Introduction
Esophageal cancer is the eighth most prevalent type
of cancer worldwide (1,2), of which, esophageal squamous cell
carcinoma (ESCC) is a predominant histological type (3). Despite advances in diagnostic tools,
surgical techniques and chemotherapy over the past few decades, the
5-year survival rate for patients with esophageal cancer ranges
between 15 and 20% (4). Therefore,
novel diagnostic tools, therapeutic strategies and molecular
prognostic markers are urgently required for this disease.
Eph receptors are the largest known family of
receptor tyrosine kinases, which participate in vital functions,
including cell migration and axon guidance during development and
homeostasis (5). Eph receptors and
ephrin ligands tend to be highly expressed during development. In
addition, Eph receptors have been reported to be aberrantly
expressed in numerous types of cancer (6-8).
Epithelial-mesenchymal transition (EMT) is an evolutionarily
conserved developmental process, during which epithelial cells lose
polarity and develop a mesenchymal phenotype. EMT progression
triggers the dissociation of carcinoma cells from the primary
tumor; these cells subsequently migrate and disseminate to distant
sites (9). Eph receptor A3 (EphA3)
is a member of the Eph family of receptors, which is highly
expressed in embryonic tissues (10,11)
and appears to serve a critical role in EMT (12). Overexpression of EphA3 has been
detected in some types of cancer, including lung cancer, leukemia,
melanoma, lymphoma and gastric carcinoma (10,13-16).
Conversely, EphA3 mutations have also been identified, thus
suggesting a tumor-suppressor role for EphA3 in other types of
cancer, including breast, colorectal and lung cancer (17-19).
Although EphA3 deletions have been identified in patients with ESCC
and in ESCC cell lines (20,21),
the role of EphA3 in ESCC remains unknown.
The present study aimed to investigate the
expression levels and functions of EphA3 in ESCC. The results
indicated that the expression levels of EphA3 were decreased in
ESCC, and its forced overexpression in the ESCC cells (KYSE450 and
KYSE510) triggered mesenchymal-epithelial transition (MET), and
inhibited cell migration and invasion. These results suggested that
EphA3 may serve a tumor-suppressor role in ESCC.
Materials and methods
Reagents and tissues
β-actin mouse monoclonal (cat. no. sc-130065) and
EphA3 rabbit polyclonal antibodies (cat. no. sc-919) were obtained
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). RhoA rabbit
antibody (cat. no. E11-0568B) was obtained from Enjing Biotech Co.,
Ltd. (Nanjing, China). Antibodies for zonula occludens (ZO)-1 (cat.
no. 5406), Snail (cat. no. 3879), Vimentin (cat. no. 3932),
E-cadherin (cat. no. 14472), N-cadherin (cat. no. 14215) and
horseradish peroxidase-linked anti-mouse/anti-rabbit immunoglobulin
G (cat. nos. 7076 and 7074) were all purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). ESCC and adjacent
non-cancerous tissue specimens were collected from patients who
underwent surgical treatment for ESCC between July 2010 and
November 2013 at Changhai Hospital (Shanghai, China). Tissue
specimens were obtained from patients who had not received
preoperative treatment, such as chemotherapy or radiotherapy. The
patients comprised eight men and seven women, with a median age of
62.2 years (range, 52-74 years). The present study was approved by
the Ethics Committee of the Second Military Medical University
(Shanghai, China). Written informed consent was obtained from each
patient. Prior to initiation of the study, histopathological
examinations were performed to confirm that there were enough
cancer cells in the tumor samples and that no cancer cells
contaminated the non-cancerous tissues.
Cell culture
The human ESCC cell lines (KYSE450, KYSE30, KYSE410
and KYSE510) were kindly provided by Professor Tao Qian (Chinese
University of Hong Kong, Hong Kong, China). These cells were
maintained in Dulbecco's modified Eagle's medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and RPMI-1640 (Gibco;
Thermo Fisher Scientific, Inc.) in a 1:1 ratio, supplemented with
10% fetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and 100 U/ml penicillin, 100 µg/ml streptomycin (Gibco;
Thermo Fisher Scientific, Inc.). All cells were cultured in a
humidified atmosphere containing 7% CO2 at 37˚C.
Generation of stable cell lines
For the production of stable cells, the full-length
coding sequence of human EphA3 (GenBank accession number,
NM_005233; https://www.ncbi.nlm.nih.gov/genbank/) was cloned into
Lenti-X lentiviral expression vectors (Clontech Laboratories, Inc.,
Mountainview, CA, USA). The recombinant lentiviruses or control
(empty vector) were then used to infect KYSE450 and KYSE510 cells
grown to 30% confluence at multiplicity of infection 50. Infected
cells were cultured for 24 h prior to selection using a
pre-optimized dose of 2 µg/ml puromycin (Invitrogen; Thermo Fisher
Scientific, Inc.) for 2 weeks. Morphological alterations of the
infected cells were observed by using an Olympus IX71 fluorescence
microscope (Olympus Corporation, Tokyo, Japan).
Plasmid construction
The full-length EphA3 cDNA was cloned into Lenti-X
lentiviral expression vectors (Clontech Laboratories, Inc.).
Tyrosine residues Y596, Y602, Y779 and lysine residue K653 were
mutated using site-directed mutagenesis by overlapping polymerase
chain reaction (PCR), as previously described (22). The recombinant lentiviruses were
then used to infect KYSE510 cells grown to 30% confluence at
multiplicity of infection 50. Infected cells were cultured for 24 h
at 37˚C prior to selection using a pre-optimized dose of 2 µg/ml
puromycin (Invitrogen; Thermo Fisher Scientific, Inc.) for 2 weeks,
in order to generate stably infected cell lines.
