Introduction
A total of ~600,000 patients annually succumb to
colorectal cancer (CRC) worldwide (1), with indications that the incidence is
on the rise, particularly in Asia (2,3) and
possibly in the younger population in the USA (4). Regular aspirin intake significantly
reduces the risk of developing CRC and other gastrointestinal tract
malignancies (5,6), which has caused debate regarding
regular aspirin use as a chemopreventive agent on a population
basis (7,8). Aspirin usage following a diagnosis of
colon cancer also has a positive outcome: Longer survival has been
reported among patients with mutated-PIK3CA CRC, but not those with
wild-type PIK3CA cancer (9).
Despite vigorous investigation, the molecular basis
for the chemopreventive effect of aspirin remains controversial and
a large number of molecular targets of aspirin and salicylates have
been suggested (10). The
molecular effects on cells and tissues include but are not limited
to: Inhibition of cyclooxygenase (COX) activity (11-13)
and protein phosphatase 2 enzymatic activity (14), inhibition of nuclear factor (NF)-κB
transcriptional activity (15),
suppression of COX-2 gene transcription (16), inhibition of IκB kinase-β (17), increased hMLH1 expression (18), selection for DNA microsatellite
stability (19), altering
mammalian target of rapamycin signalling (20) and a direct allosteric activation of
AMP kinase (21). More recently,
the breakdown product of aspirin, salicylate, has been demonstrated
to inhibit CREB-binding protein/p300 acetyltransferase activity
(22).
Therefore, it is reasonable to suggest that aspirin
may act pleiotropically and there is some evidence that aspirin and
other non-steroidal anti-inflammatory drug (NSAIDs) may also affect
the epidermal growth factor (EGF) receptor (EGFR) axis. For
example, metabolites of the NSAID sulindac can inhibit EGFR
signalling through the inhibition of EGFR phosphorylation and
decreased EGFR expression in HT29 colon cancer cells (23). A nitro-derivative of aspirin was
demonstrated to inhibit EGFR signalling in human ovarian cancer
cells (24). Furthermore, in
COX-1-positive ovarian cancer cells cultured in vitro,
aspirin inhibited EGF-associated protein kinase B (AKT) and
extracellular signal-regulated kinase (ERK) phosphorylation
(25).
In an attempt to better understand the toxicity of
aspirin to CRC cells, a number of aspirin-like analogues were
previously synthesised and diaspirins were identified as having the
capacity to induce apoptosis in a CRC cell line (26), inhibit NF-κB in vitro and
cyclin D1 expression, and suppress tumour growth in vivo in
a murine model of CRC without evidence of apparent toxicity to the
animal (27). The aim of the
present study was to investigate whether aspirin and analogues,
including fumaryldiaspirin (F-DiA), diflunisal and salicylates,
which are common breakdown products of these compounds, are able to
perturb EGF endocytosis in SW480 CRC cells (28), as these cells are known to express
relatively high levels of wild type EGFR (29) compared with normal colonic
epithelial primary cells, but exhibit decreased expression of COX-1
and negligible levels of COX-2 (30,31).
Given the role of EGF signalling in tissue repair (32), the findings of the present study
may improve our understanding of the molecular basis of the action
of aspirin as a chemopreventive agent and its inhibitory effect on
wound healing.
Materials and methods
Chemicals and reagents
Foetal bovine serum (FBS) was purchased from PAA
Laboratories (GE Healthcare Life Sciences, Little Chalfont, UK) or
Labtech International, Ltd. (Heathfield, UK). Precision Plus
Protein Colour standards and nitrocellulose were obtained from
Bio-Rad Laboratories, Inc. (Hercules, CA, USA). Human recombinant
EGF (PHG0313) was from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). Alexa Fluor 555-EGF (E-35350) was from Molecular Probes;
Thermo Fisher Scientific, Inc. EGFR (D38B1) XP® rabbit
antibody (Alexa Fluor 488-conjugate; 1:100; cat. no. 5616) and EGFR
rabbit antibody (D38B1; 1:100; cat. no. 4267) were from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Anti-early endosome
antigen 1 (EEA1) antibody (1G11) Early Endosome Marker (ab70521;
1:1,000) was from Abcam (Cambridge, UK). Anti-GAPDH antibody
(sc-25778; 1:1,000) was from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). VectaShield® mounting medium was from
Vector Laboratories, Ltd. (Peterborough, UK). All other reagents
were obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany),
unless stated otherwise. Aspirin analogues, detailed in Table I, were synthesised in-house using
previously described methods (26,27).
 | Table IList of compounds. |
Table I
List of compounds.
| Common name,
synonym | Chemical, formal
name | Laboratory
number | IC50
(mM) | Source |
|---|
| Salicylic acid,
SA | 2-hydroxybenzoic
acid | Not applicable | 2.6 | Commercially
available |
| Aspirin,
ortho-aspirin | 2-acetoxybenzoic
acid, acetylsalicylic acid | PN502 | 1.8 | Commercially
available |
| Diaspirin, DiA |
Bis-carboxyphenylsuccinate | PN508 | 0.8 | (27) |
| Fumaryldiaspirin,
F-DiA |
Bis-carboxyphenylfumarate | PN517 | 0.2 | (27) |
| Not applicable |
m-bromobenzoylsalicylic acid | PN524 | 0.4 | (27) |
| Not applicable | Isopropyl
m-bromobenzoylsalicylate | PN529 | ND | (27) |
|
meta-aspirin | 3-acetoxybenzoic
acid | PN548 | 3.8 | Synthesised in this
study |
|
para-aspirin | 4-acetoxybenzoic
acid | PN549 | 4.9 | Synthesised in this
study |
| Thioaspirin,
ortho-TASP | 2-acetylthiobenzoic
acid | PN590 | 0.2 | Synthesised in this
study |
|
m-thioaspirin,
meta-TASP | 3-acetylthiobenzoic
acid | PN591 | 0.5 | Synthesised in this
study |
|
p-thioaspirin,
para-TASP | 4-acetylthiobenzoic
acid | PN592 | 0.8 | Synthesised in this
study |
| Diflunisal,
DIF |
5-(2,4-difluorophenyl) salicylic acid | Not applicable | 0.08 | Commercially
available |
Cell culture
Colon cancer adenocarcinoma SW480 cells were
obtained from the European Collection of Authenticated Cell
Cultures (Salisbury, UK) and cultured in Leibovitz L-15 medium
(Thermo Fisher Scientific, Inc.) supplemented with 10% (v/v) FBS
supplemented with L-glutamine-penicillin-streptomycin
(Sigma-Aldrich; Merck KGaA). The OE33 and Flo-1 oesophageal cancer
cell lines were a gift from Professor Tim Underwood (University of
Southampton, Southampton, UK) and were cultured in Dulbecco’s
modified Eagle’s medium high-glucose with phenol red (Thermo Fisher
Scientific, Inc.) containing 10% (v/v) FBS supplemented with
L-glutamine-penicillin-streptomycin (Sigma-Aldrich; Merck KGaA).
Cells were cultured in tissue culture flasks (Sarstedt, Inc.,
Newton, NC, USA) at 37°C in a humidified incubator with 5%
CO2, except for SW480 cells, which were cultured in the
absence of CO2. Cells were regularly passaged at 70-80%
confluence. The OE33 (also known as JROECL33) and Flo-1 cells are
commercially available oesophageal cancer cell lines (33). The OE33 cell line expresses COX-2
and has been utilised to examine COX-2 inhibitors with respect to
proliferation (34). Both cell
lines express EGFR (35).
Additionally, the cell lines were investigated as they were derived
from oesophageal carcinomas, and thus represent a counterpoint to a
colorectal adenocarcinoma (SW480). Another aim of the present study
was to elucidate whether the sensitivity to the salicylates
observed in the SW480 cell line was specific to a cell line of CRC
origin only, given the reported chemopreventative activity of
aspirin against oesophageal cancer; reviewed in (36,37).
Cytotoxicity assay
Cell viability was assessed by MTT reduction assay
(38) with modifications (39). Briefly, SW480 cells were seeded at
a density of 104 cell/well in a 96-well plate and
incubated overnight at 37°C. Following 24 h, the culture medium was
replaced with medium containing aspirin analogues (Table I) at 0.01, 0.03, 0.1, 0.3, 1 and 3
mM, and the cells were incubated at 37°C for a further 48 h. A
no-treatment control was included in these experiments. Following
48 h of incubation, the medium was aspirated from each well, the
cells were washed once with fresh medium to remove drugs, and 300
µl MTT reagent (0.5 mg/ml) was added. The cells were
incubated at 37°C for 3 h and the solution was replaced with 200
µl dimethyl sulphoxide (DMSO). The plates were incubated at
37°C for a further 30 min and the conversion of MTT into formazan
crystals was measured by recording changes in absorbance at 540 nm
in a Multiskan Ascent microplate reader (Thermo Fisher Scientific,
Inc.). The plates were protected from light throughout the
procedure. Drug dosage ranged from 0 to 3 mM. GraphPad Prism 7
(GraphPad Software, Inc., La Jolla, CA, USA) was used to plot
dose-response curves and to determine half maximal inhibitory
concentration (IC50) values. All assays were performed
in duplicates (n=3 experiments) and viable cells were expressed as
the percentage relative to the negative control: Cell viability =
[absorbance 540 nm (treated cells-blank)/absorbance 540 nm (control
cells-blank)] ×100.
