Introduction
Glioma is the most lethal type of primary malignant
brain tumor characterized by extreme proliferation and aggressive
invasion (1,2). The invasiveness of glioma cells is
the most difficult obstacle to combat in order to improve
prognosis. Diffuse infiltrative gliomas account for ~80% of all
malignant brain tumor types, which results in a poor prognosis
despite the advancements made in developing therapeutic strategies
for malignant glioma (3,4). Furthermore, heterogeneity is the
foremost feature of glioma, which makes the diagnosis and selection
of appropriate treatments more difficult. Therefore, the
identification of genes associated with glioma invasiveness and
heterogeneity has become a research focus. Over the last decade,
substantial progress has been made in elucidating the underlying
genetic causes of glioma (5). The
expression of RAB34, member RAS oncogene family confers a poor
prognosis in patients with high-grade glioma (6). Suppressor of cytokine signaling 3
promoter methylation promotes glioma cell invasion through signal
transducer and activator of transcription 3 and focal adhesion
kinase 1 activation (7,8). However, the role of histone
deacetylase 4 (HDAC4) in glioma cells has never been
investigated.
HDAC4, a member of the class IIa family of HDACs, is
known to undergo dynamic shuttling between the nucleus and
cytoplasm and to function as a transcriptional corepressor that has
been associated with a number of cellular and epigenetic processes
(9). Cohen et al (10) revealed that the differential
phosphorylation and localization of HDAC4 was conducive to setting
up fiber type-specific transcriptional programs. Mitogen-activated
protein kinase p38 promoted the degradation of HDAC4, which
released Runt related transcription factor 2 and contributed to
chondrocyte hypertrophy and bone formation (11). Accumulating evidence indicates that
HDAC4 serves a crucial role in tumorigenesis and is frequently
dysregulated in human malignancies (12). HDAC4 served a central role in
breast cancer growth and invasion. HDAC4 knockdown by RNA
interference inhibited breast cancer cell growth, migration and
invasion through downregulating Ki-67 and matrix
metalloproteinase-2 (MMP-2) (13).
The nuclear import of HDAC4 is required for melatonin-induced
apoptosis in colorectal cancer LoVo cells (14). HDAC4 displayed a significant
upregulation in multiple myeloma, and the downregulation of HDAC4
suppressed survival and migration and promoted apoptosis and
autophagy of multiple myeloma cells compared with multiple myeloma
cells with HDAC4 overexpression (P<0.05), respectively (15). HDAC4 may bind with RELB
proto-oncogene, nuclear factor-κβ subunit (RelB)-p52 forming an
HDAC4-RelB-p52 complex, which maintains repressive chromatin around
Bcl2 modifying factor and Bcl2 interacting mediator of cell death
and regulates the survival and growth of multiple myeloma cells.
Disruption of the HDAC4-RelB-p52 complex by a HDAC4-mimetic
polypeptide blocks the growth of multiple myeloma (16). Zeng et al (17) demonstrated that HDAC4 was
overexpressed in esophageal squamous cell carcinoma, and HDAC4
overexpression was associated with an advanced clinical stage and
poor survival. HDAC4 inhibition sensitized lung cancer A549 cells
to doxorubicin resistance by reducing the phosphorylation of signal
transducers and activators of transcription-1 (STAT1) and the
expression of epidermal growth factor receptor (EGFR), which
suggested an interaction between HDAC4, STAT1 and EGFR (18). In summary, HDAC4 has been revealed
to serve an important role in tumorigenesis, metastasis and
invasion. However, little is known regarding the specific function
of HDAC4 in glioma.
In the present study, glioma cell lines (U251,
U-87MG and LN-18) and the non-cancerous human glial cell line SVG
p12 cells were used to detect the expression of HDAC4 by western
blotting. The effects of HDAC4 on U251 cell proliferation,
adenosine triphosphate (ATP) and reactive oxygen species (ROS)
generation, the cell cycle and cell invasion were analyzed. In
addition, the effects of HDAC4 on the expression of cell
cycle-associated proteins p21, p27 and CDK1 were analyzed, and
epithelial-mesenchymal transition (EMT)-associated proteins
vimentin, E-cadherin and β-catenin in U251 cells. These data may be
useful in the prediction of glioma prognosis and the establishment
of targeted therapies.
Materials and methods
Specimens
A total of 12 paired glioma tissues and adjacent
non-tumor tissues were collected from patients (7 male and 5
female; aged 12-65 years, mean age, 39.4±16.1 years and 30.5±15.2
years, respectively) with glioma that underwent routine resection
surgery at the Department of Neurosurgery of the Affiliated
Hospital of Nantong University (Nantong, China) between January
2014 and November 2016. All patients had not received any other
treatment prior to surgery, including radiotherapy and
chemotherapy. The gliomas included 1 grade I, 3 grade II, 5 grade
III and 3 grade IV [2007 World Health Organization classification
of tumors of the central nervous system (19)]. All tissue specimens were obtained
with written informed consent and ethically approved by the Human
Ethics Committee of The Affiliated Hospital of Nantong University.
All tissue samples were immediately frozen in liquid nitrogen and
stored at -80°C for further experiments.
Materials
All cell culture reagents, including fetal bovine
serum, minimum essential medium (MEM), sodium pyruvate,
L-glutamine, penicillin, streptomycin and HEPES, were purchased
from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Human glioblastoma cell lines U251 and LN-18, U-87MG (from
glioblastoma of unknown origin which was authenticated by STR
profiling) and non-cancerous human glial cell line SVG p12 cells
[identified as being infected with BK polyomavirus strain UT
(20). There was no bearing on the
results obtained using SVG p12 cells] were purchased from the
American Type Culture Collection (Manassas, VA, USA). Radio
immunoprecipitation assay (RIPA) lysis buffer, bicinchoninic acid
(BCA) assay kit, Beyo electrochemiluminesence (ECL) moon,
polyvinylidene difluoride (PVDF) membrane, ROS assay kit, cell
counting kit-8, enhanced ATP assay kit and propidium iodide (PI)
were obtained from the Beyotime Institute of Biotechnology (Haimen,
China). Transwell invasion filter was supplied by Costar (Corning
Incorporated, Corning, NY, USA). Matrigel was purchased from
Collaborative Biomedical Products (Bedford, MA, USA). The
antibodies used in the present study include: Rabbit anti-HDAC4
polyclonal antibody (cat. no. 2072S; Cell Signaling Technology,
Inc., Danvers, MA, USA), rabbit anti-p21cyclin dependent
kinase (CDK) inhibitor 1A/WAF1, anti-p27Kip1/CDK
inhibitor 1B and anti-CDK1/cell division cycle 2 poly-clonal
antibodies (cat. nos. LS-C136937-100, LS-C17115-100 and
LS-C402186-60, respectively; LifeSpan BioSciences, Inc., Seattle,
WA, USA), rabbit anti-vimentin and anti-β-catenin poly-clonal
antibodies (cat. nos. PB9359 and PA1212, respectively; Boster
Biological Technology, Pleasanton, CA, USA), rabbit anti-E-cadherin
antibody (cat. no. ab155833; Abcam, Cambridge, UK), rabbit
anti-GAPDH antibody (cat. no. orb69587; Biorbyt Ltd., Cambridge,
UK), horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G secondary antibody polyclonal antibody (cat. no.
