Introduction
Intestinal homeostasis is maintained by complex
interactions between intestinal microorganisms and the gut immune
system, and dysregulation of gut immunity may cause inflammation
and tumorigenesis (1). Interaction
between epithelial cells and stromal cells, including leukocytes
and fibroblasts, is considered to be pivotal for tumorigenesis and
cancer progression (1).
Adenocarcinoma colorectal cancer is a predominant malignancy
located in the colon and rectum, and it has been proposed to arise
from a subpopulation of self-renewing tumor stem cells located
within the tumor microenvironment (1,2).
Colorectal cancer is the third most common cancer diagnosed in USA
in 2016 (3,4) and its 5-year survival rate remains
poor at 55% (4). Colorectal cancer
is a heterogeneous group of diseases, and its prognosis remains
poor in spite of the development of novel therapeutic strategies
(5-8), and its molecular classification is
notable (5-9). The identification of novel biomarker
targets is proposed to result in prolonged survival of patients
with colorectal cancer (10).
The aryl hydrocarbon receptor (AHR) is a
ligand-activated transcription factor, which is located in manifold
types of cells (11,12). AHR forms a heterodimer with the AHR
nuclear translocator, which is transcriptionally active after
binding to xenobiotic responsive elements in various genes,
including the cytochrome P450 family 1 subfamily A member 1
(CYP1A1) gene (11,12). The AHR was initially discovered in
the process that investigates its binding to polychlorinated
aromatic hydrocarbons, including
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and
polychlorinated biphenyls (11,12).
Numerous AHR ligands, as AHR agonists, have been identified,
including synthetic and environmental chemicals, and
naturally-occurring dietary and endogenous compounds (13-18).
AHR signaling has been regulated through various signaling factors,
including nuclear factor-κB (NF-κB) p65, and it appears to serve an
important role in the regulation of diverse cellular and biological
processes (19). The canonical
target genes for AHR are well known in cytochrome P450 isoforms
(CYP1A1, CYP1A2 and CYP1B1), which are implicated in the metabolic
pathway of xenobiotics and endogenous compounds located in tissues
and cells (20,21). The AHR signaling-dependent pathway
is also implicated in manifestation of chemically-induced toxicity
and carcinogenesis, which are induced through the production of
free radicals and conversion of pro-carcinogens to ultimate
genotoxic carcinogens via metabolism that is mediated by cytochrome
P450 enzymes (20,21). Furthermore, AHR ligands are
involved in various pathologies in humans, resulting in toxic
processes, including tumor promotion, immunosuppression and
teratogenicity with disorder of the fine homeostatic regulations of
cell functions (22-26).
The physiological role of AHR in the absence of
exogenous ligand may serve a pivotal role in the regulation of cell
function, compared with cellular impacts caused by its binding of
exogenous ligand (27). Mice,
which express a constitutively active AHR, exhibited a promoted
development of hepatocarcinogenesis (28). Notably, AHR signaling may be
demonstrated to serve a role of a depressor in the development of
hepatocarcinogenesis (29).
Furthermore, AHR signaling has been demonstrated to adjust liver
repair and regeneration, and its signaling suppresses tumorigenesis
by modulating the actions of stem-like cells and β-catenin
signaling (30,31). Recently, it was demonstrated that
TCDD treatment represses the proliferation and promotes the death
of human liver cancer HepG2 cells in vitro, and that the
exhibition of these effects was implicated in AHR signaling
associated with various signaling factors, including NF-κB p65
(32).
The AHR is expressed and characterized in human
colon adenocarcinoma cells, including RKO cells (33-35),
and has been demonstrated to regulate the expression levels of
CYP1A1 (36) and CYP1A2 (37) in colorectal cancer cells in
vitro. The role of AHR thus has been reported in colon cancer
cells (38). Notably, the AHR
suppressed intestinal carcinogenesis in ApcMin/+ mice
following natural ligand treatment in vivo (39). Furthermore, the AHR is associated
with tumor prevention by regulating gut immunity in normal
intestinal tissues, and it is involved in growth suppression of
tumor cells of ApcMin/+ mice (16). Thus, the AHR may serve a repressive
role in the development of colorectal cancer. However, the
regulatory role of AHR signaling in the proliferation and death of
human colorectal cancer cells is poorly understood. Therefore, this
was investigated in RKO colorectal cancer cells in vitro. It
was demonstrated that TCDD treatment suppresses the growth and
proliferation, and stimulates the death of RKO cells, via AHR
signaling. The observations indicated that the activation of AHR
signaling serves a suppressive role in the development of human
colorectal cancer, revealing a potential novel role of AHR as a
target molecule in carcinogenesis.
Materials and methods
Materials
TCDD (>99.99% purity) was obtained from Dow
Chemicals Co. (Midland, MI, USA), and it was dissolved in dimethyl
sulfoxide (DMSO) and stored in the dark at −20°C until use.
Dulbecco’s modified Eagle’s medium (DMEM; including 4.5 g/l
glucose, L-glutamine and sodium pyruvate) and antibiotics (100
µg/ml penicillin and 100 µg/ml streptomycin; P/S)
were obtained from Corning Life Sciences (Manassas, VA, USA). Fetal
bovine serum (FBS) was purchased from Omega Scientific Inc.
(Tarzana, CA, USA). 2-methyl-2H-pyrazole-3-carboxylic acid
(2-methyl-4-o-tolylazo-phenyl)-amide (CH223191) was
purchased from Selleck Chemicals (Houston, TX, USA), and it was
dissolved in 100% DMSO. Caspase-3 inhibitor, crystal violet, and
all other chemicals were obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany).
Human colorectal cancer cells
RKO epithelial cells, which originated from male
adult patients with colorectal carcinoma, were used in the present
study. This cell line was purchased from the American Type Culture
Collection (Manassas, VA, USA). RKO cells were cultured in DMEM
including 10% FBS and 1% P/S.
Assay of colony formation of RKO
cells
RKO cells (1×103 cells/well per 2 ml of
medium in 6-well plates) were cultured in DMEM containing 10% FBS,
1% P/S and 1% fungizone in an atmosphere containing 5%
CO2 at 37°C in the presence of vehicle (1% DMSO) or TCDD
(1 or 10 nM) for 5 days, when visible clones formed on the plates
(40,41). Following the culture, the dishes
were washed with PBS (3 times with 2 ml) and fixed with 100%
methanol (adding 0.5 ml per well) for 20 min at room temperature,
and then washed 3 times with PBS (2 ml). The colonies were stained
with crystal violet. Crystal violet solution (0.5%, dissolved in
20% methanol) was added to the fixed cells for 30 min at room
temperature. Thereafter, stained cells were washed 5 times with PBS
(2 ml). After washing, the plates were air-dried for 2 h at room
temperature. The colonies (including >50 cells) were counted
under a microscope (×10; Nikon Corporation, Tokyo, Japan) using a
cell counter (Line Seiki H-102P; Line Seiki Co., Ltd., Tokyo,
Japan). Data are represented as numbers of colonies per well.
Transfection of regucalcin cDNA/pCXN2
into RKO cells
To generate the regucalcin-overexpressing RKO cells,
the RKO wild-type cells were transfected with empty pCXN2 vector
(Addgene, Inc., Cambridge, MA, USA; 600 µg/ml) or pCXN2
vector (Addgene, Inc.; 600 µg/ml) expressing a cDNA encoding
the human full-length (900 bp) regucalcin (regucalcin cDNA/pCXN2)
(42,43). For transfection, the RKO cells
(1×105/well per ml of DMEM) were grown on 24-well plates
to reach subconfluency. Regucalcin cDNA/pCXN2 (1 µg/well) or
empty pCXN2 vector (1 µg/well) alone was transfected into
the RKO cells using the synthetic cationic lipid
Lipofectamine® reagent, according to the manufacturer’s
protocols (Promega Corporation, Madison, WI, USA) (43). Following overnight incubation after
transfection, Geneticin (600 µg/ml G418; Sigma-Aldrich;
Merck KGaA) was added to the culture wells to select transfectants,
and the cells were cultured in an atmosphere containing 5%
CO2 at 37°C for 3 weeks to produce transfected cells.