Immunoblot analysis
Cells (5×104) were plated in 6-well
tissue culture plates and incubated overnight at 37°C. Cells
incubated with compounds (Table I)
for the times stated were washed with ice-cold PBS at 4°C, gently
scraped and suspended in 2X Laemmli sample buffer containing 10%
β-mercaptoethanol (40) and 0.01%
protease inhibitor cocktail (100X; 5871S; CST Biological Reagents
Co., Ltd., Shanghai, China). The samples were then heated to 100°C
for 20 min, centrifuged (18,000 × g; 5 min; room temperature),
analysed by discontinuous SDS-PAGE with a 10% resolving gel and
electro-transferred using a Bio-Rad Mini-Trans Blot electrophoretic
transfer cell (Bio-Rad Laboratories, Inc.) according to the
manufacturer’s instructions onto polyvinylidene diflouride
membranes (Immobilon®; IPVH00010; Merck KGaA). The
membranes were incubated with blocking buffer [5% (w/v) bovine
serum albumin (BSA) and 0.1% (v/v) Tween-20 in TBS (TBST), or 5%
non-fat milk (w/v) and 0.1% (v/v) TBST, for GAPDH] for 1 h at room
temperature, followed by incubation with the following specific
antibodies: EGFR rabbit-antibody (1:1,000), rabbit phosphorylated
(p)EGFR Y1068 antibody (ab5644; 1:1,000; Abcam), rabbit pEGFR Y1045
antibody (2237S; 1:1,000; Cell Signaling Technology, Inc.), rabbit
pEGFR Y1172 antibody (ab135560; 1:2,000; Abcam) and GAPDH rabbit
antibody in blocking buffer and incubated overnight at 4°C. The
membranes were washed extensively with TBST and incubated with
horseradish peroxidase-conjugated anti-rabbit immunoglobulin (Ig)G
(7074S; 1:2,000; Cell Signaling Technology, Inc.) for 3 h at room
temperature. The membranes were then washed with TBST, prior to
being visualised with Thermo Scientific™ Pierce™ ECL-plus reagent
(80196; Thermo Fisher Scientific, Inc.) according to the
manufacturer’s instructions, using CL-Xposure™ X-ray film (34088;
Thermo Fisher Scientific, Inc.). ImageJ software v1.52 (National
Institutes of Health, Bethesda, MD, USA) was employed for
densitometric analysis of western blots. Band intensities were
quantified (n=3 experiments) and pEGFR or EGFR levels were
calculated relative to GAPDH, the loading control. pEGFR levels
were also calculated relative to total EGFR. Data points represent
mean ± standard error of the mean. Statistical analysis was
performed using one-way analysis of variance followed by Dunnett’s
post hoc test.
Microscopy
For EGF clustering, SW480 cells were seeded onto
glass coverslips at ~0.3×106 cells/well and cultured in
Leibovitz L-15 medium containing 10% (v/v) FBS supplemented with
antibiotics to 70-80% confluence in 6-well tissue culture plates.
Cells were then cultured in serum-free L-15 medium with antibiotics
for 48 h. Monolayers were cooled to 4°C and incubated for 60 min in
the presence of 100-500 ng/ml Alexa Fluor 555-EGF at 4°C. For
experiments testing compounds, serum-starved cells were
pre-incubated at 37°C for 30 min in the absence or presence of
compounds (Table I), followed by
15 min at 4°C prior to the addition of an equal volume of cold
(4°C) serum-free culture medium containing 200 ng/ml Alexa Fluor
555-EGF and incubation for 60 min allowing EGF binding. Prolonged
incubation previously facilitated recruitment of ligand-receptor
complexes into clathrin-coated pits (41,42).
Cells were warmed to 37°C over 25-30 min to stimulate EGF and EGFR
internalisation, washed with PBS, fixed for 5 min with cold
acetone/methanol (-20°C; 1:1, v/v) and the coverslip was then
placed onto a drop of VectaShield mounting medium prior to
immediate epifluorescence or confocal microscopy at ×400
magnification. For preliminary experiments, cells were incubated at
37°C with 1 mM aspirin, Di-A, F-DiA or with 0.5 mM PN529 (Table I) for 30 min in serum-free medium
or with an equivalent final concentration of DMSO as described
above, prior to the addition of fluorescently-labelled EGF.
Epifluorescence microscopy was carried out using an Olympus BX61
microscope with TXRED filter set (excitation, 580-590 nm; emission,
615-630 nm; Olympus Corporation, Tokyo, Japan). For confocal
microscopy a Zeiss LSM510 Meta or a Zeiss LSM880 with Airyscan
(Zeiss GmbH, Jena, Germany), equipped with an argon excitation
laser (458, 488 and 514 nm), using a 63 × 1.4 NA oil immersion
objective. DAPI (Dako; Agilent Technologies, Inc., Santa Clara, CA,
USA) was used as a nuclear counter stain (room temperature, ≥30
min) in certain experiments
For EGFR clustering, SW480 cells were cultured on
glass coverslips in 6-well tissue culture plates to 70% confluence
and serum-starved for 48 h. For experiments testing compounds,
cells were pre-incubated at 37°C for 30 min in the absence or
presence of the drug, followed by 15 min at 4°C prior to the
addition of equivalent volume of cold (4°C) serum-free culture
medium containing 250 ng/ml (stock) recombinant (r)EGF (cat. no.
E9644; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) to achieve a
final concentration of 125 ng/ml rEGF. The cells were then
incubated for 60 min at 4°C to allow EGF binding. The cells were
warmed to 37°C over 25 min to stimulate EGF clustering and EGFR
internalisation, washed with PBS, fixed for 5 min with
acetone/methanol (-20°C; 1:1, v/v) and incubated with blocking
buffer for 60 min at room temperature. The cells were then
incubated with anti-EGFR (D38B1) XP® rabbit antibodies
(Alexa Fluor 488-conjugate; 1:100) for 60 min in blocking buffer at
37°C, washed with PBST (0.02%) and the coverslips were placed onto
a drop of Prolong Gold antifade reagent with DAPI mounting medium
(cat no. P36935; Invitrogen; Thermo Fisher Scientific, Inc.) prior
to fluorescence microscopy (×400). Aspirin, DiA, F-DiA and PN529
were added at a final concentration of 0.5 or 1 mM to independent
wells in 6-well plates. The controls without aspirin analogue were
fixed following EGF binding for 60 min (no EGF internalisation) or
following warming of the cells for 25 min at 37°C (with EGF
internalisation).
For EEA1 staining, SW480 cells were seeded onto
glass coverslips at ~0.3×106 cells/well and cultured to
~70% confluence in 6-well tissue culture plates and serum-starved
for 48 h. For experiments for preliminary testing of compounds,
serum-starved cells were preincubated at 37°C for 30 min in the
absence or presence of compounds, followed by 15 min at 4°C, prior
to addition of the equivalent volume of cold serum-free culture
medium containing 200 ng/ml Alexa Fluor 555-EGF (final
concentration of 100 ng/ml) and incubation on ice for 1 h to allow
EGF binding. In later experiments, the concentration was reduced to
20 ng/ml. Cells were warmed to 37°C over 30 min to stimulate EGF
internalisation, washed with cold (4°C) PBS thrice and fixed for 5
min with acetone/methanol (1:1, v/v) at −20°C on ice. Cells were
washed with cold PBS and blocked in blocking buffer [3% (w/v) BSA
and 0.2% (v/v) Tween-20 in PBS] for 2 h at room temperature in the
dark. Anti-EEA1 antibody was added to cells at 1:1,000 dilution and
incubated at 4°C overnight with gentle rocking. Cells were washed
with cold (4°C) blocking buffer [3% (w/v) BSA and 0.2% (v/v)
Tween-20 in PBS] thrice and incubated with goat anti-mouse IgG
H&L-fluorescein isothiocyanate (ab6785; 1:1,000; Abcam)
secondary antibody for 2 h at room temperature. Cells were then
washed with cold blocking buffer [3% (w/v) BSA and 0.2% (v/v)
Tween-20 in PBS] thrice and with cold PBS prior to placing
coverslips onto a drop of VectaShield mounting medium. Microscopy
was performed using a Zeiss LSM 880 confocal microscope, equipped
with 405 and 561 nm excitation lasers using a 40×/1.30 NA oil
immersion DIC M27 objective.
Image acquisition and processing
Images were acquired and processed with Olympus
compatible software (IPLab Imaging software v3.1; BD Biosciences,
San Jose, CA, USA) or Zeiss software [ZEN 2.3 lite (blue edition),
or Zeiss LSM Image Examiner Version 3.2.0.115; both Carl Zeiss AG,
Oberkochen, Germany] and minor brightness, contrast and tonal
alterations were performed with Adobe Photoshop CS6 (Adobe Systems,
Inc., San Jose, CA, USA) or GIMP (https://www.gimp.org/).
For semi-quantitative determination of EGF
internalisation, the goal of development of the proximity and
density quotient (PDQ) was to establish a metric that provides a
quantitative estimate of the degree of localisation of labelled EGF
to the cell nucleus. A way of doing this would be to determine the
mean density value of the protein molecules and another density
value in the vicinity of the nuclei and calculate a quotient. A
visual inspection of the data to be analysed demonstrated an
effect, whereby the clustering around one nucleus would have a
major effect on the overall density around a neighbouring nucleus.