NBP2-30348H; Novus Biologicals, LLC, Littleton, CO, USA).
Cell culture and treatment
Human glioma cell lines (U251, U-87MG and LN-18) and
non-cancerous human fetal glial (SVG p12) cells were all cultured
in MEM complemented with 10% fetal bovine serum (FBS), 1 mM sodium
pyruvate, 2 mM L-glutamine, 50 U/ml penicillin, 50 μg/ml
streptomycin and 10 mM HEPES at 37°C in a humidified atmosphere
with 5% CO2.
U251 cells were selected to perform further studies.
A total of 2×105 U251 cells were cultured in a 6-well
plate with 2 ml antibiotic-free MEM medium containing 10% FBS. U251
cells, grown at 80% confluence, were transfected with HDAC4 small
interfering RNA (siRNA; 100 nM; cat. no. stB0001595C-1-5; Guangzhou
RiboBio Co., Ltd., Guangzhou, China) or pcDNA3.1-HDAC4 (Shanghai
GeneChem Co., Ltd, Shanghai, China) for 6 h at 37°C using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer’s protocol and
negative control (cat. no. siN05815122147-1-5; Guangzhou RiboBio
Co., Ltd.). Then U251 cells were incubated at 37°C in a
CO2 incubator for 4 h. Following this, the transfection
mixture was replaced with fresh MEM medium. After 48-h
transfection, U251 cells were used for further studies.
Western blot analysis
For western blot analysis, glioma tissues or cells
were lysed for 30 min at 4°C using RIPA lysis buffer containing
ethylenediaminetetraacetic acid-free protease inhibitor cocktail
and phosphatase inhibitor cocktails 1. The total proteins were
quantified through BCA protein concentration assay kit (Beyotime
Institute of Biotechnology), and 40 μg protein per lane was
resolved on 12% sodium dodecyl-sulfate polyacrylamide gel
electrophoresis, and transferred to a PVDF membrane through
wet-type transblotting apparatus (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Then the PVDF membrane was blocked with 5%
non-fat dry milk diluted in phosphate-buffered saline solution for
1 h at 4°C, and incubated with primary antibodies directed against
HDAC4 (1:2,000), p21 (1:2,000), p27 (1:2,000), CDK1 (1:2,000),
vimentin (1:2,000), E-cadherin (1:2,000), β-catenin (1:2,000) and
GAPDH (1:1,000) at 4°C overnight. Subsequently, the PVDF membrane
was washed three times with tris-buffered saline with 0.1% Tween-20
and incubated with the corresponding secondary antibody at a
1:5,000 dilution at room temperature for 2 h. Following this, the
PVDF membrane was visualized using BeyoECL moon. The results were
analyzed through Quantity One software V4.62 (Bio-Rad Laboratories,
Inc.) to obtain the optical density ratio of target protein to
GAPDH.
Cell counting kit-8 (CCK-8) assay
A CCK-8 assay was used to assess the effects of
HDAC4 on the proliferation of U251 cells. Briefly, 200 μl
U251 cells (2×103) were seeded in 96-well plates. At 0,
1, 2 and 4 days following transfection (overexpression or
knockdown), the U251 cells were incubated with 20 μl CCK-8
solution at 37°C for 2 h. The absorbance was examined at a
wavelength of 450 nm on a 96-well micro test spectrophotometer
(BioTek Instruments, Inc., Winooski, VT, USA). The experiments were
performed in triplicate.
Cell cycle analysis
The effects of HDAC4 on the cell cycle of U251 cells
were assessed using flow cytometry (with the flow cytometer
supplied by BD Biosciences, Franklin Lakes, NJ, USA). First, U251
cells were harvested using centrifugation for 10 min at 4°C and
1,200 × g, synchronized by incubation at 4°C in serum-free MEM
medium for 24 h and incubated with complete MEM medium for 24 h at
4°C. Subsequently, U251 cells were washed twice with PBS and fixed
with cold 70% ethanol at -20°C overnight. U251 cells were washed
twice with cold PBS and resuspended in PBS containing 100
μg/ml RNase A at 4°C for 30 min. Then the
resuspended U251 cells were further incubated with 100 μg/ml
PI at 4°C for 30 min. Finally, the cell cycle was analyzed through
flow cytometry. The analysis was performed in triplicate with
CELLQuestPro v5.1 (BD Biosciences).
ATP levels assay
Intracellular ATP levels were determined using an
ATP assay kit according to the manufacturer’s protocol. Briefly,
U251 cells were lysed with ATP detection lysis buffer for 2 min at
4°C and centrifuged at 12,000 × g for 5 min at 4°C. The
supernatants were then mixed with the working solution of ATP
detection reagent, and the relative fluorescence intensity was
immediately assessed using a luminometer (Thermo Scientific
Luminoskan Ascent; Thermo Fisher Scientific, Inc.). The
fluorescence intensity was normalized by per sample protein
content, which was measured through a BCA assay kit.
ROS measurement
Production of intracellular ROS was determined using
a reactive oxygen species assay kit according to the manufacturer’s
protocol. Briefly, U251 cells were seeded in 60-mm dishes and
allowed to attach overnight. Then 10 mM 2′,7′-dichlorofluorescin
diacetate (DCFH-DA) was added and incubated with the U251 cells for
20 min at 37°C. Serum-free cell culture medium was used to wash the
U251 cells three times to fully remove the inaccessible DCFH-DA.
Once the U251 cells were collected, the fluorescence in U251 cells
was assessed through a fluorescence spectrometer at 488 nm
(excitation wavelengths) and 525 nm (emission wavelengths). ROS
levels in U251 cells were expressed as relative intensity of DCF
fluorescence per sample protein content.