Subsequently, the transfected cells were plated with limiting
dilution to isolate transfectants using 96-well plates. Surviving
clones were isolated, transferred to 35-mm dishes, and grown in
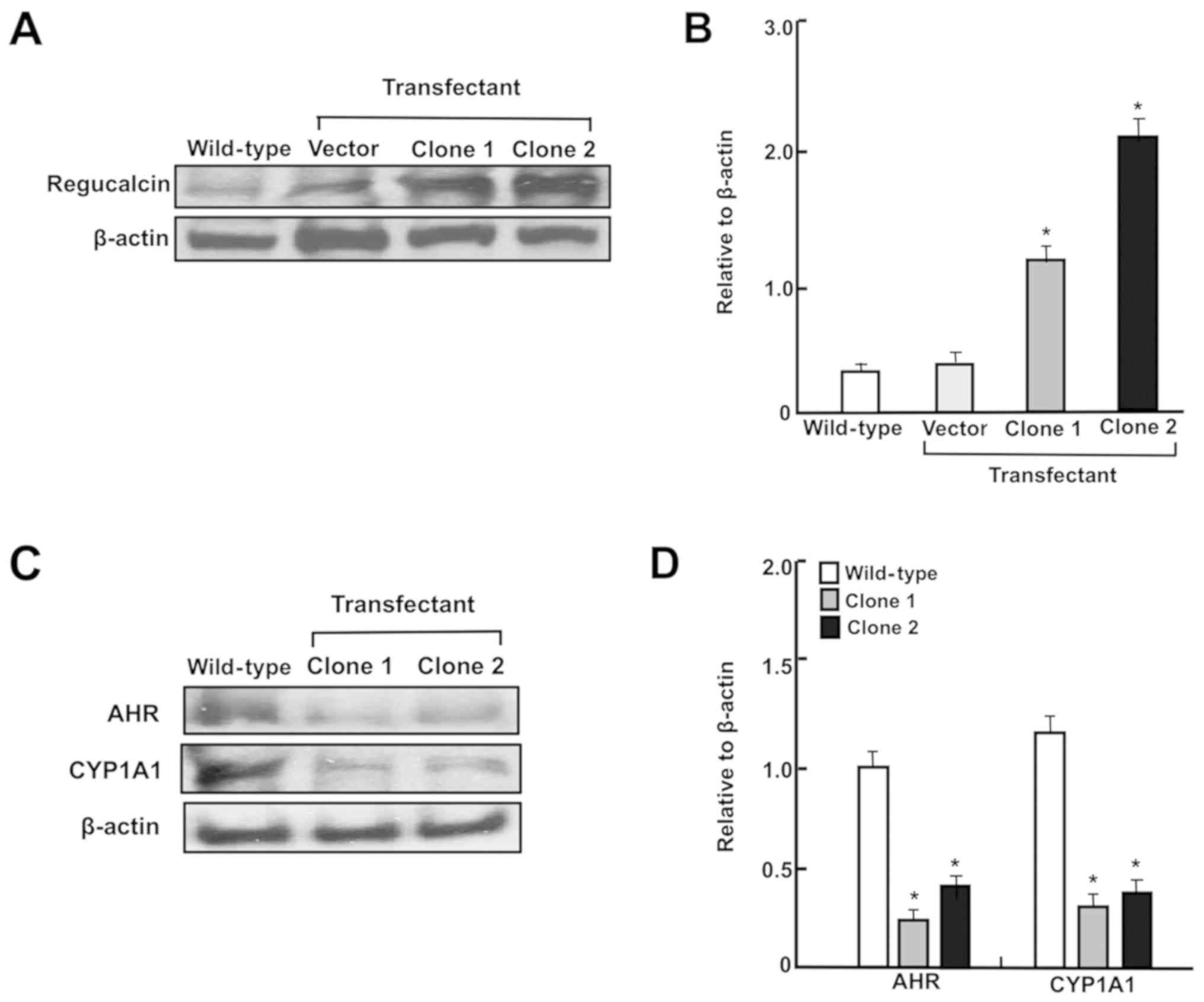
DMEM without Geneticin. The transfectant clones 1 and 2 exhibiting
stable expression of regucalcin were then obtained. The levels of
regucalcin expressed in two clones were assayed using western blot
analysis, and those exhibited an elevation expression of 7.4- or
10.9-fold in clones 1 or 2, respectively, compared with wild-type
cells, respectively, as depicted in Fig. 6A. Therefore, clone 2 was used in
the subsequent experiments.
Assay of cell proliferation
To determine the effect of TCDD on cell
proliferation, the RKO wild-type cells (1×105/ml per
well) were cultured using a 24-well plates in DMEM, containing 10%
FBS, 1% P/S and 1% fungizone, in the presence of vehicle (1% DMSO)
or TCDD (0.1, 1, 10 or 100 nM) in an atmosphere containing 5%
CO2 and 37°C for 3 or 7 days (44). In separate experiments, the RKO
wild-type cells or transfectants (1×105/ml per well)
were cultured in DMEM containing 10% FBS, 1% P/S and 1% fungizone
with or without vehicle (1% DMSO), TCDD (1, 10 or 100 nM), or
CH223191 (1 or 10 µM) with or without TCDD (10 nM) in an
atmosphere containing 5% CO2 and 37°C for 3 days. The
RKO cells were then detached from each culture dish to determine
cell number using a cell counter.
Assay of cell death
To determine the effect of TCDD on cell death, the
RKO wild-type cells (1×105/ml per well) were cultured
using 24-well plates in DMEM, containing 10% FBS, 1% P/S, and 1%
fungizone, in the absence of TCDD in an atmosphere containing 5%
CO2 and 37°C for 3 days in order to reach subconfluence.
The cultured cells at subconfluency were incubated in the presence
of vehicle (1% DMSO) or TCDD (0.1, 1, 10 or 100 nM), with or
without the caspase-3 inhibitor (10 µM) in the presence of
either vehicle or CH223191 (1 or 10 µM) for 24 h in an
atmosphere containing 5% CO2 at 37°C (45). In other experiments, the
RKO-wild-type cells or transfectants (1×105/ml per well)
were cultured in DMEM containing 10% FBS, 1% P/S and 1% fungizone
in the absence of TCDD in an atmosphere containing 5%
CO2 and 37°C for 3 days. After reaching subconfluence,
the cells were incubated in the presence of vehicle (1% DMSO), TCDD
(1, 10 or 100 nM), or CH223191 (1 or 10 µM) with or without
TCDD (10 nM) for 24 h in an atmosphere containing 5% CO2
at 37°C (45). Cells were then
detached from each culture well to determine cell number using a
cell counter.
Counting of cell number
To detach cells attached on each well after
culturing in order to assay the proliferation and death of RKO
cells, culture dishes were incubated for 2 min at 37°C with the
addition of a solution (0.1 ml per well) of 0.05% trypsin plus EDTA
in Ca2+/Mg2+-free PBS, and then cells were
detached through pipetting after the addition of DMEM (0.9 ml)
containing 10% FBS and 1% P/S into the wells (44,45).
The medium containing the suspended cells (0.1 ml) was mixed with
0.1 ml of 0.5% trypan blue staining solution (44,45).
The number of viable cells with viability was counted under a
microscope (×10; Olympus MTV-3; Olympus Corporation, Tokyo, Japan)
using a Hemocytometer plate (Sigma-Aldrich; Merck KGaA) and a cell
counter (Line Seiki H-102P; Line Seiki Co., Ltd.). The mean of two
counts was calculated for each dish. The number of cells is
presented as number per well of the plate.
Western blot analysis
To determine levels of various proteins expressed in
RKO cells, wild-type RKO cells or regucalcin-overexpressing cells
were plated in 100×21 mm dishes at a density of 1×106
cells/dish in 10 ml DMEM containing 10% FBS, 1% P/S and 1%
fungizone, and then cultured in the presence of vehicle (1% DMSO)
or TCDD (10 nM) in an atmosphere containing 5% CO2 and
37°C for 3 days. After culturing, the cells were washed three times
with ice-cold PBS and removed from the dish by scraping after the
addition of cell lysis buffer (Cell Signaling Technology, Inc.,
Danvers, MA, USA) supplemented with inhibitors of protease and
protein phosphatase (Roche Diagnostics, Indianapolis, IN, USA). The
collected lysates were centrifuged at 17,000 × g at 4°C for 10 min,
to prepare fractions including the cytoplasm and endoplasmic
reticulum of RKO cells. The concentrations of protein in
aforementioned supernatants were assayed using the Bio-Rad Protein
Assay Dye (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with
bovine serum albumin (Bio-Rad Laboratories, Inc.) as standard. The
aforementioned supernatant from cell lysate was stored at −80°C
until use for western blot assay. Samples of 40 µg
supernatant protein were applied to each lane and were separated
using SDS-PAGE (12%). After electrophoresis, the gel was
transferred onto PVDF membranes for immunoblotting with specific
antibodies. The membranes were blocked with
SuperBlock®T20 blocking buffer (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for 60 min at room temperature.
