Introduction
The World Health Organization (WHO) has published
criteria for the classification and malignancy grade of glioma as
being pilocytic (grade I), diffuse (grade II), anaplastic (grade
III) and glioblastoma multiforme (GBM; grade IV) (1). GBM is the most malignant tumor
affecting the human brain, and the overall prognosis remains very
poor. The median survival time is <15 months (2-4).
Novel biomarkers and molecular targets have yet to be identified
for the improvement of the diagnosis and treatment of human GBM.
However, the molecular mechanisms responsible for the development
of GBM, which may lead to the identification of novel therapeutic
targets, remain unclear.
3-Phosphoinositide dependent protein kinase-1 (PDK1)
is a serine-threonine kinase belonging to the AGC kinase family.
PDK1 is a transcriptional regulator of the PI3K signaling pathway
and activates several downstream proteins (5-7).
Furthermore, PDK1 regulates downstream regulators, such as protein
kinase B (PKB)/Akt (6,8), rho-associated, coiled-coil-containing
protein kinase 1 (ROCK1) (9), β3
integrin (10), phospholipase Cγ1
(PLCγ1) (11) and myotonic
dystrophy kinase–related CDC42-binding kinase-α (MRCKα) (12). In addition, the most important
evidence is derived from clinical data demonstrating that PDK1 is
frequently overexpressed in different tumor types, including
gallbladder cancer (13), acute
myeloid leukemia (14,15), melanoma (16), esophageal squamous cell carcinoma
(17) and prostate cancer
(18). PDK1 knockdown has been
shown to decrease proliferation and induce apoptosis in breast
cancer (19) and esophageal cancer
(20). PDK1 overexpression
increases tumor invasiveness (12,21,22).
In xenograft tumor models, PDK1 knockdown has also been shown to
affect tumor growth (23) and
metastasis (24-26). Notably, PDK1 overexpression is
associated with a more aggressive phenotype and a worse prognosis.
However, PDK1 expression in human GBM and its biological and
clinical significance are not yet fully understood. The
transforming growth factor- β s(TGF-β) signaling pathway plays an
important role in many cancer types, as it promotes the
proliferation of malignant tumor cells and promotes migration and
invasion by inducing epithelial-mesenchymal transition (EMT)
(27). However, the mechanisms
through which glioma cells acquire the ability to take advantage of
the TGF-β tumor-promoting effects remain elusive. Previous studies
have suggested that c-Jun and JunB share extensive homology within
the leucine zipper and basic domains, and JunB inhibits cell
proliferation and migration by antagonizing c-Jun activity
(28,29). Previous findings have also
suggested that increasing c-Jun transcriptional activity induces
glioma progression (30). Thus, we
wished to determine whether PDK1 directly regulates c-Jun to induce
EMT in human GBM.
In this study, we found that PDK1 was overexpressed
in glioma tissues and was positively associated with glioma grade.
The 5-year survival rate of patients with glioma with a high PDK1
expression was significantly lower than that of those with a low
PDK1 expression. Cox regression analysis revealed that PDK1 may be
used as an independent prognostic factor for patients with glioma.
In vivo, PDK1 promoted glioma tumor xenograft growth. In
vitro, further analyses suggested that TGF-β upregulated PDK1
protein expression and PDK1 promoted cell proliferation, migration
and invasion, and functioned as an oncogene in malignant glioma, by
upregulating c-Jun protein and inducing EMT. Furthermore, our
findings were further validated by the online Oncomine database. In
Oncomine database, PDK1 and c-Jun proteins were overexpressed in
human glioma. Taken together, these results suggest that PDK1 is
positively associated with EMT and that the upregulation of c-Jun
protein PDK1 accelerates GBM cell invasion. Thus, PDK1 inhibition
may prove to be a potential therapeutic strategy for the treatment
of GBM.
Materials and methods
Tissue microarrays and cell lines
All tumor tissues were obtained from surgery, and
were immediately frozen in liquid nitrogen and stored at −80°C
until processed. The construction of tumor tissue microarrays
(TMAs) have been described previously (31). In total, 6 non-tumorous and 113
paraffin-embedded tissues from patients with glioma of grade I to
IV were used, at the Sun Yat-Sen Memorial Hospital from January,
2005 to January, 2011. Patients with glioma, and with
clinicopathological characteristics and follow-up information
available, were included. Tissues with lost cores or insufficient
cells were excluded from this study. The TMA consisted of 2007 WHO
glioma tissues graded as follows: Grade I (n=10), grade II (n=18),
grade III (n=36) and grade IV (n=49) samples. This study was
performed in accordance with the policies of the Institutional
Research Ethics Committee of Sun Yat-Sen Memorial Hospital. Written
informed consent was obtained from the study participants at the
Sun Yat-Sen Memorial Hospital of Guangzhou City.
The human U87 (glioblastoma of unknown origin) and
U251 glioma cell lines and were purchased from the American Type
Culture Collection (ATCC). Furthermore, a previous study suggested
that the U87 cell line may be misidentified (32). Of note, however, the U87 cell line
was authenticated before use in this study. In this study, PCR
amplification of the cell samples was carried out using the STR
Multi-amplification kit, (DC2101, Promega, Madison, WI, USA) and
the data revealed that the sample cell was the U87 MG cell line
based on the ATCC database. The cells were cultured in Dulbecco’s
modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (Gibco/ Thermo Fisher
Scientific, Waltham, MA, USA) at 37°C in a 5% CO2
humidified atmosphere.
Immunohistochemistry (IHC) staining
PDK1, c-Jun, β-catenin and E-cadherin protein
expression levels were analyzed by IHC on paraffin-embedded tissue
samples as previously described (33,34).
Briefly, the specimens were cut into 5-µm-thick sections and
baked at 65°C for 30 min. The sections were deparaffinized and
antigenic retrieval. The sections were treated with 3% hydrogen
peroxide in methanol to quench the endogenous peroxidase activity
followed by incubation with 1% bovine serum albumin to block the
non-specific binding. PDK1 (ab52893; 1:100 dilution; Abcam,
Cambridge, MA, USA), c-Jun (ab32137; 1:200 dilution; Abcam),
β-catenin (ab32572; 1:500 dilution; Abcam) and E-cadherin (ab15148;
1:30 dilution; Abcam) antibodies was incubated with the sections
overnight at 4°C, respectively. After washing, the tissue sections
were treated with biotinyl-ated secondary antibody for 60 min at
room temperature. After rinsing with PBS, the slides were immersed
for 3-5 min in DAB (3, 3-diaminobenzidine) (Sigma, St. Louis, MO,
USA) solution, then monitored under a microscope. The reaction was
terminated with distilled water. The slides were then
counter-stained with hematoxylin, dehydrated and coverslipped.
Quantification of staining analysis
The degree of immunostaining of formalin-fixed,
paraffin-embedded sections was viewed and scored separately by two
experienced pathologists, and the scores were determined by
combining the proportion of positively stained tumor cells and the
intensity of staining as previously described (33,34).
Isolation of total RNA and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from the frozen samples and
cells using TRIzol reagent (Invitrogen) according to the
manufacturer’s instructions as previously described (35). RNA was treated with RNase-free
DNase I (Roche, Basel, Switzerland). The BcaBest RNA PCR kit
(Takara, Dalian, China) was then used to synthesize the cDNA
according to the manufacturer’s instructions. All primers were
synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China).
Quantitative PCR (qPCR) was carried out using the Multicolor
Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) with
Real-time PCR Master Mix (SYBR-Green). PCR reactions were performed
under the following conditions: Pre-denaturation at 94°C for 5 min,
denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec,
elongation at 72°C for 1 min and elongation at 72°C for 10 min.
β-actin was used as an internal control. The sequences of the
primers used for qPCR are listed as follows: PDK1 forward,
5′-TCAGGACGACGA GAAGCTGTAT-3′ and reverse, 5′-AACGTACTGCGCTGTT CC
CACG-3′; c-Jun forward, 5′-AGAGCGACGCGAGCC AAT-3′ and reverse,
5′-GAGCCCTTATCCAGCCCGAG-3′. Snail forward,
5′-ATGCCGCGCTCTTTCCTCGTCAG-3′ and reverse,
5′-CCTCGAGGCTCAGCGGGACAT-3′; E-cadherin forward,
5′-GTTACTGATTGGTCTACGAGA-3′ and reverse,
5′-ATTGAAATGATCCAGTGCTTG-3′; N-cadherin forward,
5′-TCATGAAATCAACTATGCAAACCC-3′ and reverse,
5′-ATTGAAATGATCCAGTGCTTG-3′; ZEB1 forward,
5′-GCACAACCAAGTGCAGAAGA-3′ and reverse, 5′-CAT
TTGCAGATTGAGGCTGA-3′; TWIST1 forward, 5′-GTCC GGAGTGTTACGAGGAG-3′
and reverse, 5′-TGGAGGACCT GGTAGAGGAA-3′ and β-actin forward,
5′-CTCCATCCTGG CCTCGCTGT-3′ and reverse, 5′-GCTGTCACCTTCAC
CGTTCC-3′. The 2−ΔΔCq method was used to quantify the
relative mRNA amount (36).