EphA3 knockdown by RNA interference
Short hairpin (sh) RNA-mediated EphA3 silencing was
performed in KYSE410 cells using BLOCK-iT™ lentiviral RNAi
expression system (Invitrogen; Thermo Fisher Scientific, Inc.)
after packaging in 293FT cells (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacture's protocol. The
recombinant lentiviruses or control were then used to infect
KYSE410 cells grown to 30% confluence at a multiplicity of
infection 50 at 37˚C for 72 h. A respective scramble control shRNA
(5'-GATCCCCGTACGCGGAATACTTCGATTCAAGAGATCGAAGTATTCCGCGTACGTTTTTA-3')
was also used. The sequences were as follows: sh1,
5'-GCCCATTTACAGTGAAGAATCcgaaGATTCTTCACTGTAAATGGGC-3'; sh2, 5'-GC
AGGTGTGAGAATAATTACTcgaaAGTAATTATTCT CACACCTGC-3'; and sh3,
5'-GGAAAGATGTTACCTTCAACAcgaaTGTTGAAGGTAACATCTTTCC-3'; lower-case
letters indicate linker sequences.
5-Aza-2'-deoxycytidine (5-Aza-dC)
treatment
KYSE510 and KYSE30 cell lines were split to obtain
cells with a low confluence (30%) 12 h prior to treatment.
Subsequently, cells were treated with 5-Aza-dC (Sigma-Aldrich;
Merck KGaA), at a concentration of 5 µM, in a humidified atmosphere
containing 7% CO2 at 37˚C. All media were replenished
daily, and cells were harvested after 3 or 4 days of treatment.
Immunocytochemistry
For immunocytochemistry, cells were grown on
coverslips to 80% confluence and were washed with PBS, fixed with
4% paraformaldehyde in PBS for 15 min at 4˚C and permeabilized with
0.2% Triton X-100 for 10 min at room temperature. Subsequently,
cells were blocked for 60 min at room temperature in PBS/0.1%
bovine serum albumin (BSA; Roche Diagnostics, Minneapolis, MN,
USA). Cells were then incubated with the EphA3 antibody (cat. no.
sc-514209; Santa Cruz Biotechnology, Inc.) at a dilution of 1:50
for 1 h at 4˚C, washed with PBS and incubated with Alexa
Fluor® 488-labeled secondary antibody (cat. no.
ab150113; Abcam) at a dilution of 1:200 for 1 h at 4°C. For actin
staining, permeabilized cells were stained with rhodamine-labeled
phalloidin (0.1 µg/ml; Cytoskeleton, Inc., Denver, CO, USA) for 1 h
at room temperature. Stained cells were viewed under a fluorescence
microscope (Olympus IX71; Olympus Corporation).
Migration and invasion assays
Cell migration and invasion assays were performed
using Transwell chambers (pore size, 8 µm; Corning Incorporated,
Corning, NY, USA) with or without a thin coating of Matrigel
(Sigma-Aldrich; Merck KGaA). Briefly, 1×105 cells were
seeded into the upper chamber of the plate in serum-free medium.
Medium containing 10% FBS was used as chemoattractant in the lower
chamber. After 28 and 50 h, respectively, migration and invasion
were evaluated by measuring the number of cells that had moved into
the lower chambers. Cells that remained on the upper chamber were
removed using a cotton swab, after which, the membranes were fixed
in ice-cold methanol for 1 h at 4°C and stained with 0.5% crystal
violet for 30 min at room temperature. Migrated/invasive cells on
the underside of the filter were counted in five random fields at
×200 magnification using an Olympus IX71 microscope (Olympus
Corporation), and the mean number of cells was calculated. The
experiments were performed in triplicate.
Protein extraction and western
blotting
Cells were lysed with ice-cold
radioimmunoprecipitation assay buffer (50 mM Tris, pH 7.4; 150 mM
NaCl; 1% sodium deoxycholate; 1% sodium dodecyl sulfate; 1% NP-40)
supplemented with protease inhibitor cocktail (Roche Diagnostics).
Cell lysates were incubated on ice for 0.5 h and insoluble proteins
were removed by centrifugation at 10,000 x g for 10 min at 4°C.
Protein concentrations were determined using the Bicinchoninic Acid
Protein Assay kit (Beyotime Institute of Biotechnology, Haimen,
China). Protein samples (20-50 µg) were separated by SDS-PAGE (5%
stacking and 10% separating gels) and were transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were then blocked for 1 h at room temperature
using 5% BSA, and the proteins were detected with specific primary
antibodies and peroxidase-conjugated secondary antibodies at the
dilutions recommended by the manufacturers, according to the
protocols (Cell Signaling Technology, Inc.). Blots were visualized
using the Chemiluminescent Substrate kit (Thermo Fisher Scientific,
Inc.) and the Gel Doc™ XR system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Detection of GTP-bound RhoA
The RhoA-binding domain (RBD) of the rhotekin
protein was amplified by PCR from the MDA-MB-231 cell line cDNA
bank (contained in our laboratory at the International Joint Cancer
Institute, Second Military Medical University), cloned into the
pGEX-4T-2 expression vector (Amersham; GE Healthcare, Chicago, IL,
USA), and was expressed as a glutathione S-transferase (GST) fusion
protein in BL21 (DE3) cells (International Joint Cancer Institute,
Second Military Medical University). The levels of GTP-bound RhoA
(active RhoA) in cell lysates were measured using
immunoprecipitation, as previously described (23). Briefly, control and
EphA3-overexpressing KYSE510 cells were lysed in M-PER Mammalian
Protein Extraction Reagent (Thermo Fisher Scientific, Inc.)