Polyclonal AHR antibody sheep IgG was obtained from R&D
Systems, Inc. (cat. no. AF6697; Minneapolis, MN, USA; dilution
1:500). Antibodies for other signaling proteins, including CYP1A1
(cat. no. sc-25304; dilution 1:1,000), NF-κB p65 (cat. no. sc-109;
dilution 1:1,000), β-catenin (cat. no. sc-39350; dilution 1:1,000)
and p53 (cat. no. sc-126; dilution 1:1,000) were obtained from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA; dilution 1:1,000),
and Ras (cat. no. 14429; dilution 1:1,000), β-actin (cat. no. 3700;
dilution 1:1,000), retinoblastoma (Rb; cat. no. 9309; dilution
1:1,000) and p21 (cat. no. 2947; dilution 1:1,000) were purchased
from Cell Signaling Technology, Inc.. Rabbit anti-regucalcin
antibody was provided from Abcam (Cambridge, MA, USA; cat. no.
ab213459; dilution 1:1,000), and it was used as described
previously (42,43,46).
For immunoblotting with the aforementioned specific antibodies, the
membranes were incubated with each primary antibody overnight at
4°C, followed by horseradish peroxidase-conjugated secondary
antibody (cat. nos. sc-2005 or sc-2305 for mouse and rabbit,
respectively; Santa Cruz Biotechnology, Inc.; dilution 1:2,000) for
60 min at 4°C. A total of 3 blots from independent experiments were
scanned on an Epson Perfection 1660 Photo scanner, and the bands
were quantified using ImageJ2 software (National Institutes of
Health, Bethesda, MD, USA).
Statistical analysis
Statistical significance was determined using
GraphPad InStat version 3 for Windows XP (GraphPad Software, Inc.,
La Jolla, CA, USA). Data are presented as the mean ± standard
deviation. Comparisons between two groups were performed using a
Student’s t-test. Furthermore, multiple comparisons were performed
using one-way analysis of variance with Tukey-Kramer multiple
comparisons post hoc test for parametric data as indicated.
P<0.05 was considered to indicate a statistically significant
difference.
Results
TCDD represses colony formation of RKO
cells
The effects of TCDD on colony formation of RKO human
colorectal cancer cells in vitro was investigated. Visible
clones of RKO cells were formed by culture for 5 days (Fig. 1). Subsequently, RKO cells were
cultured in the presence of TCDD (1 or 10 nM). The number of
colonies with >50 nuclei was significantly decreased by
treatment with TCDD (1 or 10 nM) as depicted in Fig. 1A and B. Thus, TCDD exhibited a
suppressive effect on the colony formation of RKO cells.
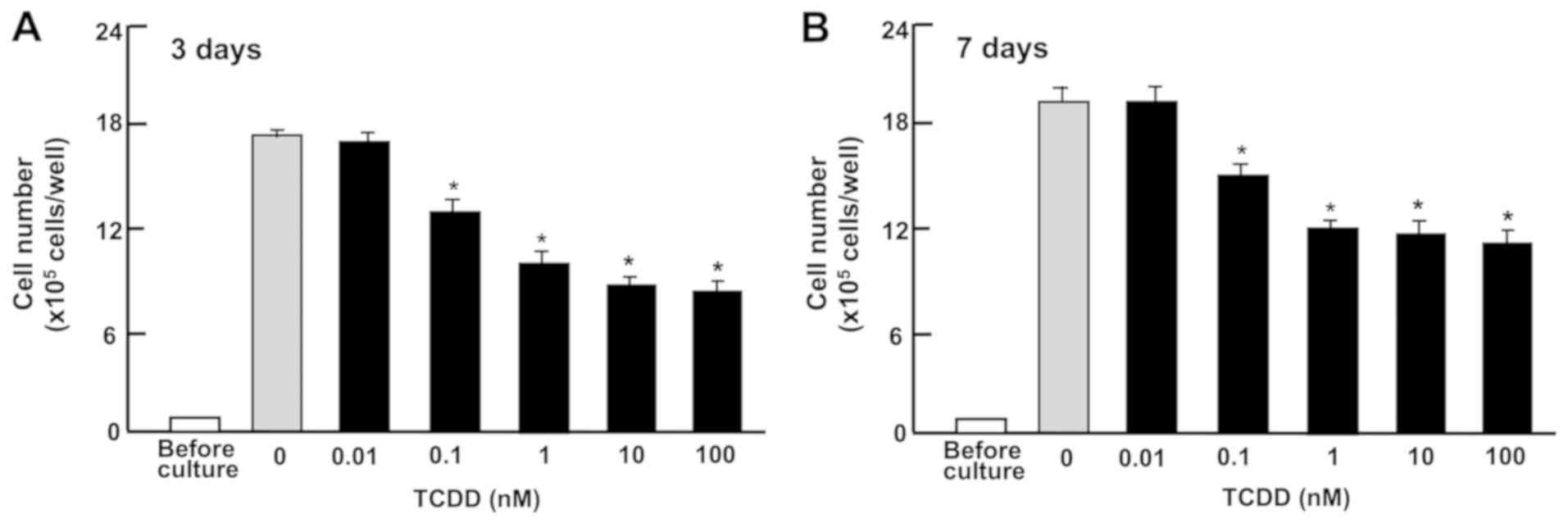
TCDD suppresses the proliferation of RKO
cells
To determine the effect of TCDD on cell growth, RKO
cells were cultured in 24-well plates in the presence of TCDD
(0.01-100 nM) for 3 or 7 days. The cells reached subconfluency
after culturing for 3 days, and they reached confluency at 4-7 days
of culture. Thus, cell growth was suppressed by the treatment with
TCDD (0.1-100 nM) for 3 (Fig. 2A)
or 7 (Fig. 2B) days.
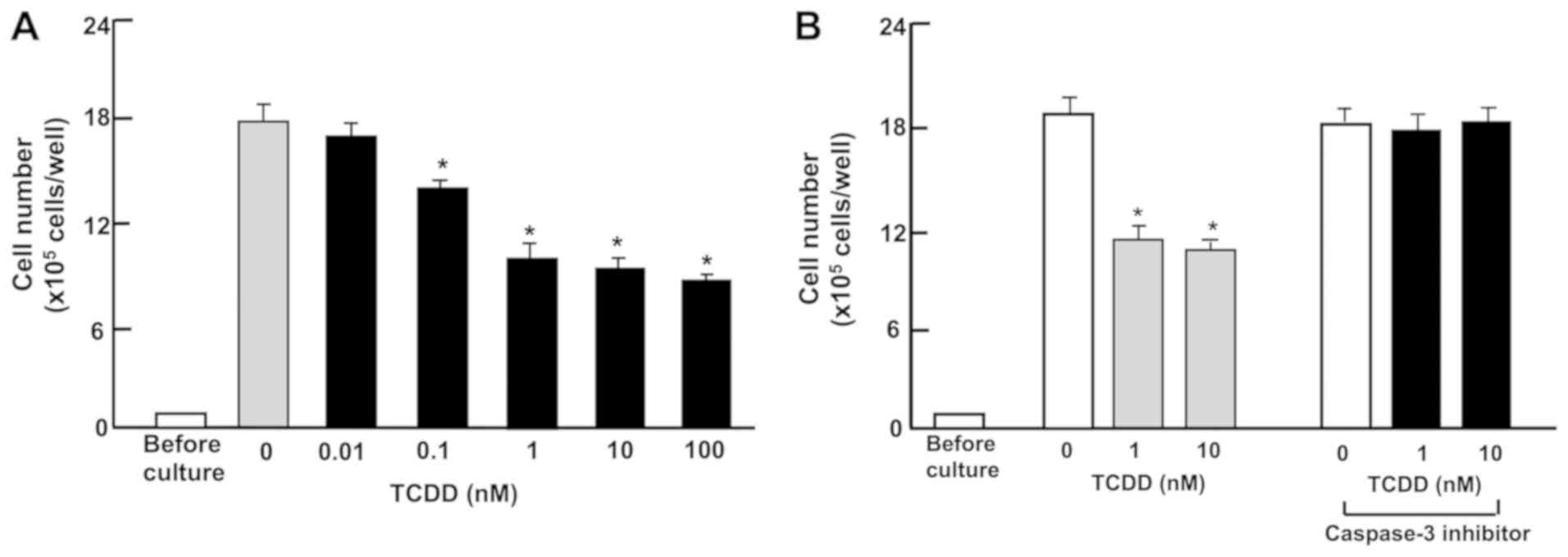
TCDD stimulates the death of RKO
cells
Subsequently, the effect of TCDD on the death of RKO
cells in vitro was investigated. The cells were cultured for
3 days to reach subconfluency, and then exposed to TCDD (0.01-100
nM) for a further 24 h. Treatment with TCDD (0.1-100 nM) resulted
in a decrease of attached cells (Fig.