Western blot analysis
For western blot assays, the total cell lysates were
prepared in high KCl lysis buffer (10 mM Tris-HCl, pH 8.0, 140 mM
NaCl, 300 mM KCl, 1 mM EDTA, 0.5% Triton X-100 and 0.5% sodium
deoxycholate) with complete protease inhibitor cocktail (Roche
Molecular Diagnostics, Branchburg, NJ, USA). The protein
concentrations were determined using a protein assay kit (Bio-Rad,
Hercules, CA, USA). Thirty micrograms of protein were separated by
10% sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto polyvinylidene fluoride membranes
(Roche, Branchburg, NJ, USA). The membranes were treated with 1%
blocking solution in TBS for 2 h, the membranes were incubated with
primary antibodies anti-PDK1 (ab52893; 1:1,000 dilution; Abcam),
c-Jun (ab32137; 1:2,000 dilution; Abcam), Snail (ab82846; 1:300
dilution; Abcam), β-catenin (ab32572; 1:5,000 dilution; Abcam),
E-cadherin (ab15148; 1:500 dilution; Abcam) and β-actin (PR0255;
1:2,000 dilution; Zhongshan Jinqiao Company, Beijing, China), at
4°C overnight, followed by anti- rabbit secondary antibody
conjugated with HRP (1:5,000; Epitomics, Burlingame, CA, USA) for 2
h. The immunolabeled proteins were detected by BM Chemiluminescence
Western Blotting kit (Roche, Branchburg, NJ, USA). The
quantification of the western blots was obtained by multiplying the
area and intensity of each band using Image J software (NIH,
Bethesda, MD, USA).
Lentivirus production and
transduction
The PDK1 sequence was amplified from normal human
genomic DNA and constructed into the lentivirus expression vector
pWPXL (Telebio Biomedical, Shanghai, China) to generate pWPXL-PDK1.
pWPXL-PDK1 was transfected into 293T cells (ATCC, Manassas, VA,
USA) using Lipofectamine 2000 (Invitrogen). The recombinant
lentiviruses were harvested from the supernatant of cell cultures
at 48 h post-transfection. The U251 and U87 cells were infected
with the recombinant lentivirus-transducing units plus 6 mg/ml
Polybrene (Sigma, St. Louis, MO, USA).
RNA interference
The selected siRNA targeting PDK1 was used in this
study. The sequences were as follows: siRNA-1 duplex (sense strand,
5′-GGUUGUUGUUGGAGAAGCATT-3′ and antisense strand,
5′-UGCUUCUCCAACAACAACCTT-3′) and siRNA-2 duplex (sense strand,
5′-GGUCAGUAGUCUUGUAAUATT-3′ and antisense strand,
5′-UAUUACAAGACUACUGACCTT-3′). The siRNAs were synthesized by
Shanghai GenePharma Co., Ltd. Approximately 2×106 cells
per well were seeded in a 6-well plates on the day prior to
transfection. Transfection with 50 nmol siRNAs was performed
according to the manufacturer’s instructions using Lipofectamine
2000 transfection reagent (Invitrogen).
MTT assays
The MTT assays were performed as previously
described (14,37). In brief, 1×105
cells/well was seeded in 96-well plates with 200 µl culture
medium. Following treatment with TGF-β1 (10 ng/ml) (Sigma) for 0,
1, 2, 3 and 5 days, the medium was replaced with 200 µl
DMEM/FBS containing 5 mg/ml MTT and incubated at 37°C for 4 h. The
supernatant was then discarded, and the cells were lysed in 200
µl DMSO for 10 min at 37°C. The optical density (OD) values
were measured at 490 nm (Epoch Microplate Spectrophotometer; BioTek
Instruments, Inc., Winooski, VT, USA).
Invasion and migration assays
The assays were performed as previously described
(38,39). The cells (1×106) were
suspended in 200 µl serum-free DMEM and seeded in the top
chambers of 24-well plates (Corning, New York, NY, USA) coated with
30 µl Matrigel (BD Biosciences, Franklin Lakes, NJ, USA).
The bottom chambers of the plates were filled with 500 µl
DMEM containing 10% FBS. The cells were allowed to migrate for 48 h
at 37°C. Following migration, cells in the top chambers were
removed using a cotton swab, and the cells which migrated to the
bottom chambers were fixed in 4% paraformaldehyde, and the cells
were stained with 0.5% (w/v) crystal violet (Sigma) for 2 h at room
temperature. The fixed and stained cells were counted in 5
independent fields under a microscope. At least, 3 chambers were
counted for each experiment. For the migration assay, a similar
protocol was followed apart from the replacement of the top chamber
of the transwell plate with an uncoated chamber. The culture medium
in the bottom chamber was replaced with DMEM containing 2.5% FBS,
and the cells were allowed to migrate for 12 h.
Wound healing assays
The cells were seeded in 6-well plates and cultured
until they reached 80% confluency, and a wound was then created by
manually scraping the cell monolayer. The cells were washed twice
with PBS to remove the floating cells, and then incubated in DMEM
supplemented with 1% FBS. Cell migration was observed at
pre-selected time points (0, 6, 24 and 48 h) as previously
described (39). Images were
acquired with a Nikon DS-5M Camera System (Nikon Instruments Inc.,
Tokyo, Japan).
Flow cytometry
The cells were cultured in 6-well plates and
detected utilizing the Gallios flow cytometer (Beckman Coulter,
Miami, FL, USA) for cell cycle assays and a BD flow cytometer (BD
Biosciences, San Jose, CA, USA) for apoptosis assays. For cell
cycle detection, the propidium iodide (PI) Detection kit (Nanjing
KeyGen Biotech. Co. Ltd., Nanjing, China) was utilized following
the manufacturer’s instructions. The cell-cycle raw date was
re-analyzed by MutiCycle for windows software (Phoenix Flow
Systems, San Diego, CA, USA). Apoptosis was measured using the
Apoptosis Detection kit (BD Pharmingen, San Diego, CA, USA)
(40). An Accuri™ C6 flow
cytometer (BD Biosciences) was utilized to quantify the percentage
of apoptotic cells.
Tumor implantation
To develop xenograft tumors, approximately
1×107 glioma U251 cells were inoculated into the mammary
fat pads of six to eight-week-old athymic female nude mice, which
were purchased from the Shanghai Experimental Animal Center
(Shanghai, China). All animals (18 mice) weighed 23-25 g, and were
maintained under SPF conditions at 20-26°C, a relative humidity of
40-70% and a 12-h light-dark cycle. All food was subjected to a
high temperature for steam disinfection (60 min, 120°C). All water
was acidified by hydrochloric acid and adjusted to a pH between 2.5
and 2.8. All efforts were made to minimize suffering. The mice were
examined by palpation for tumor formation for >60 days. After
tumors were detected, the tumor size was measured every 7 days
using calipers, and tumor volume was calculated as follows: Volume
(mm3) = length × width2 ×0.5 every 7 days for
8 weeks. The animals were sacrificed when the xenografts reached
approximately 1.5 cm in diameter, and tumor engrafts were harvested
and weighed. All animal experiments were carried out under the
guide of the Sun Yat-Sen University Committee for Use and Care of
Laboratory Animals and approved by the Animal Experimentation
Ethics Committee of Sun Yat-Sen University.
Statistical analysis
Statistical analyses were performed using
Statistical Package for Social Sciences software for Windows
version 13.0 (SPSS, Chicago, IL, USA) or Graphpad Prism software
5.0 (GraphPad Software, San Diego, CA, USA). Associations between
the patient clinicopathological characteristics and PDK1 expression
were identified using the Chi-square (χ2) test. Overall
survival (OS) was evaluated by Kaplan-Meier analysis and
differences between groups was assessed by the log-rank test, while
the prognostic significance of the clinicopathological
characteristics was determined using Cox regression analyses. OS
was calculated as the time from the date of diagnosis to the date
of death or the date of the last follow-up (if death did not
occur). The correlation between PDK1 and c-Jun protein expression
was determined using Pearson’s correlation analysis. The comparison
of two independent groups was analyzed using Student’s t-tests.
Multiple group comparisons were analyzed with one-way ANOVA and
Tukey’s HSD post hoc test. All statistical tests were two-tailed.
Errors were the SD of averaged results and P-values <0.05 were
considered to indicate statistically significant differences.