Cell invasion assays
The invasive ability of U251 cells was assessed
through 24 well Transwell permeable filters (Costar; Corning
Incorporated) with 8 μM pores. Briefly, the filter was
washed with serum-free MEM medium, and then the upper side of the
filter was evenly coated with 20 μl Matrigel (1:2 dilution
with MEM). The chamber was divided into the upper chamber and the
lower chamber. For invasion assays, subsequent to transfection with
HDAC4 siRNA, pcDNA3.1-HDAC4 or negative control, U251 cells
(1×105) were resuspended in serum free MEM medium and
seeded into the upper chamber of the Transwell invasion system. The
lower chamber was filled with complete medium. Following this, the
Transwell invasion system was incubated for 48 h at 37°C in an
incubator. Those U251 cells that invaded into the filter were
detached from filter by trypsinization and that invaded in the
lower chamber were collected and analyzed using an
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, U251 cells were incubated with MTT (5 mg/ml) for 4
h at 37°C. Then 150 μl dimethyl sulfoxide was added to
dissolve the crystals. The absorbance values were determined at 570
nm and 630 nm on a 96-well micro test spectrophotometer (BioTek
Instruments, Inc.). The analysis was performed in triplicate.
Statistical analysis
All data were presented as the mean ± standard error
of the mean from at least three independent experiments. All data
were analyzed using an unpaired Student’s t-test or one-way
analysis of variance followed by Dunnett’s test using SPSS17.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
HDAC4 displayed elevated expression in
glioma tissues and cell lines
The expression of HDAC4 protein was evaluated in
glioma by western blot analysis. First, HDAC4 expression was
analyzed in 12 paired glioma tissues and adjacent non-tumor tissues
and revealed that the expression of HDAC4 was significantly
upregulated in glioma tissues compared with glioma-adjacent normal
tissues (P<0.01; Fig. 1A).
Additionally, three glioma cell lines (U251, U-87MG and LN-18) were
analyzed. HDAC4 displayed higher expression in the three glioma
cell lines compared with the non-cancerous human glial cell line
SVG p12 (P<0.01; Fig. 1B).
These data revealed that the expression profile of HDAC4 in glioma
tissues coincided with that in glioma cell lines. In other words,
the results of the present study demonstrated that the expression
of HDAC4 was upregulated in glioma tissues and cell lines.
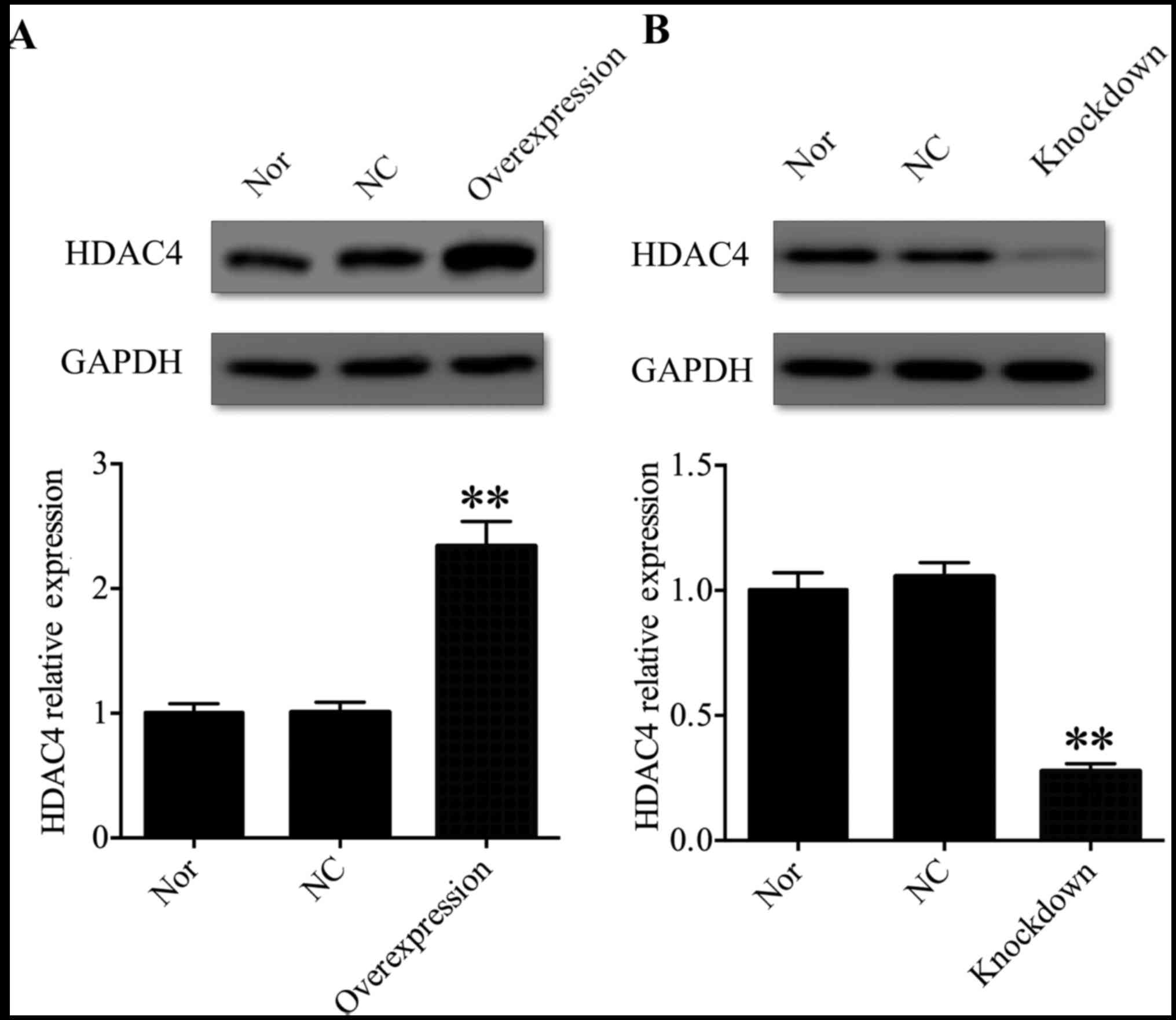
HDAC4 was successfully knocked down or
overexpressed in U251 cells
In order to address the function of HDAC4 in the
tumorigenesis and progression of glioma, U251 cells were selected
for further investigation. A pcDNA3.1-HDAC4 expression vector was
used to overexpress HDAC4 in U251 cells, and HDAC4 siRNA was used
to inhibit the expression of HDAC4 in U251 cells. Western blot
analysis was used to evaluate HDAC4 overexpression and
gene-silencing efficacy in U251 cells. The expression of HDAC4 was
significantly upregulated in the pcDNA3.1-HDAC4-transfected U251
cells compared with that in the non-transfected U251 cells or
negative control transfected U251 cells (P<0.01; Fig. 2A). Conversely, the expression of
HDAC4 was significantly downregulated in the HDAC4
siRNA-transfected U251 cells compared with the non-transfected U251
cells or negative control transfected U251 cells (P<0.01;
Fig. 2B). These data suggested
that HDAC4 was successfully knocked down or overexpressed in U251
cells.