containing protease inhibitor cocktail (Roche Diagnostics) and were
precleared with glutathione beads. The GST-RBD protein was allowed
to bind to the glutathione bead at 4°C overnight and the
supernatants of the precleared protein lysates were applied to
these beads, allowing the active form of RhoA to bind to the RBD
domain. Total RhoA levels in the cell lysate were compared to the
pulled-down active RhoA by western blotting and were measured using
Bio-Rad Quantity One 4.52 software (Bio-Rad Laboratories,
Inc.).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from cultured cells or
clinical samples using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. RT-qPCR was performed as described previously (24). Briefly, cDNA was prepared from 1
μg total RNA using AMV reverse transcriptase (Promega
Corporation, Madison, WI, USA) and random primers (Promega
Corporation), according to the manufacturer's protocol. For each
gene, PCR reactions were run three times on each sample. PCR was
performed using the Chromo4 Multicolor Real-Time Detection system
(Bio-Rad Laboratories, Inc.) in 20-µl reactions using SYBR™-Green
PCR Master Mix (Takara Bio, Inc., Otsu, Japan). The PCR thermal
cycling conditions were as follows: Predenaturation at 95°C for 30
sec, followed by 40 cycles at 95°C for 10 sec and 60°C for 40 sec,
and a final extension step at 72°C for 5 min. The relative mRNA
expression levels were normalized to the endogenous control
β-actin. Relative expression values were calculated using the
2−ΔΔCq method (25).
Because SYBR-Green binding is not sequence specific, careful design
and validation of each primer pair, as well as cautious
manipulation of RNA were undertaken to ensure that only target gene
sequence-specific, non-genomic products were amplified by qPCR. To
achieve this, primers were designed to either span or flank
introns. A dissociation curve analysis was performed at the end of
amplification, in order to verify specificity of the PCR products.
The same PCR products were also evaluated by agarose gel
electrophoresis. Data are presented as the means ± standard
deviation of three independent experiments. The primers used are as
follows: EphA3, forward 5'-ATTTTGGCAATGGGCATTTA-3', reverse
5'-ATGTATGTGGGTCAACATAAGTCC-3'; ZO-1, forward
5'-GGTCAGAGCCTTCTGATCATTC-3', reverse 5'-CATCTCTACTCCGGAGACTGC-3';
E-cadherin, forward 5'-AGGGGTCTGTCATGGAAGGT-3', reverse
5'-GCGGCATTGTAGGTGTTCA-3'; Vimentin, forward
5'-TGGTCTAACGGTTTCCCCTA-3', reverse 5'-GACCTCGGAGCGAGAGTG-3';
N-cadherin, forward 5'-GGTGGAGGAGAAGAAGACCAG-3', reverse
5'-GGCATCAGGCTCCACAGT-3'; Snail, forward
5'-TGGTTGCTTCAAGGACACAT-3', reverse 5'-GCAAATGCTCTGTTGCAGTG-3'; and
β-actin, forward 5'-CCCGCGAGTACAACCTTCT-3' and reverse 5'-CGTCAT
CCATGGCGAACT-3'.
RT-PCR
RT-PCR (Takara PCR Amplification kit, cat. no. R011;
Takara Bio, Inc.) was performed on cDNA using EphA3-specific
primers (forward, 5'-ATGTTTCCAGACACGGTACC-3' and reverse,
5'-CCATCTTCCTGAGTAGAACTGTGAGG-3'), which yielded a 269 bp product.
To serve as an internal control, a 335-bp fragment of β-actin cDNA
was co-amplified using primers (forward, 5'-TTCCTGGGCATG
GAGTCCTGTGG-3' and reverse 5'-CGCCTAGAAGCATTTGCGGTGG-3'). PCR was
conducted as follows: Predenaturation at 94°C for 3 min, followed
by 25-30 cycles at 94°C for 1 min, 55°C for 0.5 min and 72°C for
0.5 min, and followed a final extension step at 72°C for 5 min. All
PCR experiments were repeated at least twice and the products were
separated by 1.5% agarose gel electrophoresis and were stained with
ethidium bromide (cat. no. 160539; Sigma-Aldrich; Merck KGaA).
Statistical analysis
Statistical analyses were performed using Microsoft
Excel Version 2007 (Microsoft Corporation, Redmond, WA, USA). Data
are presented as the means ± standard deviation. Differences
between two groups were compared using the Student's t-test.
Differences between more than two groups were compared using
one-way analysis of variance, followed by post hoc pairwise
comparisons with the application of Dunn's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression levels of EphA3 in ESCC
To investigate EphA3 expression in ESCC, we assessed
mRNA expression levels in clinical cancer specimens, and in a
series of ESCC cell lines (Fig.
1). In the collected ESCC tissues, 11 out of 15 cancerous
tissues expressed lower levels of EphA3 than their normal
counterparts; further statistical analysis revealed that ESCC
tissues were significantly decreased compared with in adjacent
normal tissues (Fig. 1A). There
was minimal or undetectable expression of EphA3 in three of the
four cancerous cell lines (Fig. 1B and
C). However, treatment with the DNA methylation inhibitor
5-Aza-dC increased the mRNA expression levels of EphA3 in the
KYSE510 and KYSE30 cell lines (Fig.
1D).