Results
Clinicopathological characteristics of
the patients with glioma from the TMA and PDK1 expression
To investigate the function of PDK1, we used a TMA
containing 113 glioma tissues (including 10 grade I, 18 grade II,
36 grade III and 49 grade IV samples) and 6 non-tumorous tissues
and performed IHC to evaluate PDK1 expression and its ass ociation
with the clinicopathological characteristics. The
clinicopathological characteristics of the patients with glioma are
shown in Table I. From the 113
patients with glioma, there were 67 males (59.29%) and 46 females
(40.71%), with an age ranging from 21 to 79 years (median age, 53.6
years). From the 113 glioma tissues, 41 tumors (36.28%) were ≤5.0
cm in size, and the other 72 tumors (63.72%) were >5.0 cm in
size. A high PDK1 expression were found to be significantly
associated with a large tumor size (>5.0 cm) (P<0.0001) and a
higher WHO grade (P<0.0001).
 | Table IAssociation between PDK1 levels and
various clinicopathological characteristics in 113 glioma
specimens. |
Table I
Association between PDK1 levels and
various clinicopathological characteristics in 113 glioma
specimens.
| Variables | No. of cases
(n=113) | PDK1 expression
| χ2 | P-value |
|---|
| Low (n=52) | High (n=61) |
|---|
| Age (years) | | | | | |
| ≤50 | 50 | 26 (52%) | 24 (48%) | 1.292 | 0.2557 |
| ≤>50 | 63 | 26 (41.27%) | 37 (58.73%) | | |
| Sex | | | | | |
| ≤Female | 46 | 21 (45.65%) | 25 (54.35%) | 0.0042 | 0.9485 |
| ≤Male | 67 | 31 (46.27%) | 36 (53.73%) | | |
| Tumor size
(cm) | | | | | |
| ≤≤5 | 41 | 29 (70.73%) | 12 (29.27%) | 15.82 | <0.0001a |
| ≤>5 | 72 | 23 (31.94%) | 49 (68.06%) | | |
| WHO grade | | | | | |
| ≤Grade I | 10 | 9 (90.00%) | 1 (10.00%) | 28.73 | <0.0001a |
| ≤Grade II | 18 | 15 (83.33%) | 3 (16.67%) | | |
| ≤Grade III | 36 | 11 (30.56%) | 25 (69.44%) | | |
| ≤Grade IV | 49 | 13 (26.53%) | 36 (73.47%) | | |
PDK1 expression is significantly
associated with glioma progression
To investigate PDK1 expression in glioma and
non-tumorous tissues, we performed IHC staining. We found that PDK1
staining was predominant in the cytoplasm. The results revealed a
significantly higher level of PDK1 expression in glioma tissues
compared with non-tumorous tissues. In the 113 glioma tissues, we
found that a high PDK1 expression was significantly increased from
glioma grade I to IV (Fig. 1A).
Moreover, the results of the comparative quantification of the mean
optical density (MOD) of PDK1 staining among the normal tissues and
glioma specimens of different grades are summarized in Fig. 1B. The MOD of PDK1 staining
increased while glioma progressed from a lower grade to a higher
one (P<0.05). To confirm these observations, we examined the
PDK1 mRNA level in a total of 63 tissue samples (10 non-tumorous
tissues, as well as in 3 grade I, 9 grade II, 19 grade III and 22
grade IV glioma tissues) by RT-qPCR. PDK1 mRNA expression was
significantly higher in the glioma than in the normal tissues. Not
surprisingly, the PDK1 mRNA level also increased from the lower
grade to the higher grade ones (P<0.05; Fig. 1C). These results indicate that PDK1
plays a critical role in glioma initiation and progression.
 | Figure 13-Phosphoinositide dependent protein
kinase 1 (PDK1) expression is significantly associated with glioma
progression. (A) Paraffin-embedded specimens of 6 non-tumorous
brain tissues and 113 glioma specimens (including 10 grade I, 18
grade II, 36 grade III and 49 grade IV tissues) with WHO glioma
grade I to IV were stained by immunohistochemistry (IHC). The areas
in the boxed regions in the top left corner (magnification, x200)
are presented as enlarged images in the lower left corner
(magnification, x400) (scale bars, 100 µm). (B) Comparative
quantification of the mean optical density (MOD) of PDK1 staining
among nontumorous tissues and glioma specimens of different grades.
(C) PDK1 mRNA expression levels were detected by RT-qPCR in a total
of 63 tissue samples (10 non-tumorous tissues, and 3 grade I, 9
grade II, 19 grade III, and 22 grade IV glioma tissues). (D) High
PDK1 expression was associated with a significantly lower overall
survival, compared with a lower PDK1 expression (n=113,
χ2=31.71; P<0.0001). (E and F) A high PDK1 expression
was significantly associated with a lower survival compared with a
low PDK1 expression in tissues of either the grade II + III
subgroup (n=54, χ2=8.392; P=0.0038) or grade IV subgroup
(n=49, χ2=8.671, P=0.0032). (G) Images of the xenograft
tumors retrieved immediately at the end of the experiment at ~8
weeks. (H) Xenograft assay revealed that PDK1 knockdown decreased
the volume of the xenograft tumors, while PDK1 overexpression
increased significantly tumor volume, compared to the control
group. (I) PDK1 knockdown reduced the weight of the xenograft
tumors, while PDK1 overexpression significantly increased tumor
weight, compared to the control group (*P<0.05,
**P<0.01, ***P<0.001, one-way
ANOVA). |
In order to determine the prognostic value of PDK1,
Kaplan-Meier survival analyses were performed for overall survival.
It was found that patients with a high PDK1 expression had a
significantly lower overall survival, compared with those with low
PDK1 expression. In addition, the median survival time of patients
whose tumors exhibited high PDK1 expression levels was only 22
months, whereas the median survival time of those with low PDK1
expression levels was 37 months (HR, 0.2771; 95% CI, 0.1772-0.4331,
P<0.0001; Fig. 1D) The
cumulative 5-year survival rate was 26.23% (16/61) in the low PDK1
expression group, whereas it was only 5.77% (3/52) in the high PDK1
expression group. To further validate these findings, we assessed
the prognostic significance in different subgroups of patients with
glioma stratified according to the 2007 WHO glioma grade. Of note,
a high PDK1 expression was significantly associated with a shorter
survival time in the different glioma patient subgroups. The
overall survival of patients with a high PDK1 expression was
significantly decreased compared with that in those with a low PDK1
expression in either the grade II + III subgroup (n=54,
χ2=8.392, P=0.0038, log-rank; Fig. 1E) or in the grade IV subgroup
(n=49, χ2=8.671, P=0.0032, log-rank; Fig. 1F). Collectively, our data suggest
that the PDK1 expression level is significantly associated with the
disease outcome.
Univariate Cox regression analyses were used to
determine the independence of PDK1 as a prognostic marker of the
survival of patients with glioma. As shown in Table II, overall survival was strongly
associated with PDK1 expression (HR, 1.785; 95% CI, 0.685-5.296;
P<0.0001), as well as with tumor size (HR, 0.529; 95% CI,
0.332-0.895; P<0.001) and glioma grade (HR, 0.653; 95% CI,
0.298-3.762; P<0.0001). Furthermore, PDK1 expression was also
demonstrated to be a useful prognostic biomarker for patients with
glioma by multivariate analysis; the survival of the patients was
found to be associated with tumor size (HR, 0.483; 95% CI,
0.298-0.932; P<0.001), PDK1 expression (HR, 1.367; 95% CI,
0.521-5.026; P<0.0001) and glioma grade (HR, 1.173; 95% CI,
0.409-2.321; P<0.0001). Taken together, these data suggest that
PDK1 may serve as a prognostic predictor for patients with
glioma.
| Table IIUnivariate and multivariate analyses
of prognostic parameters for survival of patients with glioma. |
Table II
Univariate and multivariate analyses
of prognostic parameters for survival of patients with glioma.
| Variables | Univariate analysis
| Multivariate
analysis
|
|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Age (years) | | | | | | |
| ≤50 vs.
>50 | 0.632 | 0.358-1.369 | 0.431 | | | |
| Sex | | | | | | |
| ≤Female vs.
male | 0.751 | 0.567-3.625 | 0.512 | | | |
| Tumor size
(cm) | | | | | | |
| ≤≤5 vs. >5 | 0.529 | 0.332-0.895 | <0.001a | 0.483 | 0.298-0.932 | <0.001a |
| PDK1 level | | | | | | |
| ≤Low vs. high | 1.785 | 0.685-5.296 | <0.0001a | 1.367 | 0.521-5.026 | <0.0001a |
| WHO grade | | | | | | |
| ≤II + III vs.
IV | 0.653 | 0.298-3.762 | <0.0001a | 1.173 | 0.409-2.321 | <0.0001a |
To further determine whether PDK1 is associated with
tumor growth in vivo, we established a tumor implantation
model to assess tumor growth using glioma U251 cells. Nude mice
were subcutaneously injected with 1×107 glioma U251
cells transfected with lenti-PDK1, PDK1 siRNA or negative control.
Representative images of the xenograft tumors at 8 weeks are shown
in Fig. 1G. The data revealed that
PDK1 knockdown decreased the volume of the xenograft tumors, while
PDK1 overexpression significantly enhanced tumor volume, compared
with control group (P<0.01, Fig.
