoue T and Fujisawa M: Clinical outcome of paclitaxel
and carboplatin as second-line chemotherapy for advanced urothelial
carcinoma resistant to first-line therapy with gemcitabine and
cisplatin. Urol Int. 92:180–185. 2014. View Article : Google Scholar
|
|
34
|
Small BG, McColl BW, Allmendinger R, Pahle
J, López-Castejón G, Rothwell NJ, Knowles J, Mendes P, Brough D and
Kell DB: Efficient discovery of anti-inflammatory small- molecule
combinations using evolutionary computing. Nat Chem Biol.
7:902–908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pignata S, Cannella L, Leopardo D, Pisano
C, Bruni GS and Facchini G: Chemotherapy in epithelial ovarian
cancer. Cancer Lett. 303:73–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Murray HW, Pépin J, Nutman TB, Hoffman SL
and Mahmoud AA: Tropical medicine. BMJ. 320:490–494. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schulert AR, Rassoul AA, Mansour M, Girgis
N, McConnell E and Farid Z: Biological disposition of
antibilharzial antimony drugs. II. Antimony fate and uptake by
Schistosoma haematobium eggs in man. Exp Parasitol. 18:397–402.
1966. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Duffin J and Campling BG: Therapy and
disease concepts: The history (and future?) of antimony in cancer.
J Hist Med Allied Sci. 57:61–78. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lecureur V, Lagadic-Gossmann D and Fardel
O: Potassium antimonyl tartrate induces reactive oxygen
species-related apoptosis in human myeloid leukemic HL60 cells. Int
J Oncol. 20:1071–1076. 2002.PubMed/NCBI
|
|
40
|
Lecureur V, Le Thiec A, Le Meur A, Amiot
L, Drenou B, Bernard M, Lamy T, Fauchet R and Fardel O: Potassium
antimonyl tartrate induces caspase- and reactive oxygen
species-dependent apoptosis in lymphoid tumoral cells. Br J
Haematol. 119:608–615. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang B, Yu W, Guo J, Jiang X, Lu W, Liu M
and Pang X: The antiparasitic drug, potassium antimony tartrate,
inhibits tumor angiogenesis and tumor growth in nonsmall-cell lung
cancer. J Pharmacol Exp Ther. 352:129–138. 2015. View Article : Google Scholar
|
|
42
|
Weiss S and Hatcher RA: The mechanism of
the vomiting induced by antimony and Potassium tartrate (Tartar
emetic). J Exp Med. 37:97–111. 1923. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haldar AK, Sen P and Roy S: Use of
antimony in the treatment of leishmaniasis: Current status and
future directions. Mol Biol Int. 2011:5712422011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Robati M, Holtz D and Dunton CJ: A review
of topotecan in combination chemotherapy for advanced cervical
cancer. Ther Clin Risk Manag. 4:213–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Riemsma R, Simons JP, Bashir Z, Gooch CL
and Kleijnen J: Systematic review of topotecan (Hycamtin) in
relapsed small cell lung cancer. BMC Cancer. 10:4362010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Boabang P, Kurbacher CM, Kohlhagen H,
Waida A and Amo-Takyi BK: Anti-neoplastic activity of topotecan
versus cisplatin, etoposide and paclitaxel in four squamous cell
cancer cell lines of the female genital tract using an ATP-tumor
chemo-sensitivity assay. Anticancer Drugs. 11:843–848. 2000.
View Article : Google Scholar
|
|
47
|
Chou TC, Motzer RJ, Tong Y and Bosl GJ:
Computerized quantitation of synergism and antagonism of taxol,
topotecan, and cisplatin against human teratocarcinoma cell growth:
A rational approach to clinical protocol design. J Natl Cancer
Inst. 86:1517–1524. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fiorica JV: The role of topotecan in the
treatment of advanced cervical cancer. Gynecol Oncol. 90:S16–S21.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brave M, Dagher R, Farrell A, Abraham S,
Ramchandani R, Gobburu J, Booth B, Jiang X, Sridhara R, Justice R,
et al: Topotecan in combination with cisplatin for the treatment of
stage IVB, recurrent, or persistent cervical cancer. Oncology.
20:1401–1404. 1410–1411. 1415–1406. 2006.PubMed/NCBI
|