For this reason, it is necessary to count only those molecules,
which are unambiguously within the influence of the nucleus being
studied. This is achieved by defining the region of interest for
each nucleus. Each molecule is allocated to a region defined by the
closest nucleus. Within this region, the evenness of the
distribution of proteins is measured by counting them within an
area around each nucleus. The outer area is a torus extending from
the full radius of the measurement area to half of that radius. The
inner area is a disc within the torus. The PDQ for an individual
nucleus is the ratio of the number of molecules counted in the
outer area to those counted in the overall area. A larger PDQ
indicates reduced clustering of molecules around the nucleus.
Formally, the calculation of the PDQ is performed as follows: A set
of nuclei, N, and a set of protein molecules, M were defined. A set
of protein molecules associated with each nucleus, n, was
identified as:
with dist(m,n) as the Cartesian distance of the molecule m from the
centroid of the nucleus n.
Then, sets of molecules proximate to each nucleus
were identified as:
with rad(n) as the mean radius of the nucleus and k as an empirical
constant, selected to maximise discrimination of PDQ in the control
sets.
The PDQ for a molecule is calculated as the ratio of
the difference of the cardinalities of Pn and
Qn and the cardinality of Pn.
The tools used to calculate the PDQ were
CellProfiler (43) and the Python
programming language (https://www.python. org/). CellProfiler was used to
identify, count and locate the two objects of interest, nuclei and
protein molecules. The staining of the slides provided a clear
contrast between the red-rendered proteins and the blue-rendered
nuclei, thus the first operation was to split the image by colour
channel. The nuclei required processing in order to be reliably and
unambiguously identified, which included removal of objects <50
pixels across, smoothing and thresholding. Without the latter
steps, the segmentation algorithm would split single nuclei into
several objects. Identification of protein molecules required
suppression of objects <3 pixels. It is to be expected that
different slide processing would require tuning of the image
processing in order to reliably identify the two types of objects.
CellProfiler provided tables of data listing each of the objects
identified, its position and size. A Python program was written,
which implemented the algorithm described above. It was used to
further process these data. The value of k, which provided best
differentiation between the positive and negative control sets, was
4.
Compound synthesis
Full details of all the compounds referred to in the
present study are provided in Table
I. Aspirin analogues were, wherever possible, synthesised as
previously described (26,27). Meta-apirin (PN548) and
para-aspirin (PN549) were made by a standard aspirin
synthesis using 3-hydroxy and 4-hydroxybenzoic acids as starting
materials, in lieu of salicylic acid. The three isomeric
thioaspirin analogues (PN590, PN591 and PN592, represented as
ortho-TASP, meta-TASP and para-TASP
respectively) were prepared in good yields (~70%) from the
corresponding mercaptobenzoic acids, by reaction with acetic
anhydride in ice-cold sodium hydroxide solution, as previously
described (44). In the present
study, sodium hydroxide, rather than the potassium salt, was used
without observing any changes. While the previously published
method (39) only describes the
preparation of the meta- and para-isomers (meta-TASP and
para-TASP) it was demonstrated that the method also yields
the ortho-isomer (ortho-TASP). The crude products in each
case were demonstrated by thin layer chromatography and melting
point comparison to be suitable for use without any further
purification. Details of the additional compounds synthesised
specifically for use in the present study are provided below. All
percentage yields were comparable to the published values and
within 15%. NMR spectra were recorded on a Jeol ECS-400
instrument.
Meta-aspirin (3-acetoxybenzoic acid): White
powder; melting point (mpt), 131-134°C (literature, 131-134°C);
13C-NMR (CDCl3) δ=171.19, 169.32, 150.75,
130.88, 129.68, 127.72, 127.33, 123.53, 21.14 ppm; Infra Red (IR),
3,400-2,200, 1,758, 1,675, 1,584 cm−1.
Para-aspirin (4-acetoxybenzoic acid): White
powder; mpt, 183-185°C (literature, 187°C; 190-194°C);
13C-NMR (CDCl3) δ=171.31, 168.96, 155.06,
131.96, 126.88, 121.85, 21.25 ppm; IR 3500-2200, 1754, 1677, 1602
cm−1.
Thioaspirin (ortho-TASP): White powder; mpt,
122-125°C (literature, 125.5-126°C) (45); 13C-NMR
(CDCl3) δ=193.27, 171.48, 136.82, 132.88, 132.66,
131.92, 129.49, 129.40, 30.45 ppm; IR 3,380-1,960, 1,694, 1,676,
1,582, 1,565 cm−1.
Meta-thioaspirin (meta-TASP): White
powder; mpt, 153-156°C (literature, 152-153°C) (44); 13C-NMR
(CDCl3) δ=193.29, 171.09, 139.85, 136.16, 131.19,
130.43, 129.46, 128.88, 30.38 ppm; IR 3,380-1,990, 1,685
(overlapping thioester and -COOH C=O spectra), 1,583, 1,573
cm−1.
Para-thioaspirin (para-TASP): White
powder; mpt, 200-202°C (literature, 202.5-203.5°C) (44); 13C-NMR
(CDCl3) δ=171.31, 168.96, 155.06, 131.96, 126.88,
121.85, 21.25 ppm; IR 3,380-1,950, 1,683 (overlapping thioester and
-COOH C=O spectra), 1,592, 1,566 cm−1.
Results and Discussion
EGF and EGFR internalization
EGFR is an ~170-kDa transmembrane receptor tyrosine
kinase, belonging to the ErbB family of signalling receptors, whose
ligands include EGF and transforming growth factor-α, with roles in
cellular proliferation, survival and differentiation (46,47).
Dysfunction in the EGFR axis as a consequence of receptor
overexpression, amplification and/or mutation has been reported in
colorectal, breast, brain, lung, pancreatic, oesophageal and head
and neck neoplasias (48-52). EGFR and NF-κB signalling are
intimately linked (53,54) and it has been reported that the
aspirin analogues DiA and F-DiA can perturb NF-κB activity in SW480
cells and that salicylates can potentially antagonise wound
healing. Therefore, the aim of the present study was to investigate
the effect of aspirin and aspirin-like molecules (26,27)
on EGF endocytosis in the context of a rapid cell biological
effect, in contrast to many experiments that examine the
consequences of compound exposure of cells and tissues over
protracted time frames (days). Compound toxicity was determined by
the MTT assay and the IC50 values are presented in
Table I. The aspirin analogues
synthesised and tested contain modifications to either the number
of rings, to the position of the acetyl group and/or have a
substituted thiol group. Diflunisal is a difluorophenyl derivative
of salicylic acid and is a clinically utilised NSAID with analgesic
and anti-inflammatory properties.
In preliminary experiments to assess compound
effects on EGF internalisation, a commercially available
fluorescence-labelled, pH-insensitive conjugate of EGF was used.
Serum-starved SW480 cells were incubated with aspirin analogues,
cooled on ice to inhibit endocytosis and allowed EGF-conjugate
binding for 1 h prior to tracking the internalised EGF by confocal
and DIC microscopy (Fig. 1) and
immunofluorescence analysis (data not shown). In SW480 cells
incubated in the absence of compounds, following ~25 min of warming
to 37°C, EGF was observed to internalise with clustering often in a
perinuclear location, with the nucleus clearly visible (Fig. 1). However, in the presence of
aspirin, DiA and F-DiA, internalisation and perinuclear clustering
was inhibited. In cells incubated with DiA, substantial clustering
was noted to persist at or near the membrane, whereas with F-DiA,
staining was notably diffused. These data suggested that compounds
based on salicylate may perturb EGF internalisation and affect
signalling, given that correctly regulated EGF internalisation and
signalling via endosomes are associated (55,56).
To examine this phenomenon further, serum-starved SW480 cells were
incubated with compounds prior to stimulation with recombinant
human EGF (125 ng/ml) and EGFR localisation was probed by
immunofluorescence analysis using a commercially available
anti-EGFR monoclonal antibody. Peripheral, presumably membranous
staining, was clearly visible in the absence of warming (Fig. 2A), indicating that the EGFR is
robustly expressed in the SW480 cell line, as previously reported
(29). Internalised punctate
staining of the EGFR demonstrated close proximity to the nucleus
when the cells were warmed to 37°C within 25 min (Fig. 2B). In the presence of aspirin, DiA
and F-DiA, during the internalisation of the receptor, staining was
substantially more diffuse with all compounds (Fig. 2C-E). Furthermore, when cells were
incubated with PN529 (isopropyl m-bromobenzoylsalicylate), a
toxic brominated salicylate (27),
EGFR staining was markedly dissimilar compared with the control
cells (Fig. 2B and F). Clustering
of labelled EGFR to perinuclear locations was consistently observed
following a 25-min incubation in the absence of compounds in cells
warmed to 37°C. However, in the presence of aspirin and, more
prominently, in the presence of DiA, F-DiA and isopropyl
(m-bromobenzoylsalicylate), discrete perinuclear clustering was
substantially inhibited. These data suggest that aspirin and
analogues rapidly perturb EGFR internalisation, and this
observation may not solely be caused by a reduction in binding, as
staining observed with fluorescence-tagged EGF was robust even
following aspirin, F-DiA, PN524 or ortho-TASP treatment
(data not shown). Whilst the concentration of the tested compounds
in the experiments may appear high in comparison with other
chemotherapeutic agents, a range of 1-5 mM or above is not uncommon
in in vitro experiments investigating the molecular action
of aspirin or salicylate (21,22,57,58),
with levels of 0.5-2 mM also reported as being physiologically or
therapeutically relevant by a number of investigators (59-62).