1H). We also observed similar results regarding tumor weight.
PDK1 knockdown reduced the weight of the xenograft tumors, while
PDK1 overexpression significantly increased tumor weight, compared
with the control group (P<0.05, Fig. 1I). On the whole, these data suggest
that an increased PDK1 expression is associated with the
progression of GBM.
TGF-β upregulates PDK1, and PDK1 promotes
the proliferation and inhibits the apoptosis of glioma cells in
vitro
The TGF-β signaling pathway has been shown to play
an important role in several cancer types, as it promotes cancer
cell proliferation (27). However,
the mechanisms through which malignant glioma cells acquire the
ability to take advantage of the TGF-β tumor-promoting effects
remain elusive. In this study, we found that TGF-β
transcriptionally upregulated PDK1 expression in glioma cells. The
PDK1 mRNA level increased following treatment with TGF-β1 (10
ng/ml) for 0, 1, 2, 3, 5 days (P<0.05; Fig. 2A and B). Furthermore, cell
proliferation significantly increased following TGF-β1 treatment.
PDK1 knockdown significantly decreased cell proliferation. However,
compared with PDK1 knockdown alone, cell proliferation increased
with PDK1 knockdown and TGF-β1 treatment (P<0.05; Fig. 2C and D). These data suggest that
PDK1 promotes glioma cell proliferation, and that PDK1 knockdown
significantly inhibits cell proliferation. In addition, TGF-β
probably promotes cell viability through the activation of the PDK1
pathway.
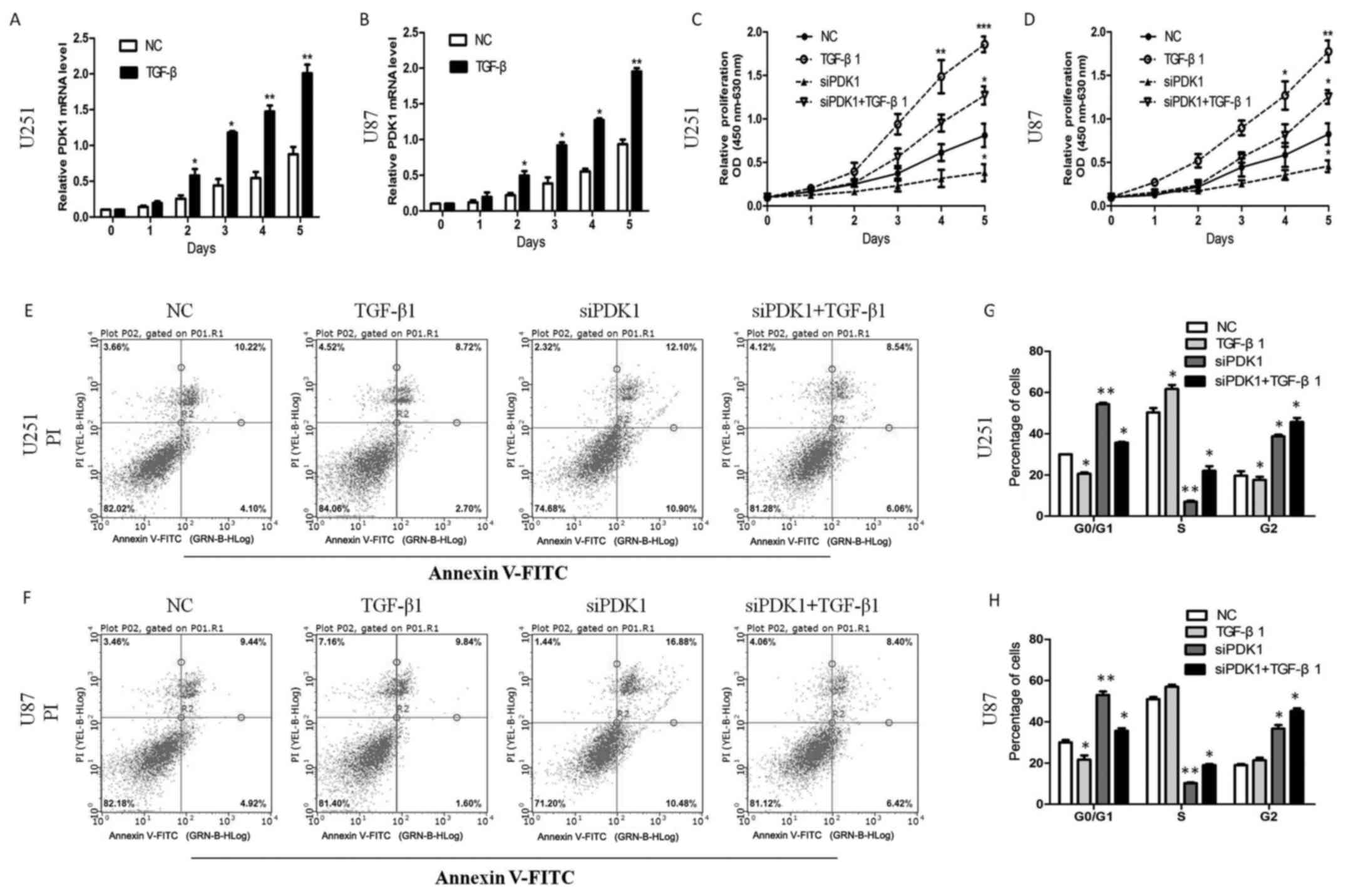 | Figure 2TGF-β upregulates 3-phosphoinositide
dependent protein kinase 1 (PDK1) and PDK1 promotes the
proliferation and inhibits the apoptosis of glioma cells in
vitro. (A and B) TGF-β transcriptionally upregulated PDK1
protein in glioma cells. And PDK1 mRNA expression increased after
TGF-β1 treatment (10 ng/ml) at 0, 1, 2, 3, 5 days
(*P<0.05, **P<0.01, Student’s t-tests).
(C and D) PDK1 knockdown significantly reduced cell proliferation,
compared to the control group. However, compared with the PDK1
knockdown group, cell proliferation increased significantly with
PDK1 knockdown and TGF-β1 treatment together *P<0.05,
**P<0.01, ***P<0.001, one-way ANOVA).
(E and F) PDK1 knockdown markedly increased the percentage of
apoptotic cells. The apoptosis of the cells decreased markedly by
TGF-β1 treatment. However, the percentage of apoptotic cells
decreased markedly following PDK1 knockdown and TGF-β1 treatment
together, compared with PDK1 knockdown alone. (G and H)
Glioblastoma cells infected with PDK1 siRNA contained more cells
ratio at the G0/G1 phase compared with control cells. In response
to TGF-β1 treatment for 24 h, the cell population in the S phase
significantly increased, whereas the accumulation of cells in the
G2 phase markedly decreased. However, the cell population in the S
phase increased through PDK1 knockdown and TGF-β1 treatment,
compared with PDK1 knockdown alone (*P<0.05,
**P<0.01, one-way ANOVA). |
To further elucidate the effects of PDK1 on glioma
cell viability, we further used the Annexin V/PI double staining
method to assess whether interference with PDK1 expression induces
cell apoptosis. The results revealed that PDK1 knockdown
markedlyincreased the percentage of apoptotic U251 cells. The
apoptotic rates of the U87 cells were similarly increased by PDK1
knockdown. As expected, cells apoptosis markedly reduced by TGF-β
treatment. However, the percentage of apoptotic cells decreased
markedly following PDK1 knockdown and TGF-β treatment, compared
with PDK1 knockdown alone (Fig. 2E and
F). Furthermore, we also used flow cytometry to assess whether
PDK1 knockdown inhibits cell cycle progression. As shown in
Fig. 2G and H, in the U251 and U87
cells transfected with PDK1 siRNA we observed a greater number of
cells in the G0/G1 phase compared with the control cells. In
response to TGF-β treatment for 24 h, the cell population in the S
phase significantly increased, whereas the accumulation of cells in
the G2 phase markedly decreased. However, the cell population in
the S phase significantly increased following PDK1 knockdown and
TGF-β treatment, compared with PDK1 knockdown alone. These data
indicate that PDK1 knockdown induces cell apoptosis by blocking
cell cycle progression at the G0/G1 phase, and the cell apoptotic
ratio significantly decreases through the activation of the
TGF-β/PDK1 pathway.
PDK1 enhances the invasive and migratory
potential of glioma cells
To determine whether the effects of PDK1 are
associated with migration and invasion, we conducted mig ration and
invasion assays using glioma U251 cells transfected with PDK1 siRNA
and/or treated with TGF-β. Transfection with PDK1 siRNA decreased
the number of migrating cells by approximately 68.5% compared with
the control group. In response to TGF-β treatment for 24 h, the
migrating cells significantly increased by approximately 81.7%
compared with the control group. However, the cells subjected to
PDK1 knockdown and TGF-β treatment exhibited a significantly
increased migration by only 46.5% compared with the control group
(Fig. 3A and B). PDK1 knockdown
reduced cell invasion by approximately 42.3% compared with the
control group. After TGF-β treatment for 24 h, the invasion of
cells increased by approximately 69.8% compared with the control
group. However, PDK1 knockdown and TGF-β treatment together
significantly increased cell invasion by only 32.6% compared with
the control group (Fig. 3C and D).