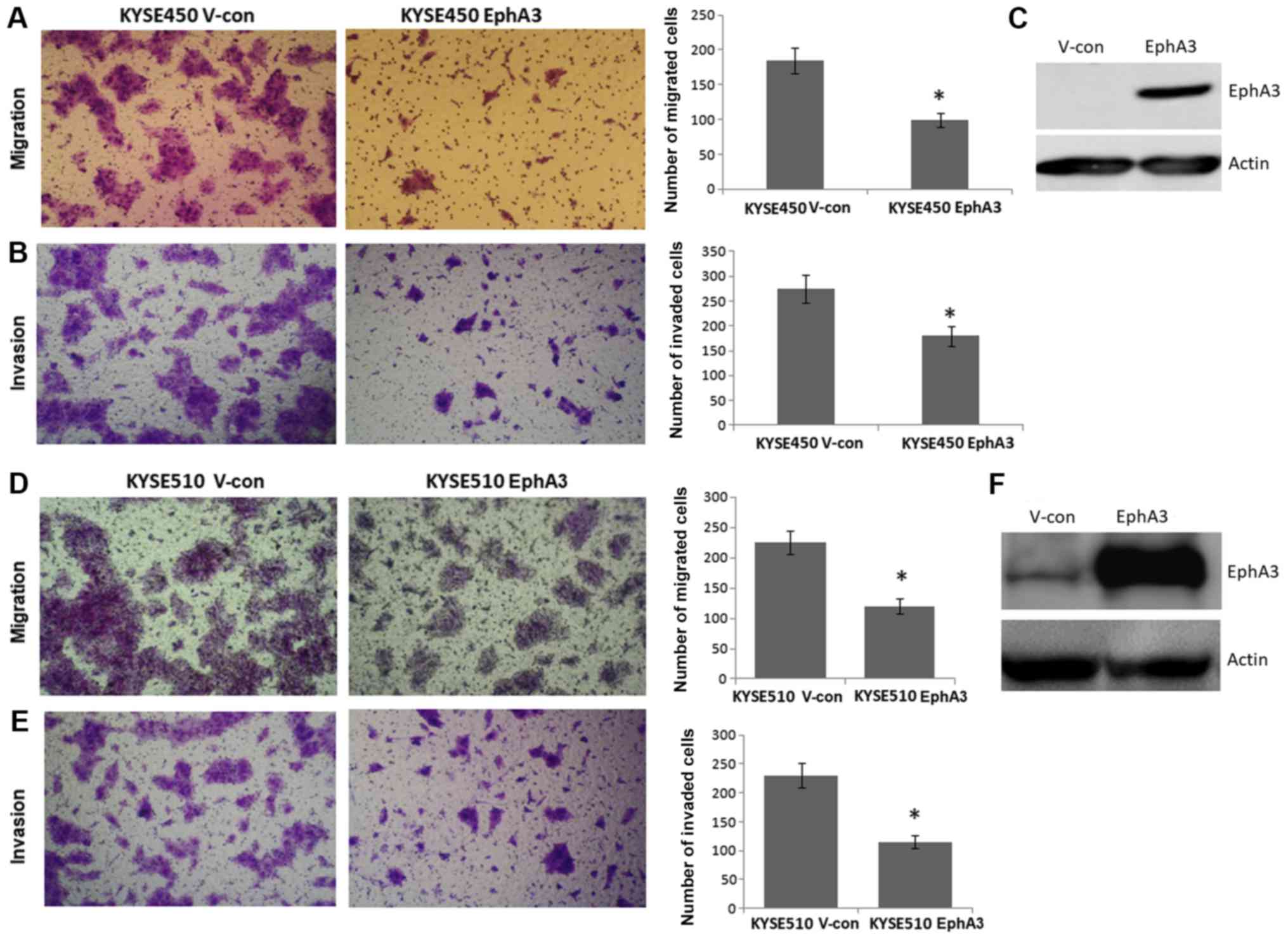
Overexpression of EphA3 inhibits the
migration and invasion of KYSE450 and KYSE510 cells
Since EphA3 was downregulated in the majority of
ESCC tissues compared with in adjacent normal tissues, the present
study aimed to investigate the role of EphA3 in ESCC. For this
purpose, KYSE450 and KYSE510 ESCC cells were infected with an
EphA3-expressing lentiviral vector or a control vector. Notably,
KYSE450 and KYSE510 cells express low endogenous levels of EphA3.
Infected cells were cultured for 24 h, after which, cell that
stably expressed EphA2 were selected using puromycin. These cell
lines were used to determine whether EphA3 could affect migration
and invasion of ESCC cells (Fig.
2).
To determine the effects of EphA3 on migration and
invasion of KYSE450 and KYSE510 cells, in vitro assays were
performed using a Transwell chamber. Overexpression of EphA3
significantly inhibited the migration of KYSE450 and KYSE510 cells
in Transwell chambers (Fig. 2A and
D). Furthermore, as determined following the addition of a thin
layer of Matrigel onto the Transwell chamber membranes, EphA3
overexpression significantly inhibited the invasion of KYSE450 and
KYSE510 cells (Fig. 2B and E).
These data indicated that overexpression of EphA3 may suppress the
migration and invasion of KYSE450 and KYSE510 cells.
EphA3 overexpression induces MET of
KYSE450 and KYSE510 cells
Cells overexpressing EphA3 were analyzed to
determine the effects of EphA3 on EMT and MET (Fig. 3). Notably, morphological
alterations characteristic of MET were observed in KYSE450 and
KYSE510 ESCC cells stably expressing EphA3. As shown in Fig. 3A and D, while the control cells
appeared elongated, which is a characteristic of mesenchymal cells,
cells overexpressing EphA3 exhibited the more rounded shape
characteristic of epithelial cells. The morphological observations
and Transwell assay results resulted in the hypothesis that EphA3
overexpression may affect the EMT process of ESCC during tumor
metastasis. In addition, the expression levels of EMT-associated
genes were detected by RT-qPCR. As shown in Fig. 3B and E, the mRNA expression levels
of the epithelial cell markers E-cadherin and ZO-1 were
upregulated, whereas the mesenchymal cell markers Vimentin and
N-cadherin were downregulated compared with in the control cells.
Furthermore, the mRNA expression levels of Snail, which is an
E-cadherin-suppressing gene, were also downregulated (Fig. 3B and E). Concordantly, western
blotting confirmed the upregulated expression of E-cadherin and
ZO-1, and downregulated expression of Vimentin compared with in the
vector control cells (Fig. 3C and
F). These data suggested that EphA3 overexpression may induce
the MET process in KYSE450 and KYSE510 cells.