3A and B), indicating that cell death is induced. In separate
experiments, RKO cells that had reached subconfluency after culture
for 3 days were incubated with a caspase-3 inhibitor (10 µM)
and TCDD. The reduction of cell number caused by the treatment with
TCDD (1 or 10 nM) was prevented in the presence of the inhibitor of
caspase-3. Activation of caspase-3 is demonstrated to induce DNA
fragmentation associated with apoptosis (45). TCDD-induced cell death may be due
to activation of caspase-3, which is known to induce DNA
fragmentation associated with cell death (41). However, this remains to be
elucidated using other methods.
Involvement of AHR signaling in the
proliferation and death of RKO cells
To characterize the TCDD-induced repression of
proliferation and promotion of death of RKO cells, the cells were
cultured in the treatment with CH223191, a suppressor of AHR
signaling (47). CH223191 (1 or 10
µM) did not have a significant effect on the proliferation
or death of RKO cells (Fig. 4A and
B). The repressive effect of TCDD (10 nM) on the proliferation
and the promoting effect of TCDD (10 nM) on the death of RKO cells
were significantly blocked by CH223191 (1 or 10 nM; Fig. 4A or B). The effects of TCDD on cell
proliferation were not completely blocked by the inhibitor
(Fig. 4A); however, the promoting
effects of TCDD on cell death were completely blocked (Fig. 4B). These results indicate that the
effects of TCDD on the proliferation and death of RKO cells are
partially mediated by AHR signaling.
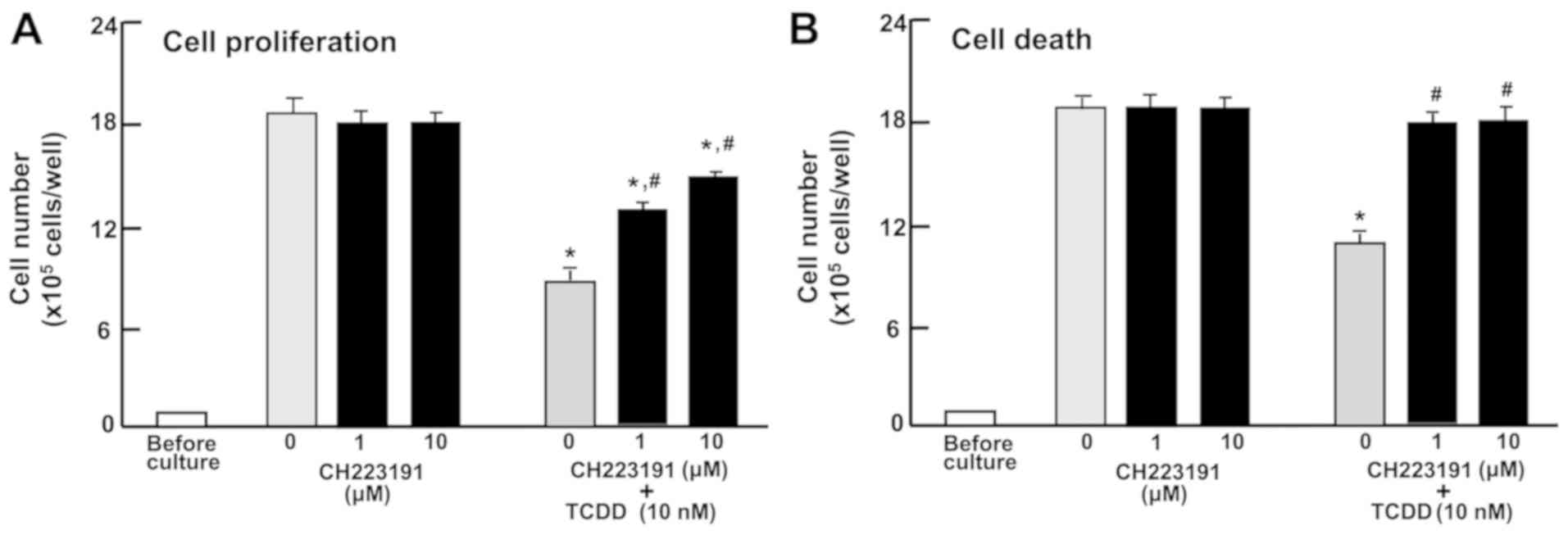 | Figure 4The AHR is involved in mediating the
effects of TCDD on the proliferation and death of RKO human
colorectal cancer cells. (A) The cells (1×105 cells/per
well in 24-well plates) were cultured in DMEM containing 10% fetal
bovine serum, 1% penicillin/streptomycin and 1% fungizone
containing vehicle (1% DMSO) or AHR inhibitor, CH223191 (1 or 10
µM), with or without TCDD (10 nM) for 3 days. (B) The cells
(1×105 cells/per well in 24-well plates) were cultured
in DMEM as aforementioned for 3 days, and once the cells reached
subconfluency, they were cultured in DMEM as aforementioned in the
presence of vehicle (1% DMSO) or CH223191 (1 or 10 µM).
After 1 h, TCDD (10 nM) was added into the medium containing
vehicle (1% DMSO) or CH223191 (1 or 10 µM), and the cells
were cultured for a further 23 h. The number of attached cells were
then counted. Data are presented as mean ± standard deviation
obtained from 8 wells of 2 replicate plates per dataset using
different dishes and cell preparations. *P<0.001 vs.
0 µM CH223191. #P<0.01 vs. 0 µM
CH223191 + 10 nM TCDD. One-way analysis of variance and
Tukey-Kramer post hoc test were used. AHR, aryl hydrocarbon
receptor; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; DMEM,
Dulbecco’s modified Eagle’s medium; DMSO, dimethyl sulfoxide. |
TCDD enhances the levels of proteins
associated with tumor suppression in RKO cells
To characterize the mechanism of TCDD action, and
determine whether or not TCDD treatment regulates the levels of key
transcription factors, western blot analysis was used. AHR and
CYP1A1 mRNAs were previously reported to be expressed in RKO cells
in vitro (34,35). It was demonstrated that the levels
of AHR and CYP1A1 were altered by TCDD in RKO cells (Fig. 5A and B). Notably, treatment with
TCDD (10 nM) significantly elevated the levels of NF-κB p65 and
β-catenin, which are crucial transcription factors associated with
cell signaling (32).
Additionally, TCDD treatment significantly elevated the levels of
p53, Rb, p21 and regucalcin, which are known as pivotal repressors
of the growth of tumor cells (48,49)
(Fig. 5C and D). TCDD (10 nM) did
not significantly alter the level of Ras, which acts upstream in
Akt signaling (32,49) (Fig. 5A
and B).