In agreement with these data, the results of wound healing assays
also revealed showed that siRNA knockdown for 48 h markedly
decreased cell migration, with less cells migrating into the gap
formed in a scratch assay. However, the number of migrating cells
increased markedly following TGF-β treatment for 48 h. Both siRNA
knockdown and TGF-β treatment had a positive effect on cell
migration and probably promoted cell migration through the
activation of the TGF-β/PDK1 pathway (Fig. 3E). These data thus suggest that
PDK1 is significantly associated with glioma cell migration and
invasion.
 | Figure 33-Phosphoinositide dependent protein
kinase 1 (PDK1) enhances the invasive and migratory potential of
glioma cells. (A and B) Transfection with PDK1 siRNA decreased the
number of migrating U251 cells. In response to TGF-β1 treatment for
24 h, the number of migrating cells increased significantly.
However, PDK1 knockdown and TGF-β1 treatment significantly
increased migration (*P<0.05, **P<0.01,
one-way ANOVA). (C and D) PDK1 knockdown reduced cell invasion. In
response to TGF-β1 treatment for 24 h, the invasion of the cells
increased significantly. However, PDK1 knockdown and TGF-β1
treatment significantly increased the invasion of cells
(*P<0.05, **P<0.01, one-way ANOVA). (E)
PDK1 knockdown markedly suppressed migration; however, the
migration of cells increased with PDK1 knockdown and TGF-β1
treatment, as assessed by wound healing assays. |
PDK1/c-Jun pathway is activated by TGF-β
and induces EMT and promotes progression in human GBM
To investigate the molecular mechanisms through
which PDK1 accelerates GBM cell invasion, the U251 glioma cells
were treated with TGF-β1 (10 ng/ml). The results revealed that the
U251 cells gradually became spindle-shaped at 0, 7, 14 and 21 days
(Fig. 4A). Moreover, the PDK1
expression level gradually increased. In addition, the expression
levels of the epithelial cell marker, E-cadherin, gradually
decreased, while those of the mesenchymal cell markers, N-cadherin,
ZEB1, SNAIL and TWIST1 gradually increased (Fig. 4B). These results indicate that
TGF-β upregulates PDK1 expression to induce EMT. In the UCSC
database (http://genome.ucsc.edu), the
transcription factor, JunB, binds to the DNA sequences downstream
of the PDK1 gene. Previous studies have also suggested that JunB
and c-Jun share extensive homology within the leucine zipper and
basic domains, and JunB inhibits cell proliferation and migration
by antagonizing c-Jun activity (28,29).
It has also been suggested that increasing c-Jun transcriptional
activity induces glioma progression (30). Therefore, in this study, we wished
to determine whether PDK1 directly regulates c-Jun to induce EMT in
human GBM.
 | Figure 4The 3-phosphoinositide dependent
protein kinase 1 (PDK1)/c-Jun pathway activated by TGF-β induces
EMT and promotes glioblastoma progression. (A) U251 glioma cells
gradually became spindle-shaped with TGF-β (10 ng/ml) treatment
after 0, 7, 14 and 21 days. (B) The PDK1 expression level gradually
increased; the expression of the epithelial cell marker,
E-cadherin, gradually decreased, while that of the mesenchymal cell
markers, N-cadherin, ZEB1, SNAIL and TWIST1, gradually increased.
(C-E) PDK1, c-Jun, SNAIL and β-catenin protein expression increased
significantly, but that of the epithelial cell marker, E-cadherin,
decreased markedly following treatment with PDK1 overexpression
plasmid or TGF-β1 in U251 and U87 cells. However, the protein
levels exhibited opposite effects following transfection with siRNA
against PDK1. (F and G) PDK1, c-Jun, SNAIL, β-catenin and
E-cadherin mRNA expression exhibited similar effects, as with
protein expression (*P<0.05, **P<0.01,
***P<0.001, one-way ANOVA). |
Subsequently, following transfection of the U251 and
U87 cells with PDK1 overexpression plasmid or treatment with
TGF-β1, the protein expression levels of PDK1 and c-Jun increased.
In addition, the protein expression levels of the epithelial cell
marker E-cadherin decreased markedly in glioma cell lines with PDK1
overexpression or treatment with TGF-β1, while those of the
mesenchymal cell markers, Snail and β-catenin, increased. PDK1 and
c-Jun protein expression was downregulated significantly when PDK1
expression was inhibited by siRNA, and E-cadherin protein
expression increased, while Snail and β-catenin protein expression
decreased (P<0.05; Fig. 4C-E).
Moreover, similar results were obtained for the mRNA expression
levels in the U251 and U87 cells (P<0.05; Fig. 4F and G).
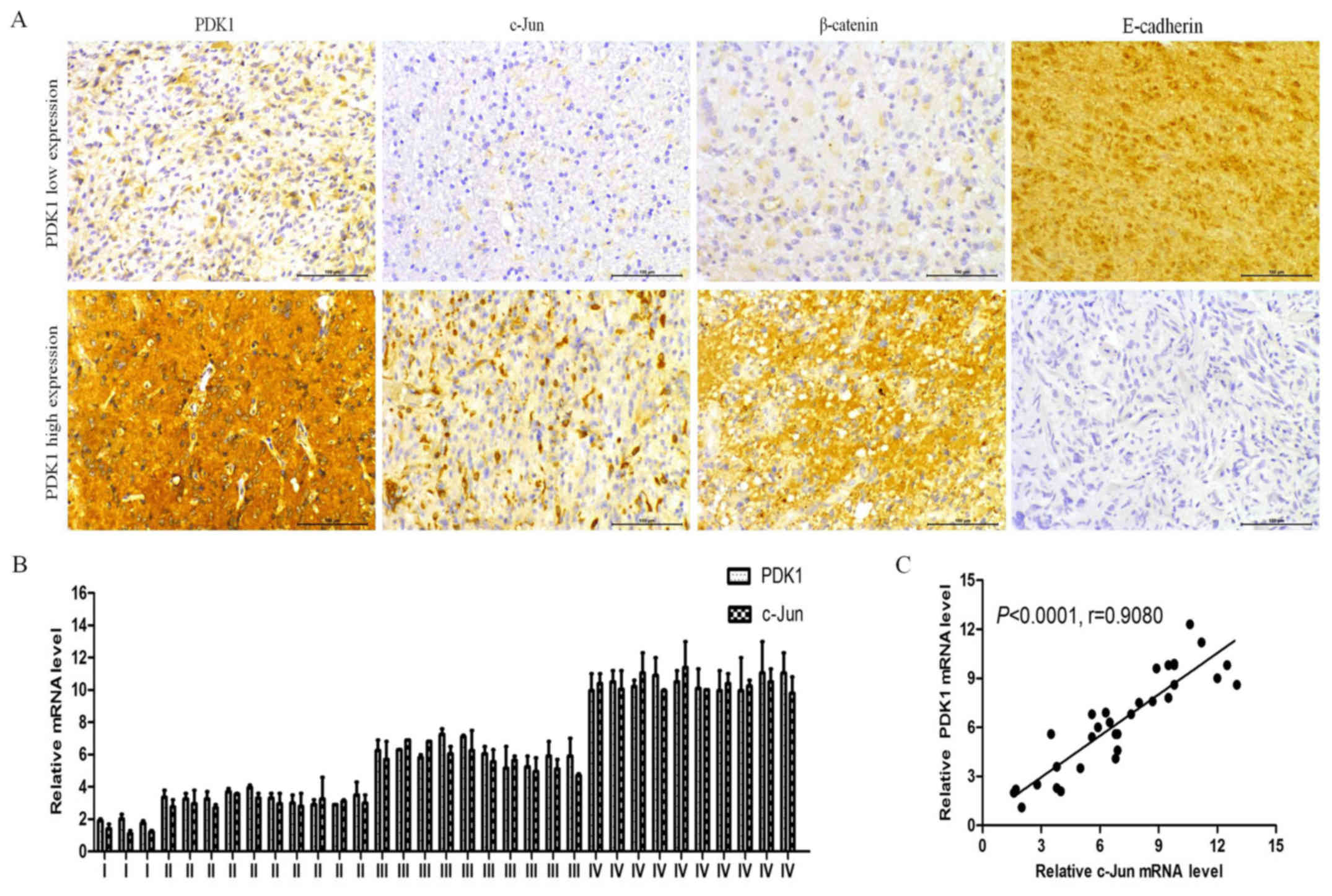
It should be noted that the PDK1, c-Jun, β-catenin
and E-cadherin expression levels in the glioma tissues with high or
low PDK1 levels were similar to those found in the cell lines
(Fig. 5A). c-Jun and β-catenin
protein expression levels were decreased in the tissues with low
PDK1 expression, and E-cadherin protein expression was increased.