EGF endocytosis in the presence of
salicylate derivatives
Ligand binding leads to conformational changes in
EGFR and EGFR dimerization (63-65),
autophosphorylation of the cytoplasmic tyrosine kinase domain with
subsequent downstream signalling through the RAS [mitogen-activated
protein kinase (MAPK)], phosphoinositide 3-kinase (AKT) and janus
kinase (JAK; signal transducer and activator of transcription
factor 3) pathways (53,66,67).
Concomitantly, the ligand receptor complex is internalised via
clathrin-dependent and -independent routes, depending on EGF
levels, with clathrin-mediated endocytosis prolonging signalling
(68-70). Signalling from endosomes with
activated EGFR may persist (71,72)
and the activated EGFR may ultimately be recycled or degraded
(73). Indeed, internalised
endosomes may be considered as signalling platforms (56). An early destination for
internalisation is the EEA1-positive endosomal compartment
depending on whether or not a clathrin-mediated endocytosis
occurred (74). Clustering of late
endosomes is at a perinuclear location and mislocalised endosomes
can compromise signalling quality (55). In addition, it has been suggested
that EGFR can translocate to the nucleus and signal therein
(75-78).
EEA1 is a Rab5 effector, intimately involved in
tethering, docking and fusion of early endosomes (79) and mediating crosstalk between
signalling pathways (80). It was
therefore examined whether spatial and temporal regulation of the
internalisation of EGF was perturbed by examining the
colocalisation of Alexa Fluor 555-labelled EGF (100 ng/ml) and EEA1
following a chase in the absence and presence of aspirin and
aspirin analogues. This was performed while additionally
controlling pH alterations with HEPES (10 mM at pH 8; Fig. 3) or phosphate buffers (data not
shown) to minimise acidification caused by the addition of the
aspirin derivatives. Buffering did not appear to attenuate the
effects exhibited by aspirin and aspirin analogues. Peripheral EGF
staining, interpreted as membrane-localised, was apparent without a
chase, whereas EGF internalisation to a perinuclear location
occurred in the absence of compounds upon warming to 37°C in the
vehicle control group (Fig. 3). In
the presence of compound at 0.5 mM, a concentration used to reduce
any non-specific effects caused by acidification, and under
buffered conditions, aspirin or aspirin analogues rapidly perturbed
EGF internalisation (Fig. 3A).
Colocalisation of EEA1 and EGF was consistently enhanced, as
evidenced by dual staining, when cells were incubated with F-DiA,
suggesting that the pathway of internalisation of EGF in the
presence of F-DiA is substantially altered in comparison to aspirin
and PN524, a brominated salicylate exhibiting an intermediate
toxicity between aspirin and F-DiA (27) to SW480 cells. Furthermore,
endosomal EEA1 staining was enhanced, when cells were incubated
with F-DiA, based on the intensity of the fluorescence in the
perinuclear location.
EGF internalisation (at 100 ng/ml) was analysed
semi-quantitatively with PDQ measurements of the labelled EGF
relative to the nucleus (Fig. 3B)
in the presence of 0.5 mM aspirin, PN517 (F-DiA) or diflunisal. A
larger PDQ indicated reduced clustering of molecules around the
nucleus. Significant perturbation of internalisation was noted with
all three compounds. It was previously demonstrated that F-DiA is
significantly more effective at inducing apoptosis in CRC cells
compared with aspirin, strongly reduces cyclin D1 levels and
inhibits NF-κB activity in vitro (27).
Given that endocytosis of the EGFR has been proposed
to occur via a number of pathways, depending on the saturability of
EGF ligand-induced receptor internalisation (42,81,82),
the cells were additionally stimulated with relatively low EGF
concentrations (20 ng/ml) under buffered conditions (HEPES). These
experiments were performed to elucidate the influence of the tested
compounds on EGF and EGFR internalisation and further utilised EEA1
staining (Fig. 4). Similar to
experiments using 100 ng/ml EGF, aspirin and F-DiA perturbed EGF
internalisation compared with the vehicle control and
colocalisation of EGF with EEA1 was markedly enhanced as a
consequence of a brief incubation with 0.5 mM F-DiA.
The universality of the phenomenon of perturbing EGF
internalisation at 20 ng/ml was examined, with a wider range of
salicylate derivatives (at 0.5 mM), including diflunisal.
Dysregulation in EGF internalisation was noted with all compounds
under HEPES-buffered conditions (Fig.
5) compared with the vehicle control, including salicylic acid
per se (Fig. 5A), aspirins
with modifications to the position of the acetyl group (Fig. 5B and F) and/or compounds with an
additional thiol group (Fig. 5D, G and
H). When internalisation occurred in the presence of compound,
it was often more diffuse than normal and the outline of the cell
membrane was visible in some instances, a phenomenon particularly
pronounced when the cells were incubated with diflunisal (Fig. 5E). Images were analysed and the PDQ
of the labelled EGF relative to the nucleus was determined
(Fig. 5I). In summary the data
strongly suggested that salicylates and derivatives, at modest
concentrations, rapidly altered EGF internalization and
endocytosis. The titratability of the inhibition of EGF
internalisation was tested for dose-dependency in the SW480 cell
line qualitatively with aspirin, F-DiA (PN517) and diflunisal
(selected because of clinical relevance and cytotoxicity) at
concentrations below 0.5 mM (Fig.
6), and although observable with aspirin and F-DiA, was most
notably observed with diflunisal, thus arguing against a
non-specific phenomenon associated with the experimental
design.
 | Figure 5Effect of salicylates and aspirin
analogues on EGF internalisation (20 ng/ml). Confocal analysis of
SW480 colorectal cancer cells incubated with Alexa Fluor 555-EGF
and preincubated in the absence/ presence of 0.5 mM of the
following compound under HEPES-buffered conditions: (A) SA, (B)
meta-aspirin, (C) DiA, (D) meta-TASP, (E) DIF, (F)
para-aspirin, (G) ortho-TASP and (H)
para-TASP. Images were acquired at 405 nm for DAPI nucleic
stain (blue) and 561 nm for Alexa Fluor-EGF (red). Representative
images were taken at ×40 magnification, with an oil/1.30 NA oil
immersion objective (n=3). Limited clustering of EGF to a
perinuclear location occurred upon warming cells (arrowheads), but
was absent with diflunisal. (I) Nuclear PDQ of labelled EGF was
determined by image analysis of three repeated experiments; data
points represent mean ± standard error of the mean. Statistical
analysis comparing the PDQ of unstimulated and warmed cells with
and without stated compounds was performed using Kruskal-Wallis
with Dunn’s post-hoc test. *P<0.05,
**P<0.01 and ***P<0.001 vs. EGF
(warmed). EGF, epidermal growth factor; PDQ, proximity and density
quotient. SA, salicylic acid; DiA, diaspirin; DIF, diflunisal;
ortho-TASP, ortho-thioaspirin; meta-TASP,
meta-thioaspirin; para-TASP,
para-thioaspirin. |
Aspirin and aspirin analogue effects on
EGFR and EGFR phosphorylation
Non-specific inhibition of kinase activity has been
proposed as a mechanism of action for aspirin and sodium salicylate
(59). Hence, compound effects on
EGFR phosphorylation, focusing on cytoplasmic phosphotyrosine
residues in the ligand-induced autophosphorylation domain were
examined, including Tyr1045, Tyr1068 and Tyr1173. Phosphorylated
Tyr1045 acts a site for c-Cbl interaction that results in receptor
ubiquitylation and degradation of EGFR, and Cbl regulates receptor
downregulation and signalling (83). Phosphorylation of Tyr1068 enhances
binding of the growth factor receptor bound protein 2 (Grb2), which
is necessary for entry into coated pits (41) and Ras/MAPK activation (84,85).
Phospho-Tyr1173 is involved in binding the tyrosine phosphatase
SHP1 and signalling via recruitment of the adaptor proteins SHC and
Grb2, resulting in control of the ERK activity (86-88).
In the present study, SW480 cells were incubated overnight with the
compounds of interest and briefly stimulated with 200 ng/ml rEGF (5
min) in serum-free medium. EGFR phosphorylation status was analysed
by immunoblotting (Fig. 7A). The
data suggested that inhibition of EGFR phosphorylation upon
stimulation, by aspirin (ortho and meta) was weak. Tyr1045 and
Tyr1173 phosphorylation was more sensitive to the presence of
diaspirins, including F-DiA (PN517) and PN524, and thioaspirins,
including ortho-TASP, meta-TASP and para-TASP,
compared with Tyr1068. Thioaspirin (ortho-TASP) was the sole
compound able to inhibit phosphorylation at all sites tested
herein. These findings suggested that aspirin and analogues at the
concentration tested (0.5 mM), even when cells are stimulated with
high rEGF concentrations (200 ng/ml), did not universally inhibit
all autophosphorylation sites, which argues against aspirin or its
analogues acting solely as a non-specific kinase inhibitor, as
suggested by Frantz et al (59). Due to the sensitivity of Tyr1045 to
the compounds and its role in receptor degradation, the compound
effects on EGF phosphorylation at this site with a 2-h incubation
period were further examined and modest changes were observed by
immunoblot analysis (Fig. 7B). As
aspirin was reported to normalise EGFR expression (89), the effect of aspirin and its
analogues on total EGFR levels in SW480 cells was further
investigated following a 24-h incubation (Fig. 7C). SW480 EGFR levels were
substantively affected by diflunisal, but weaker changes were
observed as a result of incubation with F-DiA (PN517), PN524 and
PN529, with little evidence of a significant reduction in EGFR by
aspirin or salicylate following a 24-h incubation. pEGFR levels
relative to total EGFR at pY1068 and pY1173 sites was totally
inhibited by ortho-TASP and significantly reduced at pY1045
and pY1173 phosphorylation sites by meta-TASP,
para-TASP, F-DiA, DiA and PN524. The pEGFR level at pY1068,
pY1045 and pY1173 was increased by aspirin, meta-aspirin and
para-aspirin (Fig. 7D).