EphA3 function depends on its kinase
activity and tyrosine phosphorylation status
To examine the molecular mechanisms underlying the
effects of EphA3 overexpression on cell morphology and behavior,
the present study addressed the dependence of these cellular
effects on EphA3 kinase activity and tyrosine phosphorylation
status within the EphA3 cytoplasmic domain. Briefly, the effects of
the kinase-null mutant K653R (a mutant containing a lysine to
arginine mutation at amino acid position 653, which is known to
inactivate EphA3 kinase activity) (26,27)
and the phenylalanine replacement mutants of conserved tyrosine
residues (Y596, Y602, Y779) were determined. These tyrosine
residues are believed to function together to regulate the kinase
activity of EphA3 (27,28). Wild type and mutant receptors were
expressed at comparable levels in transduced cells (Fig. 4A).
Transduction of cells with EphA3 harboring mutations
in all three tyrosines (3YF) or a kinase-null mutation (K653R) did
not induce drastic MET-like alterations (Fig. 4B). Cell migration and invasion
assays revealed that these mutations alleviated the inhibitory
effects of EphA3 (Fig. 4C and D).
These observations suggested that the functions of EphA3 were
dependent on its kinase activity and tyrosine phosphorylation
status in vitro.
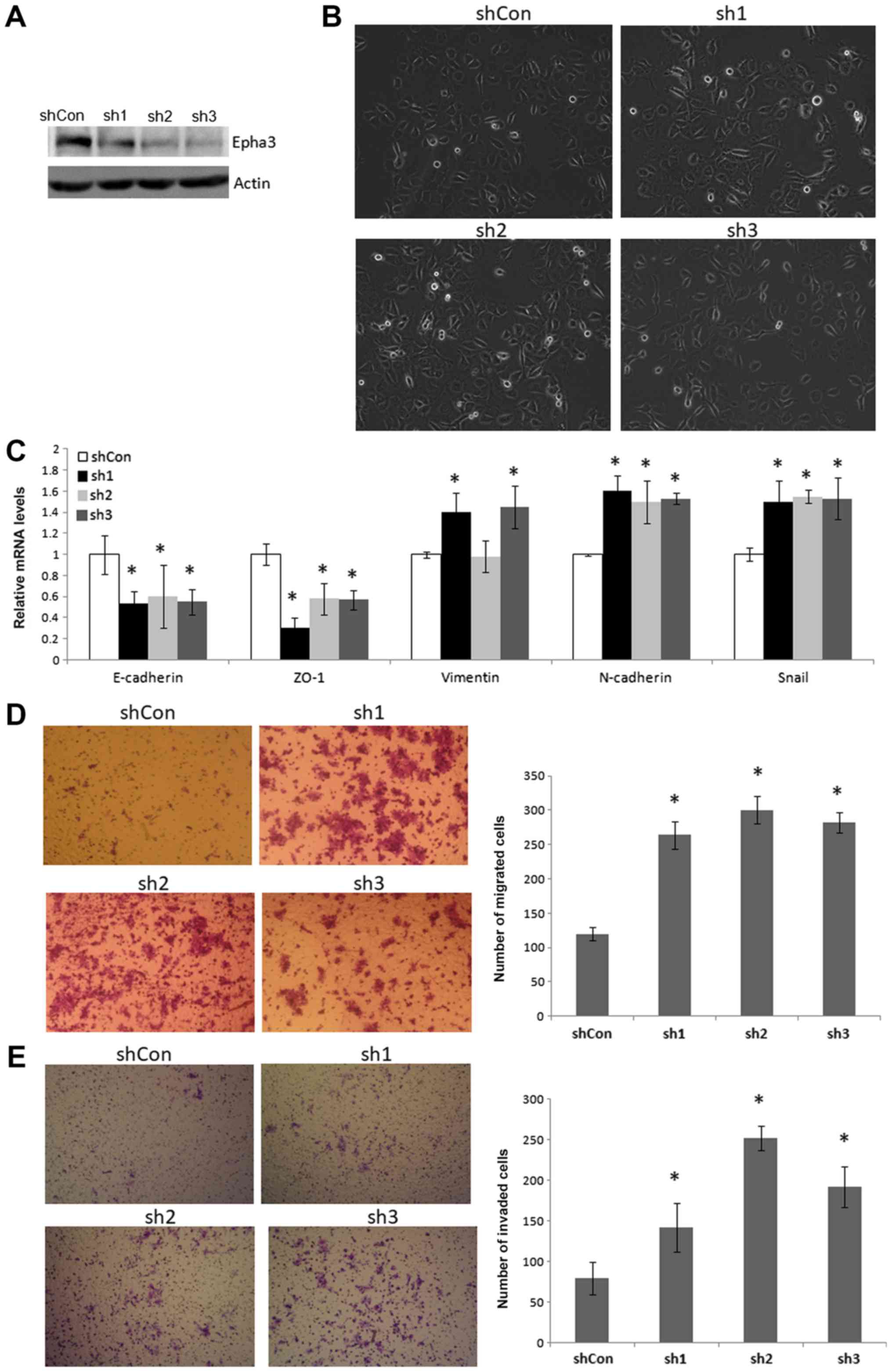
Silencing EphA3 induces EMT, and promotes
migration and invasion of KYSE410 cells
Three shRNA sequences corresponding to the EphA3
gene, and one control shRNA sequence, were designed and inserted
into a lentiviral vector, which was infected into KYSE410 cells, in
order to generate a EphA3 knockdown cell line. KYSE410 cells
express high endogenous levels of EphA3. As shown in Fig. 5, knockdown of EphA3 induced EMT
morphological alterations, and promoted cell migration and
invasion. Furthermore, the mRNA expression levels of the epithelial
cell markers E-cadherin and ZO-1 were downregulated, whereas the
mesenchymal cell markers Vimentin and N-cadherin were upregulated
compared with in the control cells (Fig. 5C). Furthermore, the mRNA expression
levels of Snail were also upregulated. These data suggested that
silencing EphA3 may induce EMT, and promote migration and invasion
of KYSE410 cells.
EphA3 functions are mediated by reducing
RhoA activity
Small Rho GTPase signaling has been reported to
contribute to invasion and metastasis, and participates in
regulating EMT, MET and cytoskeletal signaling events (29,30).