However, c-Jun and β-catenin protein expression increased, and
E-cadherin protein expression decreased when PDK1 protein highly
expressed. In agreement with these findings, the PDK1 and c-Jun
mRNA levels examined increased in the tissues with glioma grade I
to IV, with similar patterns in the glioma tissues (Fig. 5B). PDK1 protein expression also
positively correlated with c-Jun expression in the glioma tissues
(r=0.9080, P<0.0001; Fig. 5C).
Collectively, these results confirm that PDK1 promotes cell
invasion in GBM through the upregulation of c-Jun and the promotion
of EMT.
Evidence in the Oncomine database
The Oncomine database (https://www.oncomine.org/) provide solutions for
individual researchers and multinational companies, with
peer-reviewed analytical methods and a powerful set of analysis
functions that compute gene expression signatures, clusters, and
gene-set modules, automatically extracting biological insights from
the data. To further validate the findings of our study, we
searched the Oncomine database for PDK1 expression in human glioma
(data not shown). A total of 8 datasets demonstrated that PDK1
expression in human glioma was higher compared to that in normal
brain tissues. PDK1 overexpression exhibited a significant
association with a high WHO glioma grade, recurrence and treatment
response. Moreover, PDK1 overexpression was significantly
associated with MGMT methylation, EGFR amplification, LDHA mutation
and the loss of heterozygosity of chromosome 1p, 10q,19q. Not
surprisingly, 5 datasets revealed that PDK1 overexpression was
significantly associated with short survival time during follow-up.
Intriguingly, 11 datasets revealed that the protein expression
level of c-Jun was higher in human glioma, compared with normal
brain tissues, while 2 datasets demonstrated an association between
c-Jun expression and higher-grade human glioma with an
approximately 2-fold increase. Furthermore, 6 datasets indicated
that the expression of c-Jun was associated with patient survival
time during follow-up (data not shown). Collectively, these results
obtained from the Oncomine database confirm our findings that PDK1
may upregulate c-Jun protein expression to promote the progression
of human glioma.
Discussion
In this study, we examined PDK1 expression in 113
glioma and 6 normal brain tissues using TMA. We found that PDK1 was
significantly upregulated in glioma, compared with non- tumorous
tissues. Moreover, PDK1 exhibited a significant association with
WHO glioma grade, tumor size and survival time. PDK1 protein was an
independent prognostic factor. In vivo, PDK1 promoted glioma tumor
xenograft growth. TGF-β induced tumor cell EMT and increased cell
motility, which was associated with migration and invasion. In
vitro, our data confirmed that TGF-β upregulated PDK1 and PDK1
promoted cell migration and invasion, and functioned as an oncogene
in GBM, by upregulating c-Jun and inducing EMT. In glioma tissues,
c-Jun protein was increased and positively correlated with the PDK1
levels. In the Oncomine database, PDK1 and c-Jun protein levels
were overexpressed in human glioma. Of note, both PDK1 and c-Jun
protein expression levels were significantly associated with WHO
glioma grade and survival time. Taken together, these results
suggest that PDK1 is positively associated with EMT and that the
upregulation of c-Jun protein by PDK1 accelerates GBM cell
invasion.
Glioma, comprising approximately 80% of all primary
malignant brain tumors, is the most prevalent type and results in
poor clinical outcomes (41-43).
According to the WHO classification, gliomas are classified into
four histopathological grades, ranging from grade I to IV.
Glioblastoma (grade IV) is one of the most malignant forms of human
brain tumor (1). In treatment, the
approaches mainly include surgical resection, radiotherapy,
chemotherapy, targeted therapy and multiple therapies in
combination for the glioma (44).
Despite marked progression in therapies, glioma prognosis remains
unsatisfactory, and patients who suffer from GBM, the most
aggressive type of glioma, have a poor prognosis. Therefore,
identifying novel biomarkers may aid in the better understanding of
the mechanisms of carcinogenesis and in the development of targeted
treatments, which may thus improve the prognosis of patients with
glioma. However, the current methods used to characterize glioma
are inadequate. The establishment of molecular biomarkers may
identify variables required to improve tumor characterization, and
may aid in the identification of specific targets for improved
treatment with essential prognostic value for glioma patient
survival.
PDK1 is a serine-threonine kinase belonging to the
AGC kinase family. An increasing amount of data suggest that PDK1
plays a pivotal role in the regulation of cell migration and
several signaling pathways activated by growth factors and hormones
and activates members of the AGC family of protein kinases
(5,6,16,17).
PDK1 has been demonstrated to be crucial for the regulation of each
step of cell migration, by activating several proteins, such as
PKB/Akt (6,8), ROCK1 (9), β3 integrin (10), PLCγ1 (11) and MRCKα (12). Moreover, PDK1 regulates cancer cell
invasion as well, thus representing a possible target with which to
prevent cancer metastasis. Furthermore, PDK1 protein has been
recognized as a key regulator in breast cancer (45), melanoma (16), gallbladder cancer (13), ovarian cancer (46,47),
colorectal cancer (48), gastric
cancer (49), pancreatic cancer
(50,51), prostate carcinoma (52) and acute myeloid leukemia (15). In breast cancer, the activation of
SGK1 protein by PDK1 contributes to the maintenance of residual
mTORC1 activity through direct the phosphorylation and inhibition
of TSC2 (53). Moreover, PDK1
phosphorylation is frequently upregulated in breast cancer with the
concomitantly increased phosphorylation of downstream kinases,
including Akt, mTOR, p70S6K, S6 and Stat3 (54). PDK1 directly induces PLK1
phosphorylation, which in turn induces MYC phosphorylation and
protein upregulation. PDK1/PLK1/MYC signaling is critical for
cancer cell growth and survival, and PDK1/PLK1 knockdown suggests
an effective therapeutic approach (55). In human esophageal squamous cell
carcinoma (ESCC), PDK1 protein is expressed in the cytoplasm, but
is not expressed in adjacent non-cancerous tissues. In addition, a
high PDK1 expression was found to be closely associated with an
advanced tumor stage, positive lymph node metastasis and high
histological grade (17). In
summary, targeting PDK1 may prove to be an effective approach with
which to inhibit cancer progression towards a more invasive and
metastatic phenotype.
However, to the best of our knowledge, few studies
to date have investigated the expression and significance of PDK1
in glioma tissues and molecular function, particularly as regards
the PDK1 pathway regulating GBM invasion and patient prognosis. The
results of this study indicated that PDK1 was upregulated in GBM.
The PDK1 protein and mRNA levels were significantly upregulated in
glioma, compared with non-tumorous tissues. Furthermore, PDK1
overexpression was significantly associated with a large tumor size
(>5.0 cm) and higher grade, and a shorter survival time. The
overall survival of patients with a high PDK1 expression was
significantly decreased compared with that of those with a low PDK1
expression in either the grade II + III subgroup or grade IV
subgroup. Our data also indicated that PDK1 protein was an
independent prognostic factor. Xenograft assay revealed that PDK1
knockdown decreased the volume of xenograft tumors, while PDK1
overexpression significantly increased tumor volume. These results
are in agreement with those of a previous study (14), which demonstrated that PDK1
overexpression was found in >40% of patients with acute myeloid
leukemia. PDK1 overexpression occurred uniformly throughout the
leukemic population, including putative leukemia-initiating cells.
Moreover, PDK1 overexpression was closely associated with the
increased phosphorylation of PKC isoenzymes and the inhibition of
PKC strongly inhibited the survival advantage of
PDK1-overexpressing cells (14).
PDK1 has multiple complex roles in tumor biology.
PDK1 has been reported to be involved in the inhibition of
apoptosis and the promotion of growth in lung cancer (56). PDK1 protein has also been shown to
be closely associated with the proliferation, apoptosis and
invasion of esophageal cancer cells (20). Although PDK1 has been shown to be
present in multiple tumors, its functional impact on tumor
progression remains largely unknown. The findings of this study
suggested that PDK1 expression increased following TGF-β1
treatment. Furthermore, cell proliferation significantly increased
through the upregulation of PDK1 by TGF-β1. However, PDK1 knockdown
significantly reduced cell proliferation. In addition, PDK1
knockdown induced apoptosis by blocking cell cycle progression at
the G0/G1 phase.
To determine whether PDK1 is involved in migration
and invasion, we conducted migration and invasion assays using
cells transfected with PDK1 siRNA. PDK1 knockdown significantly
decreased cell migration and invasion, compared with the control
group. However, both siRNA knockdown and TGF-β treatment had a
significant positive effect on cell migration and promoted cell
migration through the activation of the TGF-β/PDK1 pathway. In
agreement with these findings, the results of wound healing assay
also demonstrated similar effects of PDK1 on cell invasion.