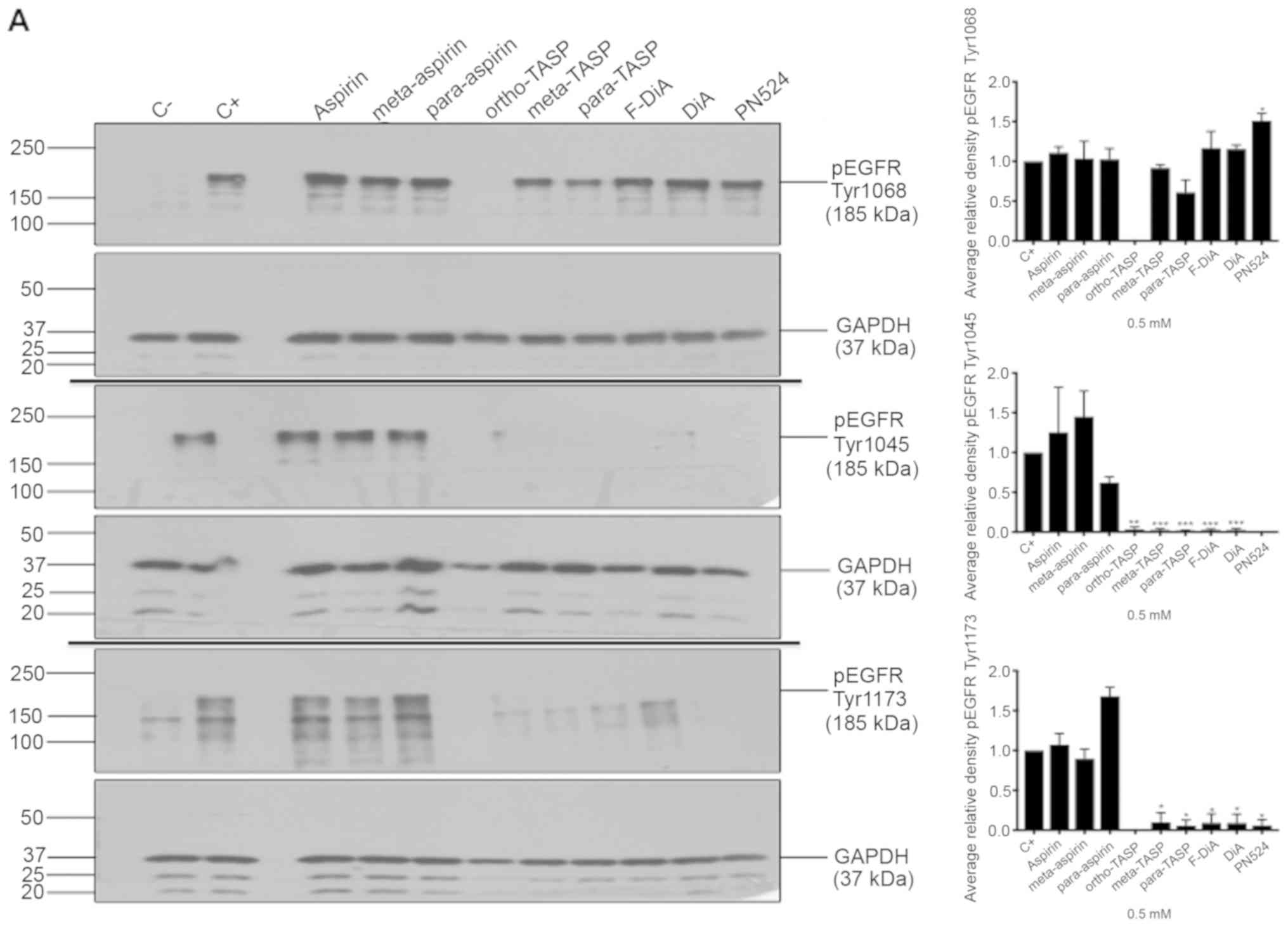 | Figure 7Effect of aspirin and analogues on
pEGFR tyrosine kinase phosphorylation sites and EGFR. SW480
colorectal cancer cells were incubated with human rEGF (200 ng/ml)
for 5 min following being treated with 0.5 mM compound (HEPES
buffered) for (A) 24 h and probed with anti-human EGFR pY1068,
pY1045 and pY1173 antibodies accompanied by corresponding
histograms, (B) 2 h and probed with anti-human EGFR pY1045 and (C)
24 h and probed with an anti-human EGFR (D38B1) XP rabbit antibody.
(D) Quantification of the effect of these compounds on pEGFR
tyrosine kinase phosphorylation sites 1,068, 1,045 and 1,173 in
relation to total EGFR expression. Cell lysates were analysed by
SDS-PAGE and immunoblotting. pEGFR and EGFR bands migrated at ~185
and ~180 kDa, respectively, molecular weight was estimated by
interpolation using Precision Plus colour standards. Band
intensities were quantified relative to GAPDH. C- represents
unstimulated and untreated cells; C+ represents untreated cells
stimulated with rEGF for 5 min. Data presented are the mean of
three individual experiments ± standard error of the mean.
*P<0.05, **P<0.01 and
***P<0.001 vs. C+ (one-way analysis of variance
followed by Dunnett’s test). EGF, epidermal growth factor; EGFR,
EGF receptor; pEGFR, phosphorylated EGFR; rEGF, recombinant EGF; Y,
tyrosine; SA, salicylic acid; DIF, diflunisal. |
Taken together, these data suggested that the EGFR
axis was sensitive to aspirin and its derivatives. These compounds
may have a rapid and profound impact on EGF endcoytosis and
autophosphorylation (particularly ortho-TASP with overnight
incubation) and, furthermore, to a limited extent, EGFR expression,
with the exception of diflunisal. Diflunisal markedly reduced EGFR
expression and perturbed EGF internalisation, an observation that,
to the best of our knowledge, has not been previously reported for
a prescription drug used in the treatment of osteoarthritis and
rheumatoid arthritis. Given that endocytosis of EGF is rapid with
migration of the ligand-receptor as a unit to a perinuclear
location and then recycled being a canonical pathway for EGF
(90) and the role of the
proteasome in the processing and degradation of EGFR (91), it would be of interest to determine
whether there is any association between diflunisal use and
proteasomal degradation of EGFR.
Compound effects on EGF internalisation
in Flo-1 and OE33 oesophageal cancer cells
It was further explored whether the phenomenon
reported herein was restricted to SW480 cells. To that end and due
to evidence that aspirin is chemopreventive in oesophageal cancer
(92,93), Alexa Fluor 555-EGF internalisation
was examined in the presence of the stated compounds with HEPES
buffering in Flo-1 and OE33 oesophageal cancer cells. It was
observed that aspirin and F-DiA perturbed EGF internalisation (at
20 ng/ml) in the oesophageal cancer cell lines (Fig. 8). EGF internalisation was markedly
affected by F-DiA (PN517) in the Flo-1 cells, whereas a modest
effect was observed with aspirin.
The present study details a rapid and hitherto
unreported effect of aspirin and analogues, which are likely to
yield salicylates upon metabolism within cells and tissues, on EGF
endocytosis. The study has relevance to wound healing, cellular
proliferation and chemoprevention. It addressed the analyses at
physiologically achievable levels (≤1 mM) of aspirin (60,61)
and analogues, and examined EGF internalisation and EGFR
phosphorylation with what is considered to be an experimentally
legitimate concentration of EGF (20 ng/ml) under buffered
conditions, as acidification of the cytosol may inhibit
endocytosis.
Given the number of compounds tested and for the
purposes of clarity the effect of compounds on EGF at unitary
values of either 0.5 or 1 mM was investigated. It is recognised
that these values are in certain cases >IC50 values
for SW480 cells reported herein but with the exception of
diflunisal the fold-differences are relatively modest; however,
plasma concentrations of diflunisal range from 150-350 µM
(22). Most significantly, the
concentrations employed for salicylate and aspirin tested were
markedly <IC50 values and within a range (0.5-2 mM)
that has been reported to be physiologically relevant.
Although a substantial body of published work
pertains to the effects of aspirin and NSAIDS on gene expression
and apoptosis in cells of cancerous origin (26,27),
experiments often focus on outcomes over hours/days. It was
observed that ligand-induced activation of receptor tyrosine
kinases is rapid (within minutes); therefore, EGF endocytosis was
investigated in the presence of aspirin/salicylates within time
frames relevant to EGF signalling and endocytosis.