Their activities are modulated by cycling between a GDP-bound
inactive form and a GTP-bound active form. To analyze the possible
signal transduction machinery in ESCC underlying EphA3-mediated
inhibition of migration and EMT, the present study focused on the
Rho signaling pathway. The activated RhoA levels were detected in
cell lysates using a GST pull-down assay, by virtue of its binding
ability to the RBD of rhotekin protein, according to Wahl et
al (31).
The RBD-bound, active RhoA-GTP level was reduced in
EphA3-overexpressing cells compared with the control cells
(Fig. 6A). Furthermore, staining
of permeabilized cells with rhodaminephalloidin revealed that the
actin cytoskeleton network was largely reorganized and
intracellular stress fiber formation was reduced in
EphA3-overexpressing cells. There were more complex actin networks
with distinct fiber bundles extending into cell protrusions and
lamellipodia in the control cells (Fig. 6B).
Discussion
The Eph receptors and their ligands, ephrins,
mediate important processes in embryonic development, particularly
in tissue organization, by modulating cell adhesion or repulsion.
Altered expression of Eph receptors and ephrins is associated with
angiogenesis and tumor vasculature in numerous types of human
cancer, including breast, lung and prostate cancer, melanoma and
leukaemia (8,32-34).
Alterations in their expression profiles in several types of cancer
have resulted in this protein family being considered prime targets
for cancer prognosis and therapy (35-38).
EphA3 (formerly known as HEK) is presently one of the most
promising therapeutic targets (39,40).
EphA3 expression is associated with tumor promotion in various
cancer types, whereas it serves a tumor-suppressor role in others
(40). EphA3 gene copy numbers
and/or expression levels have been reported to be decreased in lung
cancer, and re-expression of wild-type EphA3 in cell lines
increases apoptosis by suppressing activity of prosurvival protein
kinase B, and inhibits the growth of tumor xenografts (41).
The present study revealed that EphA3 expression was
decreased in ESCC tissues and cell lines compared with in the
normal counterparts. These findings were concordant with those of
previous studies, which revealed that the EphA3 gene is deleted in
ESCC (20,21). In addition, treatment with 5-Aza-dC
increased the mRNA expression levels of EphA3 in KYSE510 and KYSE30
cells, thus suggesting that inhibition of DNA methyltransferase can
enhance EphA3 expression. Silencing of EphA3 expression by DNA
hypermethylation also occurs in leukemia (14). In addition, irradiation of
metastatic human melanoma cells leads to decreased cell migration
alongside downregulation of EphA2 and upregulation of EphA3
(42). In this study,
overexpression of EphA3 inhibited the migration and invasion of
KYSE450 and KYSE510 cells. Furthermore, shRNA-mediated knockdown of
EphA3 in KYSE410 cells promoted cell migration and invasion, thus
suggesting that it may act as a tumor suppressor in ESCC. Future
studies are required to test and verify the tumor-suppressor role
of EphA3 in ESCC using an animal model of metastasis.
The EMT process is an important event in the tumor
invasion-metastasis cascade (43),
during which epithelial cells lose their sheet-like architecture,
cell polarity and cell to cell adhesion, and then undergo marked
cytoskeletal remodeling, and gain migratory and invasive properties
to become mesenchymal stem cells. These effects are largely
mediated through the recruitment of signaling molecules associated
with cytoskeletal organization and integrin signaling (44). During actin cytoskeletal
reorganization, important dynamic structures, such as membrane
protrusions (including lamellipodia, membrane ruffles, lamellae and
filopodia) and stress fibers are formed. It has been reported that
this process is mediated by the actions of the three
best-characterized small GTPases, including Rho, Rac and cell
division control protein 42. Furthermore, it has been indicated
that the RhoA/ROCK-dependent pathway participates in regulating
EMT, MET and cytoskeletal signaling events, and is crucial for cell
motility. EphA3-knockout mice have been reported to have a heart
valve defect, which is thought to result from defective EMT, with
fewer migrating mesenchymal cells (12). Conversely, EphA3 overexpression
promotes EMT in chick atrioventricular cushion explants (45). However, the present study observed
MET-like morphological alterations in EphA3-overexpressing ESCC
cells, as determined using phase-contrast micrographs. In addition,
overexpression of EphA3 upregulated epithelial proteins (E-cadherin
and ZO-1) and downregulated mesenchymal proteins (Vimentin,
N-cadherin and Snail). Furthermore, stress fiber formation was
reduced in EphA3-overexpressing cells, as determined by phalloidin
staining. EphA3 overexpression also decreased RhoA GTPase, thus
suggesting that EphA3 may suppress ESCC cancer migration and
invasion, at least partially through small GTPase RhoA
activation.
Together with the activation-loop tyrosine, two
conserved tyrosines in the juxtamembrane region of Eph receptors
function to regulate kinase activity, and their phosphorylation is
crucial for full enzyme activity (28,46).
These juxtamembrane tyrosines are important for signal transfer,
and have been suggested to act as docking sites for some known
signaling molecules, including Fyn, Src, RasGAP and Nck (47,48);
the majority of these molecules are involved in organization of the
cytoskeleton (49,50), thus supporting an emerging concept
that Eph receptors regulate cell adhesion and plasticity of the
actin cytoskeleton (51). It is
well known that the K653R mutant is a kinase-null mutant of EphA3
that results in inactivation of its kinase activity; in addition,
phosphorylation of the conserved residue Y596 is critical to its
kinase activity. Tyrosine phosphorylation of the other two
conserved residues, Y602 and Y779, has also been reported to
collaborate to execute the full cellular activity of EphA3
(27,28,52).