To investigate the molecular mechanisms through
which PDK1 accelerates cell invasion, we treated U251 cells with
TGF-β1. The results revealed that the U251 cells gradually became
spindle-shaped during EMT, while PDK1 expression increased
gradually. These results suggested that TGF-β may upregulate PDK1
to induce EMT. In the UCSC database (http://genome.ucsc.edu), the transcription factor JunB
binds to the DNA sequences downstream of the PDK1 gene. JunB and
c-Jun share extensive homology within the leucine zipper and basic
domains, and JunB inhibited cell proliferation and migration by
antagonizing c-Jun activity (28,29).
Therefore, we wished to determine whether PDK1 directly regulates
c-Jun to induce EMT in human GBM. In breast cancer cells, although
ATF-3 interacts with c-Jun and JunB proteins, and regulates their
expression, a heterodimer complex of only ATF-3/c-Jun forms at the
AP-1 site of the MMP-13 promoter and activates its gene expression
upon TGF-β treatment (29).
Extracellular signals can induce the post-translational
modifications of c-Jun, resulting in altered transcriptional
activity and target gene expression. This activates a number of
cellular processes, such as proliferation, apoptosis, survival,
tumorigenesis and tissue morphogenesis (57,58).
In the present study, following treatment with PDK1 overexpression
plasmid or TGF-β, PDK1 and c-Jun protein expression increased. In
addition, the protein expression of the epithelial cell marker,
E-cadherin, decreased, while that of the mesenchymal cell markers,
Snail and β-catenin, increased. PDK1 and c-Jun protein were
down-regulated significantly when PDK1 expression was inhibited by
siRNA, and E-cadherin protein increased, but Snail and β-catenin
protein decreased. IHC staining in tumor tissues with high or low
PDK1 levels revealed similar expression patterns found in the cell
lines. In agreement with these findings, the PDK1 and c-Jun mRNA
expression levels studied by RT-qPCR increased from glioma grade I
to IV with similar patterns in glioma tissues. Moreover, c-Jun
expression positively correlated with the PDK1 levels in tumor
tissues. In the Oncomine database, the data confirmed that PDK1 and
c-Jun protein promote human glioma progression.
In conclusion, the findings of this study confirmed
the prognostic potential of PDK1 expression in human GBM and
revealed the pro-migratory and pro-invasive functions of this
protein in GBM cells through the PDK1/c-Jun pathway activated by
TGF-β.
Funding
This study was funded by grants from the National
Natural Science Foundation of China (81272774, 81572497 and
81702654), the Natural Science Foundation of Guangdong Province
(2017A030313642), the Science Foundation of Guangzhou Women and
Children’s Medical Center (5001-3001023 and 5001-2150010), the Key
Laboratory of Malignant Tumor Molecular Mechanism and Translational
Medicine of Guangzhou Bureau of Science and Information Technology
[(2013)163], and the Key Laboratory of Malignant Tumor Gene
Regulation and Target Therapy of Guangdong Higher Education
Institutes (K1809001).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
DL and XX conceived and designed the experiments;
DL, XX, JL and CC performed the experiments; DL and WC analyzed the
data; DL and XX wrote the manuscript; FW and YX were involved in
data collection. FL conceived, designed and supervised the study,
and revised the manuscript. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
This study was performed in accordance with the
policies of the Institutional Research Ethics Committee of Sun
Yat-Sen Memorial Hospital. Written informed consent was obtained
from the study participants at the Sun Yat-Sen Memorial Hospital of
Guangzhou City. All animal experiments were carried out under the
guide of the Sun Yat-Sen University Committee for Use and Care of
Laboratory Animals and approved by the Animal Experimentation
Ethics Committee of Sun Yat-Sen University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations
Abbreviations:
|
PDK1
|
3-phosphoinositide dependent protein
kinase 1
|
|
EMT
|
epithelial-mesenchymal transition
|
|
TGF-β
|
transforming growth factor-β
|
|
TMA
|
tissue microarray
|
|
IHC
|
immunohistochemistry
|
|
SI
|
staining index
|
|
OS
|
overall survival
|
|
MOD
|
mean optical density
|
|
WHO
|
World Health Organization
|
Acknowledgments
Not applicable.
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS Statistical Report: Primary brain and central nervous system
tumors diagnosed in the United States in 2008-2012. Neuro-oncol.
17(Suppl 4): pp. iv1–iv62. 2015, View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van den Bent MJ and Bromberg JE:
Neuro-oncology: The many challenges of treating elderly
glioblastoma patients. Nat Rev Neurol. 11:374–375. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dirven L, Aaronson NK, Heimans JJ and
Taphoorn MJ: Health-related quality of life in high-grade glioma
patients. Chin J Cancer. 33:40–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mora A, Komander D, van Aalten DM and
Alessi DR: PDK1, the master regulator of AGC kinase signal
transduction. Semin Cell Dev Biol. 15:161–170. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pearce LR, Komander D and Alessi DR: The
nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol.
11:9–22. 2010. View
Article : Google Scholar
|
|
7
|
Raimondi C and Falasca M: Targeting PDK1
in cancer. Curr Med Chem. 18:2763–2769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pinner S and Sahai E: PDK1 regulates
cancer cell motility by antagonising inhibition of ROCK1 by RhoE.
Nat Cell Biol. 10:127–137. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
di Blasio L, Gagliardi PA, Puliafito A,
Sessa R, Seano G, Bussolino F and Primo L: PDK1 regulates focal
adhesion disassembly by modulating endocytosis of αvβ3 integrin. J
Cell Sci. 128:863–877. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Raimondi C, Chikh A, Wheeler AP, Maffucci
T and Falasca M: Correction: A novel regulatory mechanism links
PLCγ1 to PDK1. J Cell Sci. 130:10162017. View Article : Google Scholar
|
|
12
|
Gagliardi PA, di Blasio L, Puliafito A,
Seano G, Sessa R, Chianale F, Leung T, Bussolino F and Primo L:
PDK1-mediated activation of MRCKα regulates directional cell
migration and lamellipodia retraction. J Cell Biol. 206:415–434.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lian S, Shao Y, Liu H, He J, Lu W, Zhang
Y, Jiang Y and Zhu J: PDK1 induces JunB, EMT, cell migration and
invasion in human gallbladder cancer. Oncotarget. 6:29076–29086.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zabkiewicz J, Pearn L, Hills RK, Morgan
RG, Tonks A, Burnett AK and Darley RL: The PDK1 master kinase is
over-expressed in acute myeloid leukemia and promotes PKC-mediated
survival of leukemic blasts. Haematologica. 99:858–864. 2014.
View Article : Google Scholar :
|
|
15
|
Qin L, Tian Y, Yu Z, Shi D, Wang J, Zhang
C, Peng R, Chen X, Liu C, Chen Y, et al: Targeting PDK1 with
dichloroacetophenone to inhibit acute myeloid leukemia (AML) cell
growth. Oncotarget. 7:1395–1407. 2016. View Article : Google Scholar :
|
|
16
|
Scortegagna M, Ruller C, Feng Y, Lazova R,
Kluger H, Li JL, De SK, Rickert R, Pellecchia M, Bosenberg M, et
al: Genetic inactivation or pharmacological inhibition of Pdk1
delays development and inhibits metastasis of Braf(V600E):Pten(−/−)
melanoma. Oncogene. 33:4330–4339. 2014. View Article : Google Scholar
|
|
17
|
Yang Z, Wu Z, Liu T, Han L, Wang C, Yang B
and Zheng F: Upregulation of PDK1 associates with poor prognosis in
esophageal squamous cell carcinoma with facilitating tumorigenicity
in vitro. Med Oncol. 31:3372014. View Article : Google Scholar
|
|
18
|
Choucair KA, Guérard KP, Ejdelman J,
Chevalier S, Yoshimoto M, Scarlata E, Fazli L, Sircar K, Squire JA,
Brimo F, et al: The 16p13.3 (PDPK1) Genomic Gain in Prostate
Cancer: A Potential Role in Disease Progression. Transl Oncol.
5:453–460. 2012. View Article : Google Scholar
|
|
19
|
Raimondi C, Calleja V, Ferro R, Fantin A,
Riley AM, Potter BV, Brennan CH, Maffucci T, Larijani B and Falasca
M: A Small molecule inhibitor of PDK1/PLCγ1 interaction blocks
breast and melanoma cancer cell invasion. Sci Rep. 6:261422016.
View Article : Google Scholar
|
|
20
|
Yu J, Chen KS, Li YN, Yang J and Zhao L:
Silencing of PDK1 gene expression by RNA interference suppresses
growth of esophageal cancer. Asian Pac J Cancer Prev. 13:4147–4151.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie Z, Yuan H, Yin Y, Zeng X, Bai R and
Glazer RI: 3-phosphoinositide-dependent protein kinase-1 (PDK1)
promotes invasion and activation of matrix metalloproteinases. BMC
Cancer. 6:772006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maurer M, Su T, Saal LH, Koujak S, Hopkins
BD, Barkley CR, Wu J, Nandula S, Dutta B, Xie Y, et al:
3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions
on the phosphatidylinositol 3-kinase pathway in breast carcinoma.