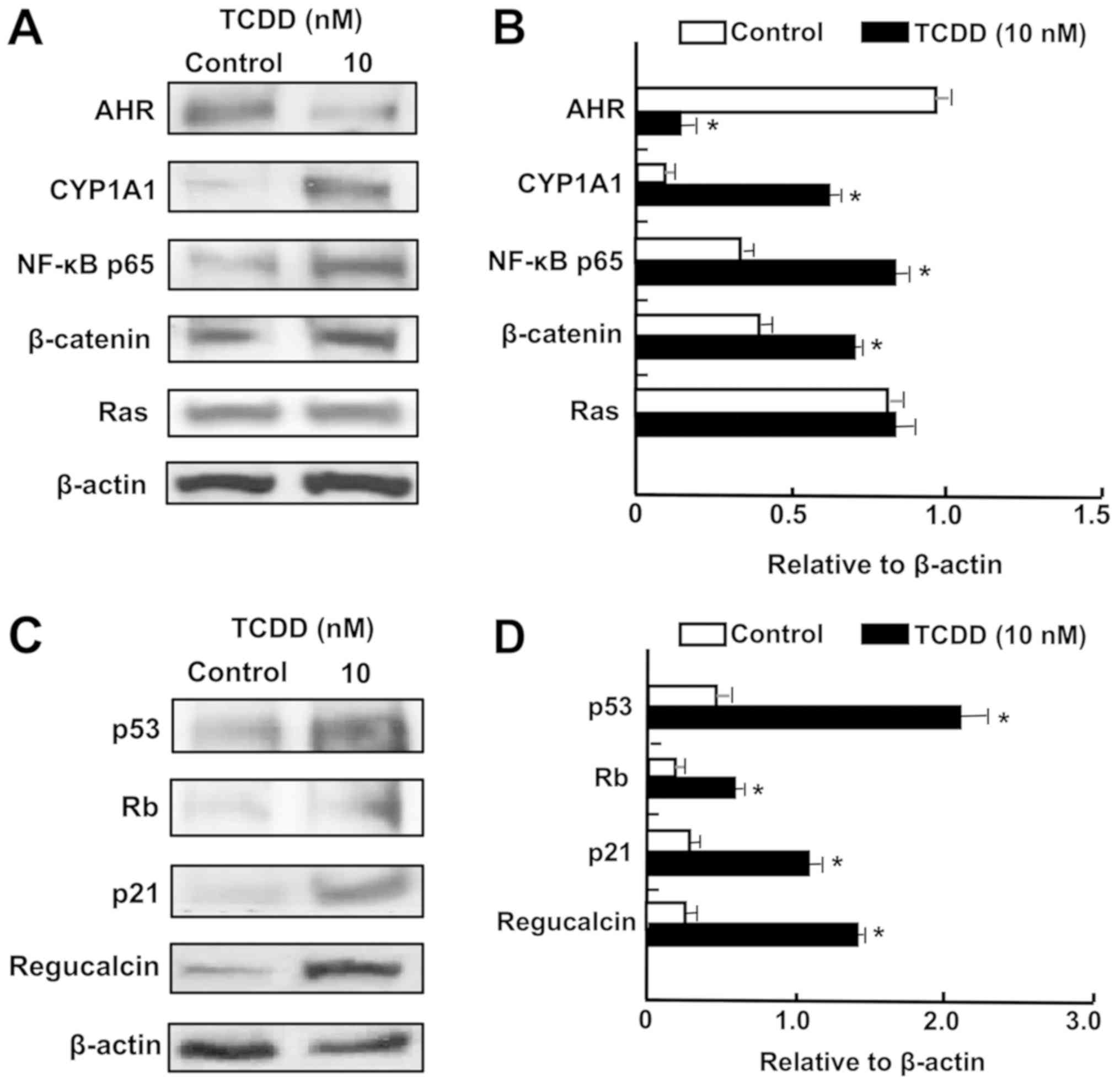 | Figure 5TCDD regulates the expression of
proteins associated with AHR signaling in RKO human colorectal
cancer cells in vitro. The cells (1×106
cells/dish) were cultured in Dulbecco’s modified Eagle’s medium
containing 10% fetal bovine serum, 1% penicillin/streptomycin and
1% fungizone in the presence of vehicle (1% dimethyl sulfoxide) or
TCDD (10 nM) for 3 days. Cell lysates were prepared and
centrifuged, and 40 µg of the supernatant protein per lane
were separated by SDS-PAGE and transferred to nylon membranes for
western blotting using specific antibodies against various proteins
as indicated. Data represent a typical figure of three independent
experiments using different cell preparations, and also are
presented as mean ± standard deviation. (A) Representative film
image for cell signaling-associated proteins. (B) Relative to
β-actin cell signaling-associated protein levels. (C)
Representative film image of tumor suppressor proteins.(D) Relative
to β-actin tumor suppressor proteins. *P<0.01 vs.
control using Student’s t-test. TCDD,
2,3,7,8-tetrachlorodibenzo-p-dioxin; AHR, aryl hydrocarbon
receptor; NF-κB, nuclear factor-κB; CYP1A1, cytochrome P450 family
1 subfamily A member 1; Rb, retinoblastoma. |
The effects of TCDD are suppressed in the
regucalcin-overexpressing RKO cells
Overexpression of regucalcin has been demonstrated
to repress the enhanced proliferation and death of RKO cells in
vitro (43). Therefore, the
present study investigated whether the effects of TCDD were
attenuated in regucalcin-overexpressing RKO cells in vitro.
These cells exhibited increased levels of regucalcin (Fig. 6A and B). Notably, regucalcin
overexpression significantly suppressed CYP1A1 and AHR levels in
RKO cells (Fig. 6C and D).
Subsequently, whether TCDD exhibits a repressive
effect on proliferation and a promoting effect on cell death in the
regucalcin-overexpressing RKO cells in vitro was
investigated. Wild-type RKO cells or regucalcin-overexpressing
cells were treated with TCDD (1, 10 or 100 nM). Proliferation of
wild-type RKO cells was significantly repressed by regucalcin
overexpression (Fig. 7A). However,
treatment with TCDD (1, 10 or 100 nM), which suppressed the
proliferation of wild-type RKO cells, did not exhibit a significant
effect on the proliferation of transfectants with or without
CH223191, an inhibitor of AHR signaling (Fig. 7B). Additionally, although treatment
with TCDD (1, 10 or 100 nM) significantly stimulated the death of
wild-type RKO cells (Fig. 7C), it
did not have a significant effect on the death of transfectants
with or without CH223191, an inhibitor of AHR signaling (Fig. 7D). These observations indicate that
regucalcin overexpression depresses AHR-dependent repression of
proliferation and promotion of death of RKO cells.
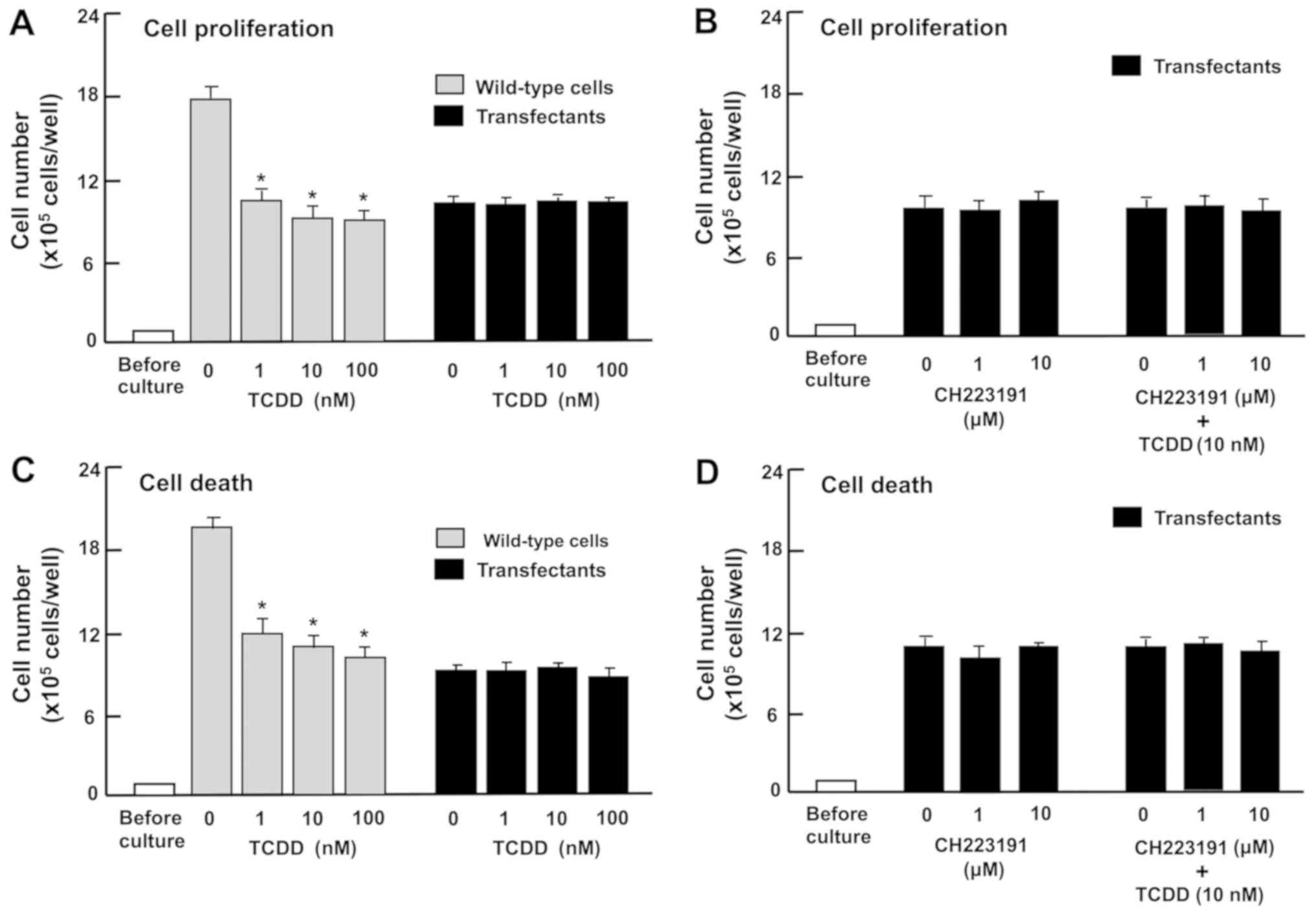 | Figure 7The effects of TCDD on the
proliferation and death of RKO human colorectal cancer cells are
attenuated by the overexpression of regucalcin in vitro.