In the present study, cells infected with EphA3 harboring mutations
in all three tyrosines (3YF) or a kinase-null mutation (K653R) did
not exhibit marked MET-like alterations, and cell migration and
invasion were not significantly inhibited. A limitation of the
present study is that kinase activity assays were not conducted and
the phosphorylation levels of these specific mutants were not
detected. The results of the present study suggested that the
functions of EphA3 were dependent on kinase activity and tyrosine
phosphorylation status. In conclusion, this study indicated that
EphA3 expression was significantly suppressed in ESCC due to
promoter methylation, whereas its overexpression prevented cancer
cells from undergoing EMT, and inhibited cell migration and
invasion via the Rho GTPase signaling pathway.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81101574
and 81570557).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XC designed the study, carried out molecular cloning
experiments and cellular experiments, and drafted the manuscript.
QM performed the immunoblotting assays. BL and CDJ participated in
study coordination, and helped to collect and analyze the data. JZL
participated in the design of the study and drafted the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Second Military Medical University. Written
informed consent was obtained from each patient or from his/her
legal guardians.
Patient consent for publication
All patients provided written informed consent.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Liu Y, Xiong Z, Beasley A, D'Amico T and
Chen XL: Personalized and targeted therapy of esophageal squamous
cell carcinoma: An update. Ann N Y Acad Sci. 1381:66–73. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
3
|
Baba Y, Watanabe M, Yoshida N and Baba H:
Neoadjuvant treatment for esophageal squamous cell carcinoma. World
J Gastrointest Oncol. 6:121–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zelinski DP, Zantek ND, Stewart JC,
Irizarry AR and Kinch MS: EphA2 overexpression causes tumorigenesis
of mammary epithelial cells. Cancer Res. 61:2301–2306.
2001.PubMed/NCBI
|
|
7
|
Easty DJ, Herlyn M and Bennett DC:
Abnormal protein tyrosine kinase gene expression during melanoma
progression and metastasis. Int J Cancer. 60:129–136. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herath NI, Spanevello MD, Doecke JD, Smith
FM, Pouponnot C and Boyd AW: Complex expression patterns of Eph
receptor tyrosine kinases and their ephrin ligands in colorectal
carcinogenesis. Eur J Cancer. 48:753–762. 2012. View Article : Google Scholar
|
|
9
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: Emt: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wicks IP, Wilkinson D, Salvaris E and Boyd
AW: Molecular cloning of HEK, the gene encoding a receptor tyrosine
kinase expressed by human lymphoid tumor cell lines. Proc Natl Acad
Sci USA. 89:1611–1615. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gale NW, Holland SJ, Valenzuela DM,
Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H,
Wilkinson DG, et al: Eph receptors and ligands comprise two major
specificity subclasses and are reciprocally compartmentalized
during embryogenesis. Neuron. 17:9–19. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stephen LJ, Fawkes AL, Verhoeve A, Lemke G
and Brown A: A critical role for the EphA3 receptor tyrosine kinase
in heart development. Dev Biol. 302:66–79. 2007. View Article : Google Scholar
|
|
13
|
Chiari R, Hames G, Stroobant V, Texier C,
Maillère B, Boon T and Coulie PG: Identification of a
tumor-specific shared antigen derived from an Eph receptor and
presented to CD4 T cells on HLA class II molecules. Cancer Res.
60:4855–4863. 2000.PubMed/NCBI
|
|
14
|
Dottori M, Down M, Hüttmann A, Fitzpatrick
DR and Boyd AW: Cloning and characterization of EphA3 (Hek) gene
promoter: DNA methylation regulates expression in hematopoietic
tumor cells. Blood. 94:2477–2486. 1999.PubMed/NCBI
|
|
15
|
Lawrenson I D, Wim mer-K lei ka mp SH, L
ock P, Schoenwaelder SM, Down M, Boyd AW, Alewood PF and Lackmann
M: Ephrin-A5 induces rounding, blebbing and de-adhesion of
EphA3-expressing 293T and melanoma cells by CrkII and Rho-mediated
signalling. J Cell Sci. 115:1059–1072. 2002.PubMed/NCBI
|
|
16
|
Xi HQ, Wu XS, Wei B and Chen L: Aberrant
expression of EphA3 in gastric carcinoma: Correlation with tumor
angiogenesis and survival. J Gastroenterol. 47:785–794. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davies H, Hunter C, Smith R, Stephens P,
Greenman C, Bignell G, Teague J, Butler A, Edkins S, Stevens C, et
al: Somatic mutations of the protein kinase gene family in human
lung cancer. Cancer Res. 65:7591–7595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stephens P, Edkins S, Davies H, Greenman
C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, et al:
A screen of the complete protein kinase gene family identifies
diverse patterns of somatic mutations in human breast cancer. Nat
Genet. 37:590–592. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wood LD, Calhoun ES, Silliman N, Ptak J,
Szabo S, Powell SM, Riggins GJ, Wang TL, Yan H, Gazdar A, et al:
Somatic mutations of GUCY2F, EPHA3, and NTRK3 in human cancers. Hum
Mutat. 27:1060–1061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brown J, Bothma H, Veale R and Willem P:
Genomic imbalances in esophageal carcinoma cell lines involve Wnt
pathway genes. World J Gastroenterol. 17:2909–2923. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen J, Guo L, Peiffer DA, Zhou L, Chan
OT, Bibikova M, Wickham-Garcia E, Lu SH, Zhan Q, Wang-Rodriguez J,
et al: Genomic profiling of 766 cancer-related genes in archived
esophageal normal and carcinoma tissues. Int J Cancer.
122:2249–2254. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li JZ, Chen X, Gong XL, Hu HY, Shi D, Lu
YM, Qiu L, Lu F, Hu ZL and Zhang JP: Identification of a functional
nuclear localization signal mediating nuclear import of the zinc
finger transcription factor ZNF24. PLoS One. 8:e799102013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren XD, Kiosses WB and Schwartz MA:
Regulation of the small GTP-binding protein Rho by cell adhesion
and the cytoskeleton. EMBO J. 18:578–585. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Chen X, Gong X, Liu Y, Feng H, Qiu
L, Hu Z and Zhang J: A transcript profiling approach reveals the
zinc finger transcription factor ZNF191 is a pleiotropic factor.