Cancer Res. 69:6299–6306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeng X, Xu H and Glazer RI: Transformation
of mammary epithelial cells by 3-phosphoinositide-dependent protein
kinase-1 (PDK1) is associated with the induction of protein kinase
Calpha. Cancer Res. 62:3538–3543. 2002.PubMed/NCBI
|
|
24
|
Du J, Yang M, Chen S, Li D, Chang Z and
Dong Z: PDK1 promotes tumor growth and metastasis in a spontaneous
breast cancer model. Oncogene. 35:3314–3323. 2016. View Article : Google Scholar
|
|
25
|
Wang Y, Fu L, Cui M, Wang Y, Xu Y, Li M
and Mi J: Amino acid transporter SLC38A3 promotes metastasis of
non-small cell lung cancer cells by activating PDK1. Cancer Lett.
393:8–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li W, Song R, Fang X, Wang L, Chen W, Tang
P, Yu B, Sun Y and Xu Q: SBF-1, a synthetic steroidal glycoside,
inhibits melanoma growth and metastasis through blocking
interaction between PDK1 and AKT3. Biochem Pharmacol. 84:172–181.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wen W, Liu G, Jin K and Hu X: TGF-β1
induces PGP9.5 expression in CAFs to promote the growth of
colorectal cancer cells. Oncol Rep. 37:115–122. 2017. View Article : Google Scholar
|
|
28
|
Eferl R and Wagner EF: AP-1: A
double-edged sword in tumorigenesis. Nat Rev Cancer. 3:859–868.
2003. View Article : Google Scholar
|
|
29
|
Gokulnath M, Swetha R, Thejaswini G,
Shilpa P and Selvamurugan N: Transforming growth factor-β1
regulation of ATF-3, c-Jun and JunB proteins for activation of
matrix metal-loproteinase-13 gene in human breast cancer cells. Int
J Biol Macromol. 94A:370–377. 2017. View Article : Google Scholar
|
|
30
|
Ohba S, Hirose Y, Kawase T and Sano H:
Inhibition of c-Jun N-terminal kinase enhances temozolomide-induced
cytotoxicity in human glioma cells. J Neurooncol. 95:307–316. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tamburrino A, Molinolo AA, Salerno P,
Chernock RD, Raffeld M, Xi L, Gutkind JS, Moley JF, Wells SA Jr and
Santoro M: Activation of the mTOR pathway in primary medullary
thyroid carcinoma and lymph node metastases. Clin Cancer Res.
18:3532–3540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:pp. 354re32016, View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luo D, Chen H, Lu P, Li X, Long M, Peng X,
Huang M, Huang K, Lin S, Tan L, et al: CHI3L1 overexpression is
associated with metastasis and is an indicator of poor prognosis in
papillary thyroid carcinoma. Cancer Biomark. 18:273–284. 2017.
View Article : Google Scholar
|
|
34
|
Luo D, Chen W, Tian Y, Li J, Xu X, Chen C
and Li F: Serpin peptidase inhibitor, clade A member 3 (SERPINA3),
is overexpressed in glioma and associated with poor prognosis in
glioma patients. OncoTargets Ther. 10:2173–2181. 2017. View Article : Google Scholar
|
|
35
|
Wang W, Wang L, Mizokami A, Shi J, Zou C,
Dai J, Keller ET, Lu Y and Zhang J: Down-regulation of E-cadherin
enhances prostate cancer chemoresistance via Notch signaling. Chin
J Cancer. 36:352017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
37
|
Zhang Y, Zhang LX, Liu XQ, Zhao FY, Ge C,
Chen TY, Yao M and Li JJ: Id4 promotes cell proliferation in
hepatocellular carcinoma. Chin J Cancer. 36:192017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Luo D, Chen H, Li X, Lu P, Long M, Peng X,
Lin S, Tan L, Zhu Y, Ouyang N, et al: Activation of the ROCK1/MMP-9
pathway is associated with the invasion and poor prognosis in
papillary thyroid carcinoma. Int J Oncol. 51:1209–1218. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu N, Zhang L, Wang Z, Cheng Y, Zhang P,
Wang X, Wen W, Yang H, Liu H, Jin W, et al: MicroRNA-101 inhibits
proliferation, migration and invasion of human glioblastoma by
targeting SOX9. Oncotarget. 8:19244–19254. 2017.
|
|
40
|
Chen H, Luo D, Zhang L, Lin X, Luo Q, Yi
H, Wang J, Yan X, Li B, Chen Y, et al: Restoration of p53 using the
novel MDM2-p53 antagonist APG115 suppresses dedifferentiated
papillary thyroid cancer cells. Oncotarget. 8:43008–43022.
2017.PubMed/NCBI
|
|
41
|
Killela PJ, Pirozzi CJ, Healy P, Reitman
ZJ, Lipp E, Rasheed BA, Yang R, Diplas BH, Wang Z, Greer PK, et al:
Mutations in IDH1, IDH2, and in the TERT promoter define clinically
distinct subgroups of adult malignant gliomas. Oncotarget.
5:1515–1525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Laws ER, Parney IF, Huang W, Anderson F,
Morris AM, Asher A, Lillehei KO, Bernstein M, Brem H, Sloan A, et
al Glioma Outcomes Investigators: Survival following surgery and
prognostic factors for recently diagnosed malignant glioma: Data
from the Glioma Outcomes Project. J Neurosurg. 99:467–473. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stetson LC, Dazard JE and Barnholtz-Sloan
JS: Protein Markers Predict Survival in Glioma Patients. Mol Cell
Proteomics. 15:2356–2365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Woolf EC and Scheck AC: Metabolism and
glioma therapy. CNS Oncol. 1:7–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dupuy F, Tabariès S, Andrzejewski S, Dong
Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al:
PDK1-dependent metabolic reprogramming dictates metastatic
potential in breast cancer. Cell Metab. 22:577–589. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lohneis P, Darb-Esfahani S, Dietel M,
Braicu I, Sehouli J and Arsenic R: PDK1 is expressed in ovarian
serous carcinoma and correlates with improved survival in
high-grade tumors. Anticancer Res. 35:6329–6334. 2015.PubMed/NCBI
|
|
47
|
Wu YH, Chang TH, Huang YF, Chen CC and
Chou CY: COL11A1 confers chemoresistance on ovarian cancer cells
through the activation of Akt/c/EBPβ pathway and PDK1
stabilization. Oncotarget. 6:23748–23763. 2015.PubMed/NCBI
|
|
48
|
Tan J, Lee PL, Li Z, Jiang X, Lim YC, Hooi
SC and Yu Q: B55β-associated PP2A complex controls PDK1-directed
myc signaling and modulates rapamycin sensitivity in colorectal
cancer. Cancer Cell. 18:459–471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang Q, Yan HB, Wang J, Cui SJ, Wang XQ,
Jiang YH, Feng L, Yang PY and Liu F: Chromatin remodeling gene
AT-rich interactive domain-containing protein 1A suppresses gastric
cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget.
7:46127–46141. 2016.PubMed/NCBI
|
|
50
|
Ferro R and Falasca M: Emerging role of
the KRAS-PDK1 axis in pancreatic cancer. World J Gastroenterol.
20:10752–10757. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kobes JE, Daryaei I, Howison CM, Bontrager
JG, Sirianni RW, Meuillet EJ and Pagel MD: Improved treatment of
pancreatic cancer with drug delivery nanoparticles loaded with a
novel AKT/PDK1 inhibitor. Pancreas. 45:1158–1166. 2016. View Article : Google Scholar :
|
|
52
|
Hu Y, Sun H, Owens RT, Gu Z, Wu J, Chen
YQ, O’Flaherty JT and Edwards IJ: Syndecan-1-dependent suppression
of PDK1/ Akt/bad signaling by docosahexaenoic acid induces
apoptosis in prostate cancer. Neoplasia. 12:826–836. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Castel P, Ellis H, Bago R, Toska E, Razavi
P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et
al: PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation
and confers resistance to PI3Kα inhibition. Cancer Cell.
30:229–242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lin HJ, Hsieh FC, Song H and Lin J:
Elevated phosphorylation and activation of PDK-1/AKT pathway in
human breast cancer. Br J Cancer. 93:1372–1381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee
ST, Feng M, Lim CZ, Lee EY, Wee ZN, et al: PDK1 signaling toward
PLK1-MYC activation confers oncogenic transformation,
tumor-initiating cell activation, and resistance to mTOR-targeted
therapy. Cancer Discov. 3:1156–1171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Han L, Zhang G, Zhang N, Li H, Liu Y, Fu A
and Zheng Y: Prognostic potential of microRNA-138 and its target
mRNA PDK1 in sera for patients with non-small cell lung cancer. Med
Oncol. 31:1292014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Meng Q and Xia Y: c-Jun, at the crossroad
of the signaling network. Protein Cell. 2:889–898. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|