HDAC4 promoted U251 cell
proliferation
In order to investigate the effect of HDAC4 on the
proliferation of glioma cells, CCK-8 analysis was performed at 24,
48 and 96 h. The proliferation curves demonstrated that when HDAC4
was overexpressed in U251 cells, the cell proliferation was
significantly increased 2.0-fold in comparison with U251 cells
transfected with empty pcDNA3.1 vector on day 4 (P<0.01;
Fig. 3A). Additionally, the effect
of HDAC4 knockdown on cell proliferation in U251 cells was
examined. The results revealed that HDAC4 knockdown significantly
suppressed the proliferation capacity of U251 cells (P<0.05;
Fig. 3B). In summary, these data
suggested that elevated HDAC4 may serve a crucial role in the
augmentation of the proliferation ability of U251 cells.
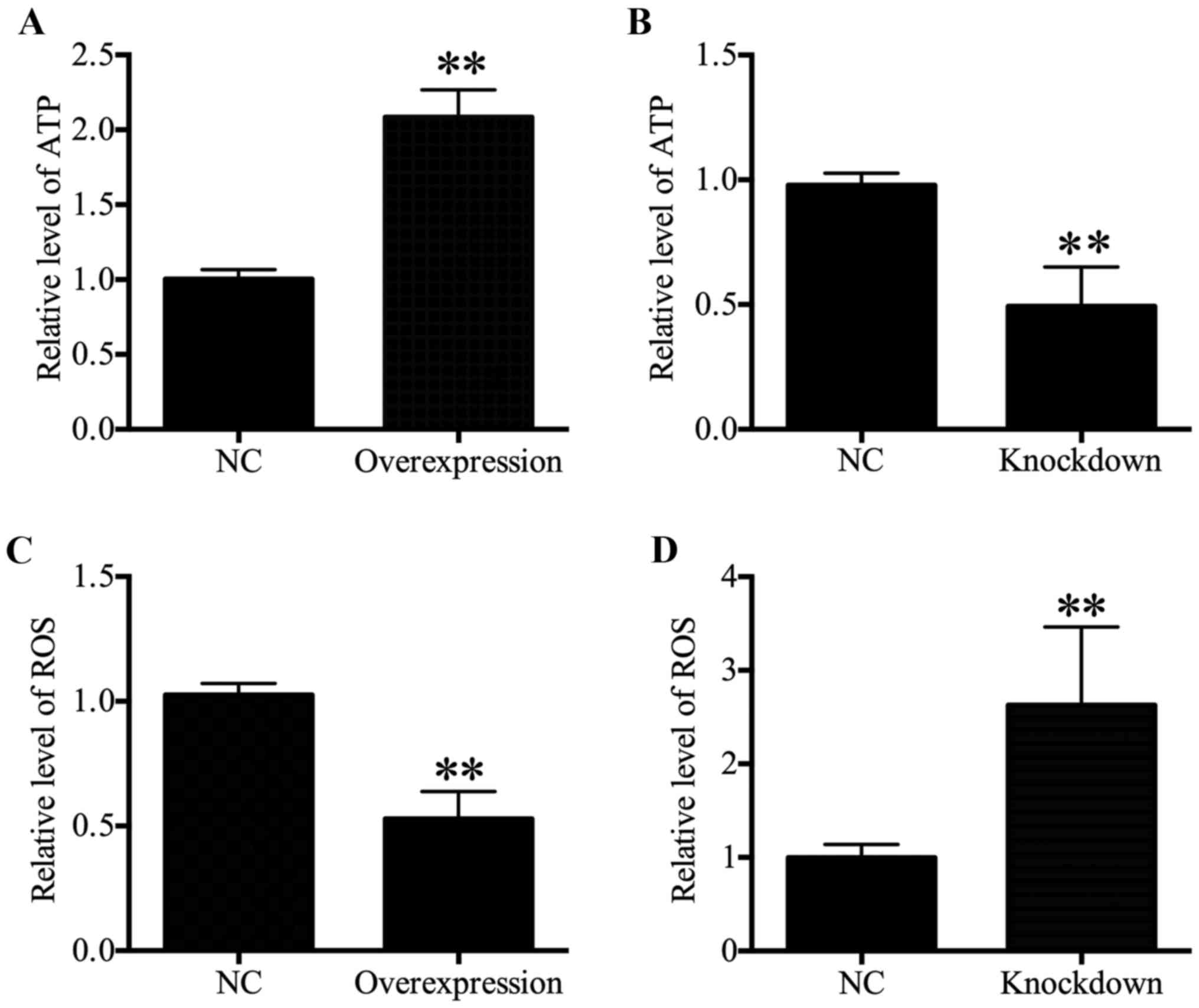
HDAC4 increased ATP levels and decreased
ROS generation
Intracellular ATP levels were associated with
proliferation (21-23). Therefore, ATP content was
determined in the present study, and results revealed that a
significant increase in the ATP content in HDAC4 overexpressing
U251 cells was observed compared with the U251 cells transfected
with empty pcDNA3.1 vector (P<0.01; Fig. 4A). Furthermore, the ATP content
reduced significantly in HDAC4 knockdown U251 cells (P<0.01;
Fig. 4B. These data indicated that
HDAC4 enhanced ATP generation. As intracellular ROS generation may
be associated with mitochondrial dysfunction and ATP generation
(24,25), the effects of HDAC4 on ROS
generation in U251 cells were further examined. The results
indicated that ROS content displayed a significant decrease in the
U251 cells transfected with pcDNA3.1-HDAC4 compared with the U251
cells transfected with empty pcDNA3.1 vector (P<0.01; Fig. 4C). Meanwhile, downregulation of
HDAC4 significantly activated ROS generation in U251 cells
(P<0.01; Fig. 4D). These
results demonstrated that HDAC4 inhibited ROS generation in U251
cells.
HDAC4 promoted U251 cell cycle
progression
In order to investigate whether the expression of
HDAC4 was associated with the cell cycle progression of glioma U251
cells, the cell cycle distribution of glioma U251 cells was
assessed by flow cytometry. Results demonstrated that HDAC4
overexpression exhibited a significant increase in the proportion
of U251 cells at the S and G2 phase, and a significant decrease in
the proportion of U251 cells at the G1 phase (P<0.05; Fig. 5A and B). On the contrary, there
were significantly fewer U251 cells at the S and G2 phase, and
significantly more U251 cells at the G1 phase in the HDAC4
knockdown group (P<0.05; Fig. 5A
and B), suggesting that HDAC4 knockdown induced U251 cells G1
arrest. The cell cycle distribution results demonstrated that HDAC4
served a central role in the promotion of U251 cell cycle
progression.