Wild-type cells or transfectants (1×105 cells/per well
of 24-well plates) were cultured in DMEM containing 10% FBS, 1% P/S
and 1% fungizone in the presence of (A) vehicle (1% DMSO) or TCDD
(1, 10 or 100 nM), or (B) vehicle (1% DMSO) or CH223191 (1 or 10
µM) with or without TCDD (10 nM). In separate experiments,
the wild-type cells or transfectants (1×105 cells/per ml
of well) were cultured in DMEM as aforementioned for 3 days, and
upon reaching subconfluence, the cells were cultured in DMEM
containing 10% FBS, 1% P/S and 1% fungizone in the presence of (C)
vehicle (1% DMSO) or TCDD (1, 10 or 100 nM), or (D) vehicle (1%
DMSO) or CH223191 (1 or 10 µM) with or without TCDD (10 nM)
for 24 h. After culture, the numbers of attached cells were
counted. Data are presented as mean ± standard deviation obtained
from 8 wells of 2 replicate plates per dataset using different
dishes and cell preparations. *P<0.001 vs. 0 nm TCDD
in wild-type cells. One-way analysis of variance and Tukey-Kramer
post hoc test were used. TCDD,
2,3,7,8-tetrachlorodibenzo-p-dioxin; DMEM, Dulbecco’s
modified Eagle’s medium; DMSO, dimethyl sulfoxide; FBS, fetal
bovine serum; P/S, penicillin/streptomycin. |
Additionally, the effects of TCDD (10 nM) on the
levels of AHR, CYP1A1, p53, Rb and p21 in the
regucalcin-overexpressing RKO cells were determined (Fig. 8A and B). TCDD treatment on
transfectants cells did not appear to have a significant effect on
CYP1A1 expression, since the effect of TCDD treatment on
AHR-dependent CYP1A1 levels were depressed by regucalcin
overexpression. Regucalcin overexpression has been demonstrated to
increase the levels of p53, Rb and p21 in RKO cells (43) and other human cancer cells
(50,51). Notably, the effects of TCDD in
increasing p53, Rb and p21 levels were potentiated by regucalcin
overexpression. Since TCDD treatment increased regucalcin levels in
wild-type RKO cells (Fig. 5), the
effects of TCDD on increasing the levels of p53, Rb and p21 in RKO
cells are likely to depend, at least in part, on the elevation in
levels of regucalcin.
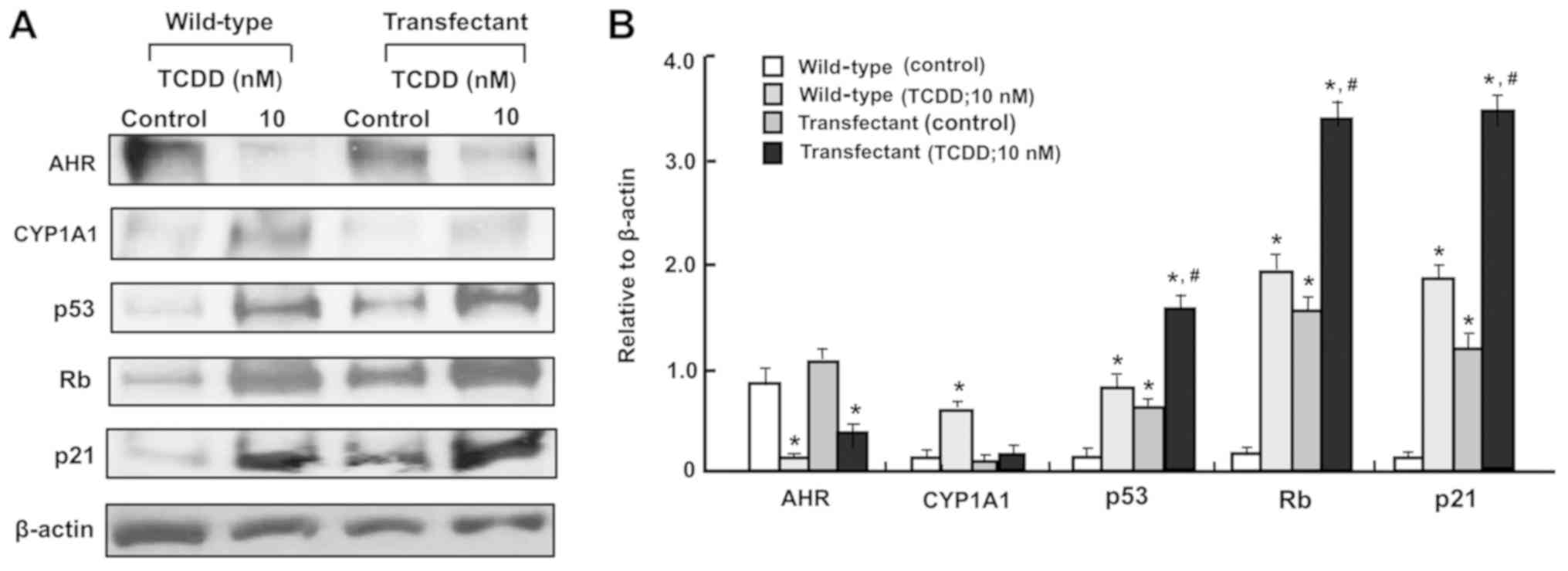 | Figure 8The TCDD-induced increase in CYP1A1
levels are suppressed in the regucalcin-overexpressing RKO human
colorectal cancer cells in vitro. The wild-type RKO cells or
regucalcin-overexpressing transfectants (1×106
cells/dish) were cultured in Dulbecco’s modified Eagle’s medium
containing 10% fetal bovine serum, 1% penicillin/streptomycin and
1% fungizone in the presence or absence of vehicle (1% dimethyl
sulfoxide) or TCDD (10 nM) for 3 days and then cell lysates were
centrifuged. Subsequently, 40 µg of the supernatant protein
per lane were separated by SDS-PAGE and transferred to nylon
membranes for western blotting using specific antibodies against
the indicated proteins. Representative data from three independent
experiments using different cell preparations are presented, and
data are presented as mean ± standard deviation. (A) Representative
film images of the TCDD effect. (B) Presented relative to β-actin
of the TCDD effect. *P<0.01, vs. wild-type (control).
#P<0.01, vs. wild-type (TCDD; 10 nM) or transfectant
(control). One-way analysis of variance and Tukey-Kramer post hoc
test were used. TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin;
Rb, retinoblastoma; AHR, aryl hydrocarbon receptor; CYP1A1,
cytochrome P450 family 1 subfamily A member 1. |
Discussion
Human colorectal cancer is diagnosed as the third
most common cancer type in USA in 2016 and its 5-year survival rate
remains poor at 55%, in spite of the promotion of novel therapeutic
strategies (3-8). Identification of novel biomarker
targets may ultimately cause the prolongation of survival of
patients with colorectal cancer (9). In the present study, it was
demonstrated that TCDD treatment suppresses the growth and
proliferation, and stimulates the death, of RKO human colorectal
cancer cells. These effects of TCDD were demonstrated to be blocked
by the treatment with CH223191, an inhibitor of AHR signaling
(47), indicating that the action
of TCDD is at least partially mediated through the AHR signaling
pathway. The observations thus demonstrate that the enhanced AHR
signaling serves a suppressive role in the development of human
colorectal cancer cells.
To investigate the mechanism of action of TCDD, it
was first demonstrated that the AHR and CYP1A1 proteins are present
in RKO cells, consistent with previous studies, demonstrating that
these mRNAs are expressed in these cells in vitro (34,35).
In the present study, TCDD treatment was demonstrated to be caused
a reduction of AHR levels and an elevation of CYP1A1 levels in the
cytosol, including endoplasmic reticulum of RKO cells. TCDD
treatment has been demonstrated to enhance the translocation of
cytoplasmic AHR into the nucleus and increases CYP1A1 expression
(11,12,32).
Notably, TCDD treatment also elevated the levels of NF-κB p65 and
β-catenin, which are crucial transcription factors implicated in
the manifold process of cell signaling, and the levels of p53, Rb,
p21 and regucalcin, which are pivotal repressors of the growth of
tumor cells (48,49). TCDD treatment did not change the
level of Ras, which acts upstream in Akt signaling. β-catenin has
been demonstrated to enhance regucalcin expression in HepG2 cells
in vitro (52). It has also
been reported that p53 modulates Hsp90 ATPase activity, which is
implicated in AHR-dependent activation of gene expression (53). These signaling factors may be
partially implicated in mediating the action of TCDD on the
proliferation and death of RKO cells. Whether or not these
molecules serve a role in the expression of the AHR gene remains to
be elucidated.