With respect to the findings presented herein, p38
MAPK was proposed as a potential rapid target, based on previously
published work. Cyclin D1 is overexpressed in cells with EGFR
mutations (94). It has previously
been reported that, in SW480 cells treated with F-DiA, cyclin D1
levels are significantly reduced and NF-κB signalling is attenuated
(27). It has previously been
reported that cyclin D1 is reduced in CRC cells treated with
aspirin, a phenomenon which has been reported to occur in response
to p38 MAPK activation within minutes (95). Furthermore, salicylate exposure to
COS-1 cells (58) can activate p38
MAPK also within minutes. A number of studies have implicated p38
MAPK as regulator of endocytic trafficking (96,97)
and EGFR internalisation (98).
p38 MAPK can regulate mu opioid receptor endocytosis, with the
early endosomal antigen EEA1 identified as a substrate for p38
MAPK, which impacts on Rab5 activity regulating endocytosis
(99). It is suggested that the
rapid dysregulation of endocytosis is caused by salicylates and
aspirin rapidly activating p38 MAPK and disrupting receptor
trafficking, perturbing EGF signalling and ultimately controlling
cyclin D1 levels. This may be facilitated by salicylate
persisting/accumulating as a result of its long half-life
determined by pharmacokinetics analysis (100), and inhibition of NF-κB and
further known downstream targets. The relatively long half-life of
salicylate may be relevant to the chemopreventive nature of
low-dose aspirin: Accumulation in tissue reduces inflammation and
may further attenuate EGF signalling. These findings suggest that
aspirin and salicylates may be useful in cancer treatment, where
EGFR amplification, overexpression and constitutive activation in a
cancer is notable, e.g., in glioblastoma (101).
With respect to the question of non-salicylate
NSAIDs also impacting on the EGFR signalling pathway,
notwithstanding concerns regarding conflicting evidence regarding
the efficacy of other NSAIDs (in comparison with aspirin) in
reducing human cancer risk, e.g., in breast cancer (102), the evidence in the literature to
date, as far as we are aware, other than work focused on sulindac
affecting EGFR activation/inhibition, directly is limited. There
are, however, reports of interrelationships between EGFR and PGE2:
for example, activation of EGFR by EGF has been reported to
stimulate PGE2 production (103).
Given the fundamental role of EGF signalling in
cancer biology, the findings reported in the present study have
relevance to the understanding of the chemopreventive activity of
aspirin, particularly with respect to COX-independent pharmacology.
A schematic illustration of the effect of selected compounds on EGF
internalisation is presented in Fig.
9, based upon the representation of the endosomal system by
Taub et al (55). It is
suggested that further research is required to evaluate the rapid
effect of salicylates on the EGFR axis, with a focus on MAPK
activation and EGF/EGFR trafficking and phosphorylation.
Furthermore, experimentation on EGF signalling in normal tissues
exposed to aspirin or diflunisal is required to establish the
physiological relevance of the observations reported herein. An
analysis of the response of other growth factors and their cognate
ligands to physiological concentrations of salicylates, with a
particular emphasis on growth factor trafficking and recycling,
appears timely. From a biochemical perspective, salicylate is a
lipophilic monohydroxybenzoic acid that can interact with lipid
membranes (104,105). It is also conceivable that
interference of salicylates with membrane structure may affect
endocytosis (106).
Funding
The present study was supported by the Research
Institute in Healthcare Science at the University of Wolverhampton
(Wolverhampton, UK).
Availability of data and materials
The analysed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors’ contributions
All the authors have read and approved the final
version of this manuscript. AIJB, CSK, STS, CJP and IDN designed
and performed the experiments, analysed the data and wrote the
manuscript. RMN wrote the Python program for proximity and density
quotient analysis, and SJ contributed to data analysis and
manuscript preparation.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
CJP and IDN are named inventors for a patent for
fumaryldiaspirin as an anti-colorectal cancer agent (US patent no.
9351980).
Acknowledgments
The authors wish to thank Dr Michael Cornes (New
Cross Hospital, Wolverhampton, UK) for discussions regarding
hydrolysis of aspirin analogues and Ms Clare Murcott (Faculty of
Science and Engineering, University of Wolverhampton,
Wolverhampton, UK) for assistance with confocal microscopy.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
EEA1
|
early endosome antigen 1
|
|
MAPK
|
mitogen-activated protein kinase
|
|
NSAID
|
non-steroidal anti-inflammatory
drug
|
References
|
1
|
Garcia-Albeniz X and Chan AT: Aspirin for
the prevention of colorectal cancer. Best Pract Res Clin
Gastroenterol. 25:461–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung JJ, Lau JY, Goh KL and Leung WK; Asia
Pacific Working Group on Colorectal Cancer: Increasing incidence of
colorectal cancer in Asia: Implications for screening. Lancet
Oncol. 6:871–876. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuriki K and Tajima K: The increasing
incidence of colorectal cancer and the preventive strategy in
Japan. Asian Pac J Cancer Prev. 7:495–501. 2006.PubMed/NCBI
|
|
4
|
Siegel RL, Fedewa SA, Anderson WF, Miller
KD, Ma J, Rosenberg PS and Jemal A: Colorectal Cancer Incidence
Patterns in the United States, 1974–2013 = J Natl Cancer Inst.
109:djw3222017.
|
|
5
|
Shaheen NJ, Straus WL and Sandler RS:
Chemoprevention of gastrointestinal malignancies with nonsteroidal
antiinflam-matory drugs. Cancer. 94:950–963. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sandler RS, Halabi S, Baron JA, Budinger
S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi
CL, et al: A randomized trial of aspirin to prevent colorectal
adenomas in patients with previous colorectal cancer. N Engl J Med.
348:883–890. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan AT, Arber N, Burn J, Chia WK, Elwood
P, Hull MA, Logan RF, Rothwell PM, Schrör K and Baron JA: Aspirin
in the chemoprevention of colorectal neoplasia: An overview. Cancer
Prev Res (Phila). 5:164–178. 2012. View Article : Google Scholar
|
|
8
|
Bibbins-Domingo K: Aspirin Use for the
Primary Prevention of Cardiovascular Disease and Colorectal Cancer:
U.S. Preventive Services Task Force Recommendation Statement. Ann
Intern Med. 164:836–845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liao X, Lochhead P, Nishihara R, Morikawa
T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, et
al: Aspirin use, tumor PIK3CA mutation, and colorectal-cancer
survival. N Engl J Med. 367:1596–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liggett JL, Zhang X, Eling TE and Baek SJ:
Anti-tumor activity of non-steroidal anti-inf lammatory drugs:
Cyclooxygenase-independent targets. Cancer Lett. 346:217–224. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thun MJ, Henley SJ and Patrono C:
Nonsteroidal anti-inflammatory drugs as anticancer agents:
Mechanistic, pharmacologic, and clinical issues. J Natl Cancer
Inst. 94:252–266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brown JR and DuBois RN: COX-2: A molecular
target for colorectal cancer prevention. J Clin Oncol.
23:2840–2855. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan AT, Ogino S and Fuchs CS: Aspirin and
the risk of colorectal cancer in relation to the expression of
COX-2. N Engl J Med. 356:2131–2142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bos CL, Kodach LL, van den Brink GR, Diks
SH, van Santen MM, Richel DJ, Peppelenbosch MP and Hardwick JC:
Effect of aspirin on the Wnt/beta-catenin pathway is mediated via
protein phosphatase 2A. Oncogene. 25:6447–6456. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kopp E and Ghosh S: Inhibition of NF-kappa
B by sodium salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu XM, Sansores-Garcia L, Chen XM,
Matijevic-Aleksic N, Du M and Wu KK: Suppression of inducible
cyclooxygenase 2 gene transcription by aspirin and sodium
salicylate. Proc Natl Acad Sci USA. 96:5292–5297. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin MJ, Yamamoto Y and Gaynor RB: The
anti-inflammatory agents aspirin and salicylate inhibit the
activity of I(kappa)B kinase-beta. Nature. 396:77–80. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goel A, Chang DK, Ricciardiello L, Gasche
C and Boland CR: A novel mechanism for aspirin-mediated growth
inhibition of human colon cancer cells. Clin Cancer Res. 9:383–390.
2003.PubMed/NCBI
|
|
19
|
Rüschoff J, Wallinger S, Dietmaier W,
Bocker T, Brockhoff G, Hofstädter F and Fishel R: Aspirin
suppresses the mutator phenotype associated with hereditary
nonpolyposis colorectal cancer by genetic selection. Proc Natl Acad
Sci USA. 95:11301–11306. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Din FV, Valanciute A, Houde VP, Zibrova D,
Green KA, Sakamoto K, Alessi DR and Dunlop MG: Aspirin inhibits
mTOR signaling, activates AMP-activated protein kinase, and induces
autophagy in colorectal cancer cells. Gastroenterology.