As p21, p27, CDK1 and CDK2 are associated with cell
proliferation and cell cycle (26-28),
the expression of p21, p27, CDK1 and CDK2 were determined in U251
cells. The present results revealed that cyclin-dependent kinase
inhibitors p21 and p27 displayed a significant decrease in the
HDAC4 overexpressing group, and a significant increase in HDAC4
knockdown group compared with their respective negative controls
(P<0.05; Fig. 6). On the
contrary, cyclin-dependent kinases CDK1 and CDK2 displayed an
opposite expression pattern to p21 and p27, which demonstrated that
the expression of CDK1 and CDK2 was significantly enhanced in the
HDAC4 overexpressing group, and was significantly inhibited in the
HDAC4 knockdown group (P<0.05; Fig.
6). These data indicated that HDAC4 enhanced the expression of
cyclin-dependent kinases CDK1 and CDK2, and suppressed the
expression of cyclin-dependent kinase inhibitors p21 and p27.
 | Figure 6Effect of HDAC4 on the expression of
p21, p27, CDK1 and CDK2 in U251 cells. (A) Expression of p21, p27,
CDK1 and CDK2 in U251 cells transfected with pcDNA3.1-HDAC4 or
empty pcDNA3.1 vector, and (B) quantified western blot analysis
results. (C) Expression of p21, p27, CDK1 and CDK2 in U251 cells
transfected with HDAC4 siRNA or siRNA negative control, and (D)
quantified western blot analysis results. Overexpression indicates
the U251 cells transfected with pcDNA3.1-HDAC4 and knockdown
indicates the U251 cells transfected with HDAC4 siRNA. Data were
expressed as the mean ± standard error of the mean.
*P<0.05 and **P<0.01 vs. NC. HDAC4,
histone deacetylase 4; NC, negative control; CDK1, cyclin-dependent
kinase 1; CDK2, cyclin-dependent kinase 2; siRNA, small interfering
RNA. |
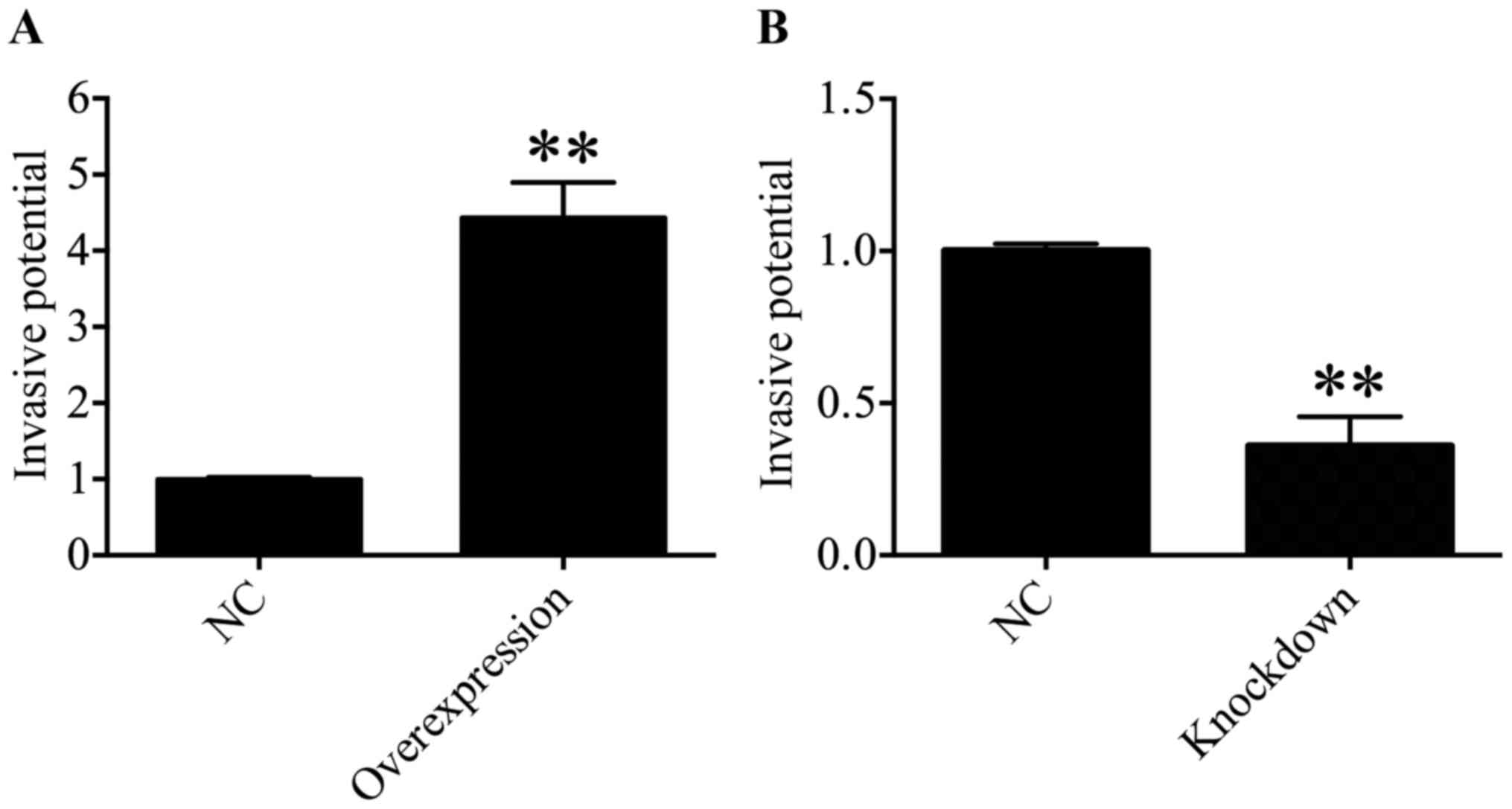
HDAC4 promoted the invasiveness of U251
cells
Subsequently, the function of HDAC4 in the
invasiveness of U251 cells was determined through a Transwell
invasion assay. The results suggested that the invasive ability of
U251 cells was significantly enhanced in the HDAC4 overexpression
group compared with in the empty pcDNA3.1 vector group (P<0.01;
Fig. 7A). On the contrary, the
downregulation of HDAC4 exhibited a significant decrease in the
invasive capacity of U251 cells compared with the siRNA negative
control group (P<0.01; Fig.