BMC Genomics. 10:2412009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Brennaman LH, Moss ML and Maness PF:
EphrinA/EphA-induced ectodomain shedding of neural cell adhesion
molecule regulates growth cone repulsion through ADAM10
metalloprotease. J Neurochem. 128:267–279. 2014. View Article : Google Scholar
|
|
27
|
Hu T, Shi G, Larose L, Rivera GM, Mayer BJ
and Zhou R: Regulation of process retraction and cell migration by
EphA3 is mediated by the adaptor protein Nck1. Biochemistry.
48:6369–6378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Binns KL, Taylor PP, Sicheri F, Pawson T
and Holland SJ: Phosphorylation of tyrosine residues in the kinase
domain and juxtamembrane region regulates the biological and
catalytic activities of Eph receptors. Mol Cell Biol. 20:4791–4805.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sahai E and Marshall CJ: RHO-GTPases and
cancer. Nat Rev Cancer. 2:133–142. 2002. View Article : Google Scholar
|
|
31
|
Wahl S, Barth H, Ciossek T, Aktories K and
Mueller BK: Ephrin-A5 induces collapse of growth cones by
activating Rho and Rho kinase. J Cell Biol. 149:263–270. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dodelet VC and Pasquale EB: Eph receptors
and ephrin ligands: Embryogenesis to tumorigenesis. Oncogene.
19:5614–5619. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Clevers H and Batlle E: EphB/EphrinB
receptors and Wnt signaling in colorectal cancer. Cancer Res.
66:2–5. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Surawska H, Ma PC and Salgia R: The role
of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev.
15:419–433. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oricchio E, Nanjangud G, Wolfe AL, Schatz
JH, Mavrakis KJ, Jiang M, Liu X, Bruno J, Heguy A, Olshen AB, et
al: The Eph-receptor A7 is a soluble tumor suppressor for
follicular lymphoma. Cell. 147:554–564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hatano M, Eguchi J, Tatsumi T, Kuwashima
N, Dusak JE, Kinch MS, Pollack IF, Hamilton RL, Storkus WJ and
Okada H: EphA2 as a glioma-associated antigen: A novel target for
glioma vaccines. Neoplasia. 7:717–722. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee JW, Han HD, Shahzad MM, Kim SW,
Mangala LS, Nick AM, Lu C, Langley RR, Schmandt R, Kim HS, et al:
EphA2 immunoconjugate as molecularly targeted chemotherapy for
ovarian carcinoma. J Natl Cancer Inst. 101:1193–1205. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kiewlich D, Zhang J, Gross C, Xia W,
Larsen B, Cobb RR, Biroc S, Gu JM, Sato T, Light DR, et al:
Anti-EphA2 antibodies decrease EphA2 protein levels in murine CT26
colorectal and human MDA-231 breast tumors but do not inhibit tumor
growth. Neoplasia. 8:18–30. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boyd AW, Ward LD, Wicks IP, Simpson RJ,
Salvaris E, Wilks A, Welch K, Loudovaris M, Rockman S and Busmanis
I: Isolation and characterization of a novel receptor-type protein
tyrosine kinase (hek) from a human pre-B cell line. J Biol Chem.
267:3262–3267. 1992.PubMed/NCBI
|
|
40
|
Janes PW, Slape CI, Farnsworth RH,
Atapattu L, Scott AM and Vail ME: EphA3 biology and cancer. Growth
Factors. 32:176–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhuang G, Song W, Amato K, Hwang Y, Lee K,
Boothby M, Ye F, Guo Y, Shyr Y, Lin L, et al: Effects of
cancer-associated EPHA3 mutations on lung cancer. J Natl Cancer
Inst. 104:1182–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mosch B, Pietzsch D and Pietzsch J:
Irradiation affects cellular properties and Eph receptor expression
in human melanoma cells. Cell Adhes Migr. 6:113–125. 2012.
View Article : Google Scholar
|
|
43
|
Grant CM and Kyprianou N: Epithelial
mesenchymal transition (EMT) in prostate growth and tumor
progression. Transl Androl Urol. 2:202–211. 2013.
|
|
44
|
Miao H, Burnett E, Kinch M, Simon E and
Wang B: Activation of EphA2 kinase suppresses integrin function and
causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol.
2:62–69. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Frieden LA, Townsend TA, Vaught DB,
Delaughter DM, Hwang Y, Barnett JV and Chen J: Regulation of heart
valve morphogenesis by Eph receptor ligand, ephrin-A1. Dev Dyn.
239:3226–3234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zisch AH, Pazzagli C, Freeman AL,
Schneller M, Hadman M, Smith JW, Ruoslahti E and Pasquale EB:
Replacing two conserved tyrosines of the EphB2 receptor with
glutamic acid prevents binding of SH2 domains without abrogating
kinase activity and biological responses. Oncogene. 19:177–187.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mellitzer G, Xu Q and Wilkinson DG:
Control of cell behaviour by signalling through Eph receptors and
ephrins. Curr Opin Neurobiol. 10:400–408. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu Q, Mellitzer G and Wilkinson DG: Roles
of Eph receptors and ephrins in segmental patterning. Philos Trans
R Soc Lond B Biol Sci. 355:993–1002. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schoenwaelder SM and Burridge K:
Bidirectional signaling between the cytoskeleton and integrins.
Curr Opin Cell Biol. 11:274–286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schlaepfer DD and Hunter T: Integrin
signalling and tyrosine phosphorylation: Just the FAKs? Trends Cell
Biol. 8:151–157. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schmucker D and Zipursky SL: Signaling
downstream of Eph receptors and ephrin ligands. Cell. 105:701–704.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shi G, Yue G and Zhou R: EphA3 functions
are regulated by collaborating phosphotyrosine residues. Cell Res.
20:1263–1275. 2010. View Article : Google Scholar : PubMed/NCBI
|