Furthermore, it was determined that the effects of
TCDD are attenuated in the regucalcin-overexpressing RKO cells.
Overexpression of regucalcin has been demonstrated to repress the
proliferation and death of RKO cells in vitro (43). Notably, regucalcin overexpression
was demonstrated to decrease AHR and CYP1A1 levels in RKO cells,
indicating that overexpressed regucalcin suppresses AHR signaling
in RKO cells. Regucalcin has been indicated to translocate from the
cytoplasm to nucleus in various types of normal and cancer cells,
including liver and kidney (48,49),
and it regulates the gene expressions of various proteins,
including p53 and Rb, apparently acting as a novel transcriptional
factor via binding nuclear DNA (48,49,54).
Thus, it was considered that regucalcin serves a crucial role as a
novel suppressor of AHR signaling. Notably, it was considered that
TCDD treatment, which exhibits a repressive effect on the
proliferation and a promoting effect on the death of wild-type RKO
cells, did not have such effects in regucalcin-overexpressing RKO
cells. These results support the view that AHR signaling is
depressed by regucalcin overexpression in RKO cells in
vitro.
Subsequently, whether the action of TCDD on the
levels of AHR, CYP1A1, p53, Rb and p21 was attenuated in
regucalcin-overexpressing RKO cells was investigated. Whereas TCDD
treatment decreased AHR levels and increased CYP1A1 levels, these
effects were determined to be depressed by regucalcin
overexpression, indicating that AHR signaling activated by TCDD, an
agonist, is suppressed by regucalcin overexpression. Notably, the
effects of TCDD in increasing p53, Rb and p21 levels were
demonstrated to be potentiated by regucalcin overexpression.
Overexpression of regucalcin has been demonstrated to increase the
levels of p53, Rb and p21 in RKO cells (43), and in other types of human cancer
cells (50,51). Additionally, TCDD treatment
increased regucalcin levels in wild-type RKO cells. These
observations indicate that the action of TCDD in increasing the
levels of p53, Rb and p21 in RKO cells are mediated, at least in
part, via increases in regucalcin. Additionally, regucalcin
overexpression suppressed the activation of AHR signaling
associated with CYP1A1 expression. It is not known whether the
deficiency of regucalcin enhances AHR signaling, although this
remains to be elucidated using regucalcin siRNA. It is possible
that the activation of AHR signaling enhances regucalcin gene
expression, and that increased regucalcin suppresses AHR signaling
pathways. This suppressive effect may result in inhibition of
metabolic pathways associated with CYP1A1. The schematic diagram to
demonstrate the mechanistic association between TCDD, AHR and
regucalcin is depicted in Fig. 9.
Collectively, AHR signaling may serve a crucial role in suppression
of the growth of colorectal cancer cells, probably mediated via
manifold molecules linked to tumor suppression.
 | Figure 9Schematic diagram of the mechanistic
association between TCDD, AHR, CYP1A1, RGN and other molecules in
RKO human colorectal cancer cells. TCDD activates AHR signaling by
binding to ARNT. The complex is translocated into the nucleus and
regulates expression of various genes. TCDD-activated AHR signaling
enhances expression of various genes, including CYP1A1, RGN, p53,
Rb and p21. Overexpressed RGN regulates the suppression of pathways
of AHR signaling associated with CYP1A1, resulting in inhibition of
metabolic pathways. Furthermore, overexpressed RGN enhances the
expressions of p53, Rb and p21, which is increased via
TCDD-activated AHR signaling, revealing a potential suppressive
effect of cell proliferation and stimulatory effect of cell death.
TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; Rb,
retinoblastoma; AHR, aryl hydrocarbon receptor; CYP1A1, cytochrome
P450 family 1 subfamily A member 1; RGN, regucalcin; ARNT, AHR
nuclear translocator. |
In conclusion, the present study demonstrates that
the agonist of AHR signaling, TCDD, suppresses the growth of human
colorectal cancer cells and stimulates their death, via AHR
signaling, probably as the result of stimulation of manifold
molecules in regulating various signaling pathways. Therefore,
targeting AHR signaling may cause an antitumor effect in
vivo, providing a novel strategic tool for therapy of various
cancer types.
Funding
The present study was supported in part from a NIH
grant (grant no. 1 RO1ES024434; received by OH).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors’ contributions
MY conceived and designed the study. MY performed
the experiment, and MY and OH discussed the data. MY wrote the
manuscript, and OH reviewed and edited the manuscript. All authors
read and approved the manuscript, and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the study are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Medema JP and Vermeulen L:
Microenvironmental regulation of stem cells in intestinal
homeostasis and cancer. Nature. 474:318–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Porter MG and Stoeger SM: Atypical
colorectal neoplasms. Surg Clin North Am. 97:641–656. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
American Cancer Society: Cancer facts and
figures 2016. American Cancer Society; Atlanta, GA: 2016,
https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2016.html.
Accessed January 24, 2017.
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
6
|
Alnabulsi A and Murray GI: Integrative
analysis of the colorectal cancer proteome: Potential clinical
impact. Expert Rev Proteomics. 13:1–11. 2016. View Article : Google Scholar
|
|
7
|
Alnabulsi A, Swan R, Cash B, Alnabulsi A
and Murray GI: The differential expression of omega-3 and omega-6
fatty acid metabolising enzymes in colorectal cancer and its
prognostic significance. Br J Cancer. 116:1612–1620. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carini F, Mazzola M, Rappa F, Jurjus A,
Geagea AG, Al Kattar S, Bou-Assi T, Jurjus R, Damiani P, Leone A,
et al: Colorectal carcinogenesis: Role of oxidative stress and
antioxidants. Anticancer Res. 37:4759–4766. 2017.PubMed/NCBI
|
|
9
|
Colussi D, Brandi G, Bazzoli F and
Ricciardiello L: Molecular pathways involved in colorectal cancer:
Implications for disease behavior and prevention. Int J Mol Sci.
14:16365–16385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kudryavtseva AV, Lipatova AV, Zaretsky AR,
Moskalev AA, Fedorova MS, Rasskazova AS, Shibukhova GA, Snezhkina
AV, Kaprin AD, Alekseev BY, et al: Important molecular genetic
markers of colorectal cancer. Oncotarget. 7:53959–53983. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hankinson O: The aryl hydrocarbon receptor
complex. Annu Rev Pharmacol Toxicol. 35:307–340. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hankinson O: Role of coactivators in
transcriptional activation by the aryl hydrocarbon receptor. Arch
Biochem Biophys. 433:379–386. 2005. View Article : Google Scholar
|
|
13
|
Denison MS and Nagy SR: Activation of the
aryl hydrocarbon receptor by structurally diverse exogenous and
endogenous chemicals. Annu Rev Pharmacol Toxicol. 43:309–334. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nguyen LP and Bradfield CA: The search for
endogenous activators of the aryl hydrocarbon receptor. Chem Res
Toxicol. 21:102–116. 2008. View Article : Google Scholar
|
|
15
|
Ronnekleiv-Kelly SM, Nukaya M, Díaz-Díaz
CJ, Megna BW, Carney PR, Geiger PG and Kennedy GD: Aryl hydrocarbon
receptor-dependent apoptotic cell death induced by the flavonoid
chrysin in human colorectal cancer cells. Cancer Lett. 370:91–99.
2016. View Article : Google Scholar :
|
|
16
|
Ikuta T, Kurosumi M, Yatsuoka T and
Nishimura Y: Tissue distribution of aryl hydrocarbon receptor in
the intestine: Implication of putative roles in tumor suppression.
Exp Cell Res. 343:126–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pastorková B, Vrzalová A, Bachleda P and
Dvořák Z: Hydroxystilbenes and methoxystilbenes activate human aryl
hydrocarbon receptor and induce CYP1A genes in human hepatoma cells
and human hepatocytes. Food Chem Toxicol. 103:122–132. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Safe S, Lee SO and Jin UH: Role of the
aryl hydrocarbon receptor in carcinogenesis and potential as a drug
target. Toxicol Sci. 135:1–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mulero-Navarro S and Fernandez-Salguero
PM: New trends in aryl hydrocarbon receptor biology. Front Cell Dev
Biol. 4:452016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stejskalova L and Pavek P: The function of
cytochrome P450 1A1 enzyme (CYP1A1) and aryl hydrocarbon receptor
(AhR) in the placenta. Curr Pharm Biotechnol. 12:715–730. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nukaya M, Moran S and Bradfield CA: The
role of the dioxin-responsive element cluster between the Cyp1a1
and Cyp1a2 loci in aryl hydrocarbon receptor biology. Proc Natl
Acad Sci USA. 106:4923–4928. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pierre S, Chevallier A, Teixeira-Clerc F,
Ambolet-Camoit A, Bui LC, Bats AS, Fournet JC, Fernandez-Salguero
P, Aggerbeck M, Lotersztajn S, et al: Aryl hydrocarbon
receptor-dependent induction of liver fibrosis by dioxin. Toxicol
Sci. 137:114–124. 2014. View Article : Google Scholar
|
|
23
|
Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz
S, Van Winkle L, Matsumura F and Vogel CF: Activation of aryl
hydrocarbon receptor induces vascular inflammation and promotes
atherosclerosis in apolipoprotein E−/− mice.