7B). These data suggested that HDAC4 strengthened the invasive
ability of U251 cells.
Altogether, these results indicated thee functional
importance of HDAC4 in the invasiveness of U251 cells. Therefore,
whether there exists any evidence for these molecular events in the
invasiveness of U251 cells was questioned. Park et al
(29) demonstrated that HDAC4 may
activate the EMT in airway epithelial cells, and that HDAC4
knockdown may reduce EMT. Therefore, EMT markers, including
vimentin, β-catenin and E-cadherin, were investigated and results
indicated that the expression of vimentin was significantly
upregulated in the HDAC4 overexpression group and was significantly
downregulated in the HDAC4 knockdown group compared with their
respective negative controls (P<0.01; Fig. 8). Nevertheless, the expression of
E-cadherin and β-catenin was negatively associated with the
expression of vimentin expression, which revealed that the
expression of E-cadherin and β-catenin was significantly reduced in
the HDAC4 overexpression group and was significantly enhanced in
the HDAC4 knockdown group compared with the negative control
(P<0.01; Fig. 8). These data
indicated that the aberrant expression of vimentin, β-catenin and
E-cadherin were potential markers of the malignant transformation
of glioma.
Discussion
Glioma is the most lethal type of primary malignant
brain tumor. Although the current treatment modalities have
progressed substantially, the recurrence of glioma remains a
therapeutic challenge (30).
Therefore, the survival of patients with glioma remains
unsatisfactory. This is mainly due to the fact that the molecular
mechanisms of glioma are unclear (31,32).
Growing evidence has revealed that HDAC4 was frequently
dysregulated in human malignancies (12-15).
HDAC5 are increased in human glioma tissues and promoted the
proliferation of glioma cells by the upregulation of Notch 1
(33). However, the involvement of
HDAC4 in gliomagenesis, migration and invasiveness remains
elusive.
In the present study, the results revealed that the
expression of HDAC4 was upregulated in glioma tissues and cell
lines, which was consistent with the expression profile of HDAC4 in
gastric cancer, colorectal cancer, lung cancer and ovarian cancer
(14,34-37).
These data indicated that elevated HDAC4 may serve an important
role in gliomagenesis and progression. In order to address the
function of HDAC4 in glioma cells, the glioma U251 cells were
selected for further investigation. Furthermore, HDAC4
overexpression or knockdown was performed in U251 cells. Western
blot analysis revealed that HDAC4 displayed a higher expression in
the HDAC4 overexpression group and a lower expression in the HDAC4
knockdown group, which suggested that HDAC4 was successfully
overexpressed or knockdown in U251 cells. Following this, the
effects of HDAC4 on the cell proliferation, ATP level, ROS
generation, cell cycle and invasion of U251 cells were determined.
The results of the present study revealed that the HDAC4 may serve
a crucial role in augmenting the proliferation ability of U251
cells and in the promotion of U251 cell cycle progression, which
was consistent with and confirmed by previous studies from Zeng
et al (17) and Kang et
al (17,34). Zeng et al revealed that
HDAC4 was overexpressed in esophageal squamous cell carcinoma, and
that HDAC4 overexpression was associated with advanced clinical
stage and poor survival (17).
Kang et al revealed that HDAC4 was upregulated in gastric
cancer and that elevated HDAC4 promoted the proliferation of
SGC-7901 cells, but HDAC4 knockdown inhibited the proliferation of
SGC-7901 cells, resulting in cell cycle arrest at G0/G1 phase in
SGC-7901 cells (34). As
intracellular ATP levels were associated with cell proliferation
and intracellular ROS generation was associated with mitochondrial
dysfunction and served an important regulatory role in cell death
and proliferation (21-25,38),
the present study therefore examined the effects of HDAC4 on
intracellular ATP levels and ROS generation in U251 cells. The
results of the present study indicated that HDAC4 enhanced ATP
generation, but inhibited ROS generation in U251 cells, which was
also consistent with and confirmed by a previous study on gastric
cancer (34). Elevated HDAC4 in
gastric cancer promoted the ATP levels in gastric cancer cells, but
HDAC4 knockdown inhibited the ATP levels of gastric cancer cells,
resulting in cell cycle arrest at the G0/G1 phase and ROS increase
in gastric cancer cells (34).
Thus, it was speculated that lower ATP levels and higher ROS levels
may prevent the proliferation and tumorigenesis. The present data
indicated that HDAC4 improved the cell proliferation and cycle
progression of U251 cells through enhancing the ATP generation and
suppressing the ROS generation.
Cyclin-dependent kinase inhibitors (p21 and p27) and
cyclin-dependent kinases (CDK1 and CDK2) were associated with cell
proliferation and the cell cycle (26-28).
The present data indicated that HDAC4 enhanced the expression of
cyclin-dependent kinases CDK1 and CDK2, and suppressed the
expression of cyclin-dependent kinase inhibitors p21 and p27, which
was also consistent with and confirmed by previous studies. Zeng
et al (17) demonstrated
that HDAC4 promoted the proliferation of esophageal carcinoma cell
and G1/S cell cycle progression by inhibiting p21 and p27 and
upregulating CDK2 and CDK4. Mottet et al (39) reported that HDAC4 participated in
the repression of p21 in a human glioblastoma model. These data
suggested that HDAC4 improved the cell proliferation and cycle
progression of U251 cells through enhancing the expression of CDK1
and CDK2 and suppressing the expression of p21 and p27, and was
accompanied by an increase in ATP levels and reduced ROS levels.
However, the potential regulatory mechanism of HDAC4 in the
expression of CDK1, CDK2, p21 and p27 also needs to be investigated
further.
Glioma is characterized by diffuse infiltration
(1,2,4).
Therefore, the present study determined the effects of HDAC4 on the
invasiveness of U251 cells and the results revealed that HDAC4
strengthened the invasive ability of U251 cells. The results were
confirmed by the study by Park et al (40), which demonstrated that the high
expression of HDAC4 was associated with the high invasion of
MDA-MB-231 cells. Ahn et al (41) revealed that the repression of MMP-2
was associated with the reduction of HDAC4 in ovarian cancer.