142:1504–1515.e3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hawley SA, Fullerton MD, Ross FA,
Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green
KA, Mustard KJ, et al: The ancient drug salicylate directly
activates AMP-activated protein kinase. Science. 336:918–922. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shirakawa K, Wang L, Man N, Maksimoska J,
Sorum AW, Lim HW, Lee IS, Shimazu T, Newman JC, Schröder S, et al:
Salicylate, diflunisal and their metabolites inhibit CBP/p300 and
exhibit anticancer activity. Elife. 5:pii: e11156. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pangburn HA, Kraus H, Ahnen DJ and Rice
PL: Sulindac metabolites inhibit epidermal growth factor receptor
activation and expression. J Carcinog. 4:162005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Selvendiran K, Bratasz A, Tong L, Ignarro
LJ and Kuppusamy P: NCX-4016, a nitro-derivative of aspirin,
inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in
cisplatin-resistant human ovarian cancer cells and xenografts. Cell
Cycle. 7:81–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cho M, Kabir SM, Dong Y, Lee E, Rice VM,
Khabele D and Son DS: Aspirin Blocks EGF-stimulated Cell Viability
in a COX-1 Dependent Manner in Ovarian Cancer Cells. J Cancer.
4:671–678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deb J, Dibra H, Shan S, Rajan S, Manneh J,
Kankipati CS, Perry CJ and Nicholl ID: Activity of aspirin
analogues and vanillin in a human colorectal cancer cell line.
Oncol Rep. 26:557–565. 2011.PubMed/NCBI
|
|
27
|
Claudius AK, Kankipati CS, Kilari RS,
Hassan S, Guest K, Russell ST, Perry CJ, Stark LA and Nicholl ID:
Identification of aspirin analogues that repress NF-κB signalling
and demonstrate anti-proliferative activity towards colorectal
cancer in vitro and in vivo. Oncol Rep. 32:1670–1680. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahmed D, Eide PW, Eilertsen IA, Danielsen
SA, Eknæs M, Hektoen M, Lind GE and Lothe RA: Epigenetic and
genetic features of 24 colon cancer cell lines. Oncogenesis.
2:e712013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shigeta K, Hayashida T, Hoshino Y,
Okabayashi K, Endo T, Ishii Y, Hasegawa H and Kitagawa Y:
Expression of epidermal growth factor receptor detected by
cetuximab indicates its efficacy to inhibit in vitro and in vivo
proliferation of colorectal cancer cells. PLoS One. 8:e663022013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Richter M, Weiss M, Weinberger I,
Fürstenberger G and Marian B: Growth inhibition and induction of
apoptosis in colorectal tumor cells by cyclooxygenase inhibitors.
Carcinogenesis. 22:17–25. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin PC, Lin YJ, Lee CT, Liu HS and Lee JC:
Cyclooxygenase-2 expression in the tumor environment is associated
with poor prognosis in colorectal cancer patients. Oncol Lett.
6:733–739. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Werner S and Grose R: Regulation of wound
healing by growth factors and cytokines. Physiol Rev. 83:835–870.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boonstra JJ, van Marion R, Beer DG, Lin L,
Chaves P, Ribeiro C, Pereira AD, Roque L, Darnton SJ, Altorki NK,
et al: Verification and unmasking of widely used human esophageal
adenocar-cinoma cell lines. J Natl Cancer Inst. 102:271–274. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheong E, Ivory K, Doleman J, Parker ML,
Rhodes M and Johnson IT: Synthetic and naturally occurring COX-2
inhibitors suppress proliferation in a human oesophageal
adenocarcinoma cell line (OE33) by inducing apoptosis and cell
cycle arrest. Carcinogenesis. 25:1945–1952. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song S, Honjo S, Jin J, Chang SS, Scott
AW, Chen Q, Kalhor N, Correa AM, Hofstetter WL, Albarracin CT, et
al: The Hippo Coactivator YAP1 Mediates EGFR Overexpression and
Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res.
21:2580–2590. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bosetti C, Talamini R, Franceschi S, Negri
E, Garavello W and La Vecchia C: Aspirin use and cancers of the
upper aerodigestive tract. Br J Cancer. 88:672–674. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bosetti C, Gallus S and La Vecchia C:
Aspirin and cancer risk: An updated quantitative review to 2005.
Cancer Causes Control. 17:871–888. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: Assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
40
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang X, Huang F, Marusyk A and Sorkin A:
Grb2 regulates internalization of EGF receptors through
clathrin-coated pits. Mol Biol Cell. 14:858–870. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang F, Khvorova A, Marshall W and Sorkin
A: Analysis of clathrin-mediated endocytosis of epidermal growth
factor receptor by RNA interference. J Biol Chem. 279:16657–16661.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Carpenter AE, Jones TR, Lamprecht MR,
Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA,
Moffat J, et al: CellProfiler: Image analysis software for
identifying and quantifying cell phenotypes. Genome Biol.
7:R1002006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bordwell FG and Boutan PJ: Conjugative
Effects in Divalent Sulfur Groupings1. J Am Chem Soc.
78:854–860. 1956. View Article : Google Scholar
|
|
45
|
Nelander L, Johansson G, Toplin I, Melera
A and Nilsson L: The heats of hydrolysis of aspirin, thioaspirin,
and their p-analogues. Acta Chem Scand. 18:973–984. 1964.
View Article : Google Scholar
|
|
46
|
Schneider MR and Wolf E: The epidermal
growth factor receptor ligands at a glance. J Cell Physiol.
218:460–466. 2009. View Article : Google Scholar
|
|
47
|
Yarden Y; The EGFR family and its ligands
in human cancer: signalling mechanisms and therapeutic
opportunities. Eur J Cancer. 37(Suppl 4): S3–S8. 2001. View Article : Google Scholar
|
|
48
|
Radinsky R, Risin S, Fan D, Dong Z,
Bielenberg D, Bucana CD and Fidler IJ: Level and function of
epidermal growth factor receptor predict the metastatic potential
of human colon carcinoma cells. Clin Cancer Res. 1:19–31.
1995.PubMed/NCBI
|
|
49
|
Voldborg BR, Damstrup L, Spang-Thomsen M
and Poulsen HS: Epidermal growth factor receptor (EGFR) and EGFR
mutations, function and possible role in clinical trials. Ann
Oncol. 8:1197–1206. 1997. View Article : Google Scholar
|
|
50
|
Citri A and Yarden Y: EGF-ERBB signalling:
Towards the systems level. Nat Rev Mol Cell Biol. 7:505–516. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Spano JP, Lagorce C, Atlan D, Milano G,
Domont J, Benamouzig R, Attar A, Benichou J, Martin A, Morere JF,
et al: Impact of EGFR expression on colorectal cancer patient
prognosis and survival. Ann Oncol. 16:102–108. 2005. View Article : Google Scholar
|
|
52
|
Normanno N, De Luca A, Bianco C, Strizzi
L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F and
Salomon DS: Epidermal growth factor receptor (EGFR) signaling in
cancer. Gene. 366:2–16. 2006. View Article : Google Scholar
|
|
53
|
Shostak K and Chariot A: EGFR and NF-κB:
Partners in cancer. Trends Mol Med. 21:385–393. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Habib AA, Chatterjee S, Park SK, Ratan RR,
Lefebvre S and Vartanian T: The epidermal growth factor receptor
engages receptor interacting protein and nuclear factor-kappa B
(NF-kappa B)-inducing kinase to activate NF-kappa B. Identification
of a novel receptor-tyrosine kinase signalosome. J Biol Chem.
276:8865–8874. 2001. View Article : Google Scholar
|
|
55
|
Taub N, Teis D, Ebner HL, Hess MW and
Huber LA: Late endosomal traffic of the epidermal growth factor
receptor ensures spatial and temporal fidelity of mitogen-activated
protein kinase signaling. Mol Biol Cell. 18:4698–4710. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Murphy JE, Padilla BE, Hasdemir B,
Cottrell GS and Bunnett NW: Endosomes: A legitimate platform for
the signaling train. Proc Natl Acad Sci USA. 106:17615–17622. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Din FV, Dunlop MG and Stark LA: Evidence
for colorectal cancer cell specificity of aspirin effects on NF
kappa B signalling and apoptosis. Br J Cancer. 91:381–388. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Schwenger P, Bellosta P, Vietor I,
Basilico C, Skolnik EY and Vilcek J: Sodium salicylate induces
apoptosis via p38 mitogen-activated protein kinase but inhibits
tumor necrosis factor-induced c-Jun N-terminal
kinase/stress-activated protein kinase activation. Proc Natl Acad
Sci USA. 94:2869–2873. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Frantz B, O’Neill EA, Ghosh S and Kopp E:
The effect of sodium salicylate and aspirin on NF-kappa B. Science.
270:2017–2019. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Amann R and Peskar BA: Anti-inflammatory
effects of aspirin and sodium salicylate. Eur J Pharmacol. 447:1–9.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nordt SP, Clark RF, Castillo EM and Guss
DA: Comparison of three aspirin formulations in human volunteers.