Arterioscler Thromb Vasc Biol. 31:1260–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brito JS, Borges NA, Esgalhado M, Magliano
DC, Soulage CO and Mafra D: Aryl hydrocarbon receptor activation in
chronic kidney disease: Role of uremic toxins. Nephron. 137:1–7.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Esser C: The aryl hydrocarbon receptor in
immunity: Tools and potential. Methods Mol Biol. 1371:239–257.
2016. View Article : Google Scholar
|
|
26
|
Murray IA, Patterson AD and Perdew GH:
Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat
Rev Cancer. 14:801–814. 2014. View Article : Google Scholar
|
|
27
|
Barouki R, Coumoul X and
Fernandez-Salguero PM: The aryl hydrocarbon receptor, more than a
xenobiotic-interacting protein. FEBS Lett. 581:3608–3615. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moennikes O, Loeppen S, Buchmann A,
Andersson P, Ittrich C, Poellinger L and Schwarz M: A
constitutively active dioxin/aryl hydrocarbon receptor promotes
hepatocarcinogenesis in mice. Cancer Res. 64:4707–4710. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fan Y, Boivin GP, Knudsen ES, Nebert DW,
Xia Y and Puga A: The aryl hydrocarbon receptor functions as a
tumor suppressor of liver carcinogenesis. Cancer Res. 70:212–220.
2010. View Article : Google Scholar
|
|
30
|
Mathew LK, Simonich MT and Tanguay RL:
AHR-dependent misregulation of Wnt signaling disrupts tissue
regeneration. Biochem Pharmacol. 77:498–507. 2009. View Article : Google Scholar :
|
|
31
|
Jackson DP, Li H, Mitchell KA, Joshi AD
and Elferink CJ: Ah receptor-mediated suppression of liver
regeneration through NC-XRE-driven p21Cip1 expression.
Mol Pharmacol. 85:533–541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamaguchi M and Hankinson O:
2,3,7,8-Tetrachlorodibenzo- p-dioxin suppresses the growth of human
liver cancer HepG2 cells in vitro: Involvement of cell signaling
factors. Int J Oncol. 53:1657–1666. 2018.PubMed/NCBI
|
|
33
|
Harper PA, Prokipcak RD, Bush LE, Golas CL
and Okey AB: Detection and characterization of the Ah receptor for
2,3,7,8-tetra-chlorodibenzo-p-dioxin in the human colon
adenocarcinoma cell line LS180. Arch Biochem Biophys. 290:27–36.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Megna BW, Carney PR, Nukaya M, Geiger P
and Kennedy GD: Indole-3-carbinol induces tumor cell death:
Function follows form. J Surg Res. 204:47–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Megna BW, Carney PR, Depke MG, Nukaya M,
McNally J, Larsen L, Rosengren RJ and Kennedy GD: The aryl
hydrocarbon receptor as an antitumor target of synthetic
curcuminoids in colorectal cancer. J Surg Res. 213:16–24. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wohak LE, Krais AM, Kucab JE, Stertmann J,
Øvrebø S, Seidel A, Phillips DH and Arlt VM: Carcinogenic
polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a
p53-dependent mechanism. Arch Toxicol. 90:291–304. 2016. View Article : Google Scholar :
|
|
37
|
Li W, Harper PA, Tang BK and Okey AB:
Regulation of cytochrome P450 enzymes by aryl hydrocarbon receptor
in human cells: CYP1A2 expression in the LS180 colon carcinoma cell
line after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin or
3-methylcholanthrene. Biochem Pharmacol. 56:599–612. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xie G and Raufman J-P: Role of the aryl
hydrocarbon receptor in colon neoplasia. Cancers (Basel). 7. pp.
1436–1446. 2015, View Article : Google Scholar
|
|
39
|
Kawajiri K, Kobayashi Y, Ohtake F, Ikuta
T, Matsushima Y, Mimura J, Pettersson S, Pollenz RS, Sakaki T,
Hirokawa T, et al: Aryl hydrocarbon receptor suppresses intestinal
carcinogenesis in ApcMin/+ mice with natural ligands.
Proc Natl Acad Sci USA. 106:13481–13486. 2009. View Article : Google Scholar
|
|
40
|
Fang Z, Tang Y, Fang J, Zhou Z, Xing Z,
Guo Z, Guo X, Wang W, Jiao W, Xu Z and Liu Z: Simvastatin inhibits
renal cancer cell growth and metastasis via AKT/mTOR, ERK and
JAK2/STAT3 pathway. PLoS One. 8:e628232013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang K, Li Y, Jiang YZ, Dai CF, Patankar
MS, Song JS and Zheng J: An endogenous aryl hydrocarbon receptor
ligand inhibits proliferation and migration of human ovarian cancer
cells. Cancer Lett. 340:63–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Misawa H, Inagaki S and Yamaguchi M:
Suppression of cell proliferation and deoxyribonucleic acid
synthesis in the cloned rat hepatoma H4-II-E cells overexpressing
regucalcin. J Cell Biochem. 84:143–149. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yamaguchi M, Osuka S and Murata T:
Prolonged survival of patients with colorectal cancer is associated
with a higher regucalcin gene expression: Overexpression of
regucalcin suppresses the growth of human colorectal carcinoma
cells in vitro. Int J Oncol. 53:1313–1322. 2018.PubMed/NCBI
|
|
44
|
Yamaguchi M and Daimon Y: Overexpression
of regucalcin suppresses cell proliferation in cloned rat hepatoma
H4-II-E cells: Involvement of intracellular signaling factors and
cell cycle-related genes. J Cell Biochem. 95:1169–1177. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Izumi T and Yamaguchi M: Overexpression of
regucalcin suppresses cell death in cloned rat hepatoma H4-II-E
cells induced by tumor necrosis factor-alpha or thapsigargin. J
Cell Biochem. 92:296–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yamaguchi M and Isogai M: Tissue
concentration of calcium-binding protein regucalcin in rats by
enzyme-linked immunoadsorbent assay. Mol Cell Biochem. 122:65–68.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Choi E-Y, Lee H, Dingle RWC, Kim KB and
Swanson HI: Development of novel CH223191-based antagonists of the
aryl hydrocarbon receptor. Mol Pharmacol. 81:3–11. 2012. View Article : Google Scholar :
|
|
48
|
Tsurusaki Y and Yamaguchi M: Role of
regucalcin in liver nuclear function: Binding of regucalcin to
nuclear protein or DNA and modulation of tumor-related gene
expression. Int J Mol Med. 14:277–281. 2004.PubMed/NCBI
|
|
49
|
Yamaguchi M: Suppressive role of
regucalcin in liver cell proliferation: Involvement in
carcinogenesis. Cell Prolif. 46:243–253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamaguchi M, Osuka S, Weitzmann MN,
El-Rayes BF, Shoji M and Murata T: Prolonged survival in pancreatic
cancer patients with increased regucalcin gene expression:
Overexpression of regucalcin suppresses the proliferation in human
pancreatic cancer MIA PaCa-2 cells in vitro. Int J Oncol.
48:1955–1964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yamaguchi M, Osuka S, Weitzmann MN,
El-Rayes BF, Shoji M and Murata T: Prolonged survival in
hepatocarcinoma patients with increased regucalcin gene expression:
HepG2 cell proliferation is suppressed by overexpression of
regucalcin in vitro. Int J Oncol. 49:1686–1694. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nejak-Bowen KN, Zeng G, Tan X, Cieply B
and Monga SP: Beta-catenin regulates vitamin C biosynthesis and
cell survival in murine liver. J Biol Chem. 284:28115–28127. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kochhar A, Kopelovich L, Sue E, Guttenplan
JB, Herbert BS, Dannenberg AJ and Subbaramaiah K: p53 modulates
Hsp90 ATPase activity and regulates aryl hydrocarbon receptor
signaling. Cancer Prev Res (Phila). 7:596–606. 2014. View Article : Google Scholar
|
|
54
|
Yamaguchi M: Role of regucalcin in cell
nuclear regulation: Involvement as a transcription factor. Cell
Tissue Res. 354:331–341. 2013. View Article : Google Scholar : PubMed/NCBI
|