Generally speaking, these data suggested the functional importance
of HDAC4 in the invasiveness of U251 cells. One previous study
demonstrated that HDAC4 may activate EMT in airway epithelial
cells, and that HDAC4 knockdown may reduce EMT (29). Therefore, EMT markers, including
vimentin, β-catenin and E-cadherin, were investigated and the
results revealed that HDAC4 enhanced the expression of vimentin and
suppressed the expression of E-cadherin and β-catenin in U251
cells. These data indicated that the aberrant expression of
vimentin, β-catenin and E-cadherin were potential markers of the
malignant transformation of glioma. However, the potential
regulatory mechanism of HDAC4 in the expression of vimentin,
β-catenin and E-cadherin also needs to be investigated further.
In addition, there are a number of shortcomings in
the present study. First, the small sample size is a limitation of
the study, and hence it is not possible to analyze the association
between different grades and the expression of HDAC4 in glioma.
Future studies will be conducted with a larger sample size to
investigate this, which will substantiate the present study.
Second, the expression of HDAC4 was detected only by western blot
analysis; a morphological examination, for example
immunohistochemistry, will be performed in future studies.
Additionally, the present study only examined the protein
expression of HDAC4 and relevant genes. In future studies, the mRNA
level of the HDAC4 and relevant genes will be detected, which may
further clarify whether abnormalities of these genes emerge in
transcription and translation, or the translation only. All of this
will render the present research conclusions more reliable.
In summary, the data generated by the present study
demonstrated that HDAC4 was significantly upregulated in glioma,
and promoted glioma cell proliferation and invasion mediated
through, at least partially, the repression of p21, p27, E-cadherin
and β-catenin, and by the potentiation of CDK1, CDK2 and vimentin
(Fig. 9). Altogether, the present
study revealed that HDAC4 overexpression was central for the
tumorigenesis of glioma, which may serve as a useful prognostic
biomarker and potential therapeutic target for glioma.
Funding
The present study was supported by the Fund of
Doctoral Start-up of Nantong University (grant no. 15B27) and the
Nantong Science and Technology Department (key technology research
project no. MS2201515110).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
JYC and XJN designed the study. JYC, TTX, YW, JJC,
JL, XYC, XC and YFY performed the experiments. JJC, JL, XYC, XC and
YFY collected and assembled the data. JYC and XJN performed the
data analysis. XJN provided scientific expertise. JYC and XJN wrote
the manuscript.
Ethics approval and consent to
participate
This study was ethically approved by the Human
Ethics Committee of The Affiliated Hospital of Nantong University
(Nantong, China).
Patient consent for publication
All patients agreed to publication and provided
written informed consent.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Farshidfar Z, Faeghi F, Mohseni M,
Seddighi A, Kharrazi HH and Abdolmohammadi J: Diffusion tensor
tractography in the presurgical assessment of cerebral gliomas.
Neuroradiol J. 27:75–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kazakova MH, Staneva DN, Koev IG, Staikov
DG, Mateva N, Timonov PT, Miloshev GA and Sarafian VS: Protein and
mRNA levels of YKL-40 in high-grade glioma. Folia Biol (Praha).
60:261–267. 2014.
|
|
3
|
Chen S, Han M, Chen W, He Y, Huang B, Zhao
P, Huang Q, Gao L, Qu X and Li X: KIF1B promotes glioma migration
and invasion via cell surface localization of MT1-MMP. Oncol Rep.
35:971–977. 2016. View Article : Google Scholar
|
|
4
|
Ortega-Aznar A, Jimenez-Leon P, Martinez E
and Romero-Vidal FJ: Clinico-pathological and molecular aspects of
diagnostic and prognostic value in gliomas. Rev Neurol. 56:161–170.
2013.In Spanish. PubMed/NCBI
|
|
5
|
Waitkus MS, Diplas BH and Yan H:
Isocitrate dehydrogenase mutations in gliomas. Neurooncol.
18:16–26. 2016.
|
|
6
|
Lin M, Zhu Q, Wang J, Yang W, Fan H, Yi J
and Jiang M: Molecules involved in acrosomal exocytosis and
cortical granule exocytosis. Biotarget. 1:112017. View Article : Google Scholar
|
|
7
|
Lindemann C, Hackmann O, Delic S, Schmidt
N, Reifenberger G and Riemenschneider MJ: SOCS3 promoter
methylation is mutually exclusive to EGFR amplification in gliomas
and promotes glioma cell invasion through STAT3 and FAK activation.
Acta Neuropathol. 122:241–251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fang Q, Xu T, Wu C, Zhou S and Sun H:
Biotargets in neural regeneration. Biotarget. 1:62017. View Article : Google Scholar
|
|
9
|
Crow M, Khovanov N, Kelleher JH, Sharma S,
Grant AD, Bogdanov Y, Wood JN, McMahon SB and Denk F: HDAC4 is
required for inflammation-associated thermal hypersensitivity.
FASEB J. 29:3370–3378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cohen TJ, Choi MC, Kapur M, Lira VA, Yan Z
and Yao TP: HDAC4 regulates muscle fiber type-specific gene
expression programs. Mol Cells. 38:343–348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou J, Li P, Chen Q, Wei X, Zhao T, Wang
Z and Wei L: Mitogen-activated protein kinase p38 induces HDAC4
degradation in hypertrophic chondrocytes. Biochim Biophys Acta.
1853:370–376. 2015. View Article : Google Scholar :
|
|
12
|
Xiao H, Jiao J, Wang L, O’Brien S, Newick
K, Wang LC, Falkensammer E, Liu Y, Han R, Kapoor V, et al: HDAC5
controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells.
Int J Cancer. 138:2477–2486. 2016. View Article : Google Scholar
|
|
13
|
Hsieh TH, Hsu CY, Tsai CF, Long CY, Chai
CY, Hou MF, Lee JN, Wu DC, Wang SC and Tsai EM: miR-125a-5p is a
prognostic biomarker that targets HDAC4 to suppress breast
tumorigenesis. Oncotarget. 6:494–509. 2015. View Article : Google Scholar :
|
|
14
|
Wei JY, Li WM, Zhou LL, Lu QN and He W:
Melatonin induces apoptosis of colorectal cancer cells through
HDAC4 nuclear import mediated by CaMKII inactivation. J Pineal Res.
58:429–438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Amodio N, Stamato MA, Gullà AM, Morelli E,
Romeo E, Raimondi L, Pitari MR, Ferrandino I, Misso G, Caraglia M,
et al: Therapeutic targeting of miR-29b/HDAC4 epigenetic loop in
multiple myeloma. Mol Cancer Ther. 15:1364–1375. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vallabhapurapu SD, Noothi SK, Pullum DA,
Lawrie CH, Pallapati R, Potluri V, Kuntzen C, Khan S, Plas DR,
Orlowski RZ, et al: Transcriptional repression by the
HDAC4-RelB-p52 complex regulates multiple myeloma survival and
growth. Nat Commun. 6:84282015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeng LS, Yang XZ, Wen YF, Mail SJ, Wang
MH, Zhang MY, Zheng XF and Wang HY: Overexpressed HDAC4 is
associated with poor survival and promotes tumor progression in
esophageal carcinoma. Aging (Albany NY). 8:1236–1249. 2016.