West J Emerg Med. 12:381–385. 2011. View Article : Google Scholar
|
|
62
|
Borthwick GM, Johnson AS, Partington M,
Burn J, Wilson R and Arthur HM: Therapeutic levels of aspirin and
salicylate directly inhibit a model of angiogenesis through a
Cox-independent mechanism. FASEB J. 20:2009–2016. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dawson JP, Berger MB, Lin CC, Schlessinger
J, Lemmon MA and Ferguson KM: Epidermal growth factor receptor
dimerization and activation require ligand-induced conformational
changes in the dimer interface. Mol Cell Biol. 25:7734–7742. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hubbard SR and Miller WT: Receptor
tyrosine kinases: Mechanisms of activation and signaling. Curr Opin
Cell Biol. 19:117–123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Arkhipov A, Shan Y, Das R, Endres NF,
Eastwood MP, Wemmer DE, Kuriyan J and Shaw DE: Architecture and
membrane interactions of the EGF receptor. Cell. 152:557–569. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Huang L and Fu L: Mechanisms of resistance
to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 5:390–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sigismund S, Woelk T, Puri C, Maspero E,
Tacchetti C, Transidico P, Di Fiore PP and Polo S:
Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl
Acad Sci USA. 102:2760–2765. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sigismund S, Argenzio E, Tosoni D,
Cavallaro E, Polo S and Di Fiore PP: Clathrin-mediated
internalization is essential for sustained EGFR signaling but
dispensable for degradation. Dev Cell. 15:209–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Barbieri E, Di Fiore PP and Sigismund S:
Endocytic control of signaling at the plasma membrane. Curr Opin
Cell Biol. 39:21–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Burke P, Schooler K and Wiley HS:
Regulation of epidermal growth factor receptor signaling by
endocytosis and intracellular trafficking. Mol Biol Cell.
12:1897–1910. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Goh LK, Huang F, Kim W, Gygi S and Sorkin
A: Multiple mechanisms collectively regulate clathrin-mediated
endocytosis of the epidermal growth factor receptor. J Cell Biol.
189:871–883. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mellman I and Yarden Y: Endocytosis and
cancer. Cold Spring Harb Perspect Biol. 5:a0169492013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Villaseñor R, Kalaidzidis Y and Zerial M:
Signal processing by the endosomal system. Curr Opin Cell Biol.
39:53–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang YN, Yamaguchi H, Hsu JM and Hung MC:
Nuclear trafficking of the epidermal growth factor receptor family
membrane proteins. Oncogene. 29:3997–4006. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang YN and Hung MC: Nuclear functions and
subcellular trafficking mechanisms of the epidermal growth factor
receptor family. Cell Biosci. 2:132012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang YN, Lee HH, Lee HJ, Du Y, Yamaguchi H
and Hung MC: Membrane-bound trafficking regulates nuclear transport
of integral epidermal growth factor receptor (EGFR) and ErbB-2. J
Biol Chem. 287:16869–16879. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Brand TM, Iida M, Li C and Wheeler DL: The
nuclear epidermal growth factor receptor signaling network and its
role in cancer. Discov Med. 12:419–432. 2011.PubMed/NCBI
|
|
79
|
Jovic M, Sharma M, Rahajeng J and Caplan
S: The early endosome: A busy sorting station for proteins at the
crossroads. Histol Histopathol. 25:99–112. 2010.
|
|
80
|
Pálfy M, Reményi A and Korcsmáros T:
Endosomal crosstalk: Meeting points for signaling pathways. Trends
Cell Biol. 22:447–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Henriksen L, Grandal MV, Knudsen SL, van
Deurs B and Grøvdal LM: Internalization mechanisms of the epidermal
growth factor receptor after activation with different ligands.
PLoS One. 8:e581482013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Capuani F, Conte A, Argenzio E, Marchetti
L, Priami C, Polo S, Di Fiore PP, Sigismund S and Ciliberto A:
Quantitative analysis reveals how EGFR activation and
downregulation are coupled in normal but not in cancer cells. Nat
Commun. 6:79992015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pennock S and Wang Z: A tale of two Cbls:
Interplay of c-Cbl and Cbl-b in epidermal growth factor receptor
downregulation. Mol Cell Biol. 28:3020–3037. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Rojas M, Yao S and Lin YZ: Controlling
epidermal growth factor (EGF)-stimulated Ras activation in intact
cells by a cell-permeable peptide mimicking phosphorylated EGF
receptor. J Biol Chem. 271:27456–27461. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Nyati MK, Morgan MA, Feng FY and Lawrence
TS: Integration of EGFR inhibitors with radiochemotherapy. Nat Rev
Cancer. 6:876–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Downward J, Waterfield MD and Parker PJ:
Autophosphorylation and protein kinase C phosphorylation of the
epidermal growth factor receptor. Effect on tyrosine kinase
activity and ligand binding affinity. J Biol Chem. 260:14538–14546.
1985.PubMed/NCBI
|
|
87
|
Keilhack H, Tenev T, Nyakatura E,
Godovac-Zimmermann J, Nielsen L, Seedorf K and Böhmer FD:
Phosphotyrosine 1173 mediates binding of the protein-tyrosine
phosphatase SHP-1 to the epidermal growth factor receptor and
attenuation of receptor signaling. J Biol Chem. 273:24839–24846.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY,
Lin CY, Lee HJ, Wang YN, Liu M, Liao HW, et al: Crosstalk between
Arg 1175 methylation and Tyr 1173 p hosphorylation negatively
modulates EGFR-mediated ERK activation. Nat Cell Biol. 13:174–181.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Li H, Zhu F, Boardman LA, Wang L, Oi N,
Liu K, Li X, Fu Y, Limburg PJ, Bode AM, et al: Aspirin Prevents
Colorectal Cancer by Normalizing EGFR Expression. EBioMedicine.
2:447–455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Murthy U, Basu M, Sen-Majumdar A and Das
M: Perinuclear location and recycling of epidermal growth factor
receptor kinase: Immunofluorescent visualization using antibodies
directed to kinase and extracellular domains. J Cell Biol.
103:333–342. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kesarwala AH, Samrakandi MM and
Piwnica-Worms D: Proteasome inhibition blocks ligand-induced
dynamic processing and internalization of epidermal growth factor
receptor via altered receptor ubiquitination and phosphorylation.
Cancer Res. 69:976–983. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Farrow DC, Vaughan TL, Hansten PD,
Stanford JL, Risch HA, Gammon MD, Chow WH, Dubrow R, Ahsan H, Mayne
ST, et al: Use of aspirin and other nonsteroidal anti-inflammatory
drugs and risk of esophageal and gastric cancer. Cancer Epidemiol
Biomarkers Prev. 7:97–102. 1998.PubMed/NCBI
|
|
93
|
Funkhouser EM and Sharp GB: Aspirin and
reduced risk of esophageal carcinoma. Cancer. 76:1116–1119. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kobayashi S, Shimamura T, Monti S, Steidl
U, Hetherington CJ, Lowell AM, Golub T, Meyerson M, Tenen DG,
Shapiro GI, et al: Transcriptional profiling identifies cyclin D1
as a critical downstream effector of mutant epidermal growth factor
receptor signaling. Cancer Res. 66:11389–11398. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Thoms HC, Dunlop MG and Stark LA:
p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4
stimulates nucleolar translocation of RelA and apoptosis in
colorectal cancer cells. Cancer Res. 67:1660–1669. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Cavalli V, Vilbois F, Corti M, Marcote MJ,
Tamura K, Karin M, Arkinstall S and Gruenberg J: The stress-induced
MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5
complex. Mol Cell. 7:421–432. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Fratti RA, Chua J and Deretic V: Induction
of p38 mitogen-activated protein kinase reduces early endosome
auto-antigen 1 (EEA1) recruitment to phagosomal membranes. J Biol
Chem. 278:46961–46967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Vergarajauregui S, San Miguel A and
Puertollano R: Activation of p38 mitogen-activated protein kinase
promotes epidermal growth factor receptor internalization. Traffic.
7:686–698. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Macé G, Miaczynska M, Zerial M and Nebreda
AR: Phosphorylation of EEA1 by p38 MAP kinase regulates mu opioid
receptor endocytosis. EMBO J. 24:3235–3246. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Higgs GA, Salmon JA, Henderson B and Vane
JR: Pharmacokinetics of aspirin and salicylate in relation to
inhibition of arachidonate cyclooxygenase and antiinflammatory
activity. Proc Natl Acad Sci USA. 84:1417–1420. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hatanpaa KJ, Burma S, Zhao D and Habib AA:
Epidermal growth factor receptor in glioma: Signal transduction,
neuropathology, imaging, and radioresistance. Neoplasia.
12:675–684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Moris D, Kontos M, Spartalis E and
Fentiman IS: The Role of NSAIDs in Breast Cancer Prevention and
Relapse: Current Evidence and Future Perspectives. Breast Care
(Basel). 11. pp. 339–344. 2016, View Article : Google Scholar
|
|
103
|
Taub M, Parker R, Mathivanan P, Ariff MA
and Rudra T: Antagonism of the prostaglandin E2 EP1 receptor in
MDCK cells increases growth through activation of Akt and the
epidermal growth factor receptor. Am J Physiol Renal Physiol.
307:F539–F550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Barrett MA, Zheng S, Roshankar G, Alsop
RJ, Belanger RK, Huynh C, Kučerka N and Rheinstädter MC:
Interaction of aspirin (acetylsalicylic acid) with lipid membranes.
PLoS One. 7:e343572012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Alsop RJ, Toppozini L, Marquardt D,
Kučerka N, Harroun TA and Rheinstädter MC: Aspirin inhibits
formation of cholesterol rafts in fluid lipid membranes. Biochim
Biophys Acta. 1848.805–812. 2015.
|
|
106
|
Puri C, Tosoni D, Comai R, Rabellino A,
Segat D, Caneva F, Luzzi P, Di Fiore PP and Tacchetti C:
Relationships between EGFR signaling-competent and
endocytosis-competent membrane microdomains. Mol Biol Cell.
16:2704–2718. 2005. View Article : Google Scholar : PubMed/NCBI
|





