View Article : Google Scholar
|
|
18
|
Kaowinn S, Jun SW, Kim CS, Shin DM, Hwang
YH, Kim K, Shin B, Kaewpiboon C, Jeong HH, Koh SS, et al: Increased
EGFR expression induced by a novel oncogene, CUG2, confers
resistance to doxorubicin through Stat1-HDAC4 signaling. Cell Oncol
(Dordr). 40:549–561. 2017. View Article : Google Scholar
|
|
19
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Henriksen S, Tylden GD, Dumoulin A, Sharma
BN, Hirsch HH and Rinaldo CH: The human fetal glial cell line SVG
p12 contains infectious BK polyomavirus. J Virol. 88:7556–7568.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
George J, Gonçalves FQ, Cristóvão G,
Rodrigues L, Meyer Fernandes JR, Gonçalves T, Cunha RA and Gomes
CA: Different danger signals differently impact on microglial
proliferation through alterations of ATP release and extracellular
metabolism. Glia. 63:1636–1645. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kühn F: New insights into the interaction
between ADP-ribose and human TRPM2 channel. Biotarget. 1:142017.
View Article : Google Scholar
|
|
23
|
Almontashiri NA, Chen HH, Mailloux RJ,
Tatsuta T, Teng AC, Mahmoud AB, Ho T, Stewart NA, Rippstein P,
Harper ME, et al CARDIoGRAM Consortium: SPG7 variant escapes
phosphorylation-regulated processing by AFG3L2, elevates
mitochondrial ROS, and is associated with multiple clinical
phenotypes. Cell Reports. 7:834–847. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang K, Wang W, Jin X, Wang Z, Ji Z and
Meng G: Silibinin, a natural flavonoid, induces autophagy via
ROS-dependent mitochondrial dysfunction and loss of ATP involving
BNIP3 in human MCF7 breast cancer cells. Oncol Rep. 33:2711–2718.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Van de Wouwer M, Couzinié C,
Serrano-Palero M, González-Fernández O, Galmés-Varela C,
Menéndez-Antolí P, Grau L and Villalobo A: Activation of the
BRCA1/Chk1/p53/p21(Cip1/ Waf1) pathway by nitric oxide and cell
cycle arrest in human neuroblastoma NB69 cells. Nitric Oxide.
26:182–191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang J, Wang G and Khan MF: Disorder of
G2-M Checkpoint control in aniline-induced cell proliferation in
rat spleen. PLoS One. 10:e01314572015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yi X, Li Y, Zai H, Long X and Li W: KLF8
knockdown triggered growth inhibition and induced cell phase arrest
in human pancreatic cancer cells. Gene. 585:22–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park IH, Kang JH, Shin JM and Lee HM:
Trichostatin A inhibits epithelial mesenchymal transition induced
by TGF-β1 in airway epithelium. PLoS One. 11:e01620582016.
View Article : Google Scholar
|
|
30
|
Tu Y, Gao X, Li G, Fu H, Cui D, Liu H, Jin
W and Zhang Y: MicroRNA-218 inhibits glioma invasion, migration,
proliferation, and cancer stem-like cell self-renewal by targeting
the polycomb group gene Bmi1. Cancer Res. 73:6046–6055. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Le Rhun E, Taillibert S and Chamberlain
MC: Anaplastic glioma: Current treatment and management. Expert Rev
Neurother. 15:601–620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wirsching HG, Happold C, Roth P and Weller
M: Management of diffusely infiltrating glioma in the elderly. Curr
Opin Oncol. 27:502–509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Q, Zheng JM, Chen JK, Yan XL, Chen HM,
Nong WX and Huang HQ: Histone deacetylase 5 promotes the
proliferation of glioma cells by upregulation of Notch 1. Mol Med
Rep. 10:2045–2050. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kang ZH, Wang CY, Zhang WL, Zhang JT, Yuan
CH, Zhao PW, Lin YY, Hong S, Li CY and Wang L: Histone deacetylase
HDAC4 promotes gastric cancer SGC-7901 cells progression via p21
repression. PLoS One. 9:e988942014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kaewpiboon C, Srisuttee R, Malilas W, Moon
J, Oh S, Jeong HG, Johnston RN, Assavalapsakul W and Chung YH:
Upregulation of Stat1-HDAC4 confers resistance to etoposide through
enhanced multidrug resistance 1 expression in human A549 lung
cancer cells. Mol Med Rep. 11:2315–2321. 2015. View Article : Google Scholar
|
|
36
|
Isaacs JT, Antony L, Dalrymple SL, Brennen
WN, Gerber S, Hammers H, Wissing M, Kachhap S, Luo J, Xing L, et
al: Tasquinimod Is an aAllosteric modulator of HDAC4 survival
signaling within the compromised cancer microenvironment. Cancer
Res. 73:1386–1399. 2013. View Article : Google Scholar
|
|
37
|
Stronach EA, Alfraidi A, Rama N, Datler C,
Studd JB, Agarwal R, Guney TG, Gourley C, Hennessy BT, Mills GB, et
al: HDAC4-regulated STAT1 activation mediates platinum resistance
in ovarian cancer. Cancer Res. 71:4412–4422. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen EI: Mitochondrial dysfunction and
cancer metastasis. J Bioenerg Biomembr. 44:619–622. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mottet D, Pirotte S, Lamour V, Hagedorn M,
Javerzat S, Bikfalvi A, Bellahcène A, Verdin E and Castronovo V:
HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells
through a Sp1-dependent, p53-independent mechanism. Oncogene.
28:243–256. 2009. View Article : Google Scholar
|
|
40
|
Park SY, Jun JA, Jeong KJ, Heo HJ, Sohn
JS, Lee HY, Park CG and Kang J: Histone deacetylases 1, 6 and 8 are
critical for invasion in breast cancer. Oncol Rep. 25:1677–1681.
2011.PubMed/NCBI
|
|
41
|
Ahn MY, Kang DO, Na YJ, Yoon S, Choi WS,
Kang KW, Chung HY, Jung JH, Min S and Kim HS: Histone deacetylase
inhibitor, apicidin, inhibits human ovarian cancer cell migration
via class II histone deacetylase 4 silencing. Cancer Lett.
325:189–199. 2012. View Article : Google Scholar : PubMed/NCBI
|