Introduction
Glioblastoma multiforme (GBM) is the most common and
aggressive form of primary malignant brain tumor (1-3).
Although multiple treatments have emerged in recent years, such as
surgery, radiotherapy, and chemotherapy, the overall survival of
patients with glioblastoma has not changed significantly (4,5).
Glioma stem cells (GSCs) are increasingly recognized as a driving
force supporting glioma development, resistance to treatment, and
malignant recurrence (6). GSCs
possess self-renewal capacity and multipotency, and can induce
tumorigenesis (7). The elucidation
of the molecular mechanism controlling GSCs may provide novel
information for development of targeted treatment for GBM (3).
Bromodomain and extraterminal domain (BET) proteins
are epigenetic readers that regulate a variety of important
biologic processes, such as transcriptional regulation, chromatin
remodeling, and DNA damage repair (7-9). BET
proteins were revealed to have oncogenic effects for the first time
in NUT midline carcinoma (10). By
screening a library of small hairpin RNAs targeting known chromatin
regulators in acute myeloid leukemia (AML), a subsequent study
identified bromodomain-containing protein 4 (Brd4) as an important
factor in the maintenance of AML (11). Recently, BET proteins, especially
Brd4, have emerged as valuable therapeutic targets for treating
several cancers, such as breast, prostate, and liver cancers
(12-14). These findings indicated Brd4 could
be a therapeutic target for treating GBM.
The small-molecule BET inhibitor JQ1 can
competitively bind to acetylated lysine residues in the BET
bromodomain to prevent binding of the BET protein with chromatin
(15). BET bromodomain inhibitors
can inhibit the transcription of c-Myc, and thus, there are
potential targets for the clinical treatment of Myc-driven cancer,
including GBM (16,17). In several cancers, JQ1 has notable
anti-inflammatory and anti-tumor effects in vitro and in
vivo (18,19). Although some studies have examined
Brd4, its role in GBM tumorigenesis is yet to be determined, and
the potential of JQ1 for treating GBM is largely unexplored.
To resolve these uncertainties, we investigated the
effects of Brd4 in GSCs using JQ1 or small interfering RNAs
(siRNAs) in vitro and in vivo. Transcriptome
sequencing analysis of JQ1-treated GSCs revealed that Brd4 is
closely associated with the PI3K-AKT pathway. Furthermore, our
findings revealed the mechanism by which the vascular endothelial
growth factor (VEGF)/PI3K/AKT axis regulates cell cycle progression
and apoptosis of GSCs, which would in part explain the involvement
of Brd4 in the anti-tumor effects of JQ1. We aimed to confirm the
role of Brd4 in the tumorigenesis and progression of GBM, and
reveal the underlying molecular mechanisms. Furthermore, our study
identified JQ1 as a novel promising inhibitor for treating GBM.
Materials and methods
Cell culture and transfection
Primary murine glioma stem cells (CSC2078 and
CSC1589) were isolated from human glial fibrillary acidic protein
(hGFAP)-Cre+ p53L/L phosphatase tensin homolog
(Pten)L/+ mice as previously described (20); cell isolation was not conducted in
the present study. The numbers of GSCs were encoded as the same as
the code number of transgenic mice. The hGFAP-Cre transgene to
delete p53 alone or in combination with Pten in all CNS lineages
using conditional p53 and Pten alleles as previously described
(20). Mice (n=42, 15-40 weeks
old, 20-22 g, Dana-Farber Cancer Institute) were interbred and
maintained on FvB/C57Bl6 hybrid background. 73% of
hGFAP-Cre+; P53lox/lox; Ptenlox/+
mice (between ages 15-40 weeks) presented with acute-onset
neurological symptoms in clinical analysis. All 42 neurologically
symptomatic mice harbored malignant gliomas were classified as
anaplastic astrocytomas [World Health Organization (WHO) III, n=28,
66%] or GBM (WHO IV, n=14, 34%). In this study, all manipulations
were performed with IACUC approval (20). Patient-derived glioma stem cells
(GSCs) TS543 were a gift from Dr Cameron W. Brenner (Memorial Sloan
Kettering Cancer Center) (21).
CSC2078 and CSC1589 were cultured in neurobasal media (Dulbecco's
Modified Eagle's medium/F12, HyClone; GE Healthcare Life Sciences)
with epidermal growth factor (20 ng/ml, cat. no. 315-09, PeproTech,
Inc.) and basic fibroblast growth factor (10 ng/ml, cat. no.
450-33, PeproTech, Inc.) or differentiation medium (neurobasal
media with 1% FBS). TS543 cells were cultured in NeuroCult NS-A
proliferation media (human; Stem Cell Technologies) supplemented
with epidermal growth factor (20 ng/ml, cat. no. GMP100-15,
PeproTech, Inc.) and basic fibroblast growth factor (10 ng/ml, cat.
no. 100-18B, PeproTech, Inc.). Cells were cultured at 37°C in a 5%
CO2.
CSC2078 cells were seeded into a 6-well plate at
2×105 cells per well overnight. The following day,
CSC2078 were transfected with three independent Brd4-specific
siRNAs (20 µM; siBrd4s-475, siBrd4s-1648, siBrd4s-3820) and
a negative control siRNAs (20 µM) for 48 h. siRNAs was
purchased from Gene Pharma and the sequences were described in
Table SI. The transfection agent
INTERFERin (Thermo Fisher Scientific, Inc.) was used according to
the manufacturer's protocol. The efficiency of transfection was
determined by western blotting.
Cell viability, cell proliferation, and
self-renewal assay
To determine the dose response to JQ1, cells were
seeded into a 96-well plate in triplicate at the density of 10,000
cells per well and adhered overnight. Cells were treated with
serial dilutions of JQ1 at 37°C for 24 or 48 h. JQ1 was added by
2-fold serial dilutions (the concentration of JQ1 for CSC2078 and
TS543: 0.05, 0.1, 0.2, 0.4 and 0.8 µM; the concentration of
JQ1 for CSC1589: 0.0625, 0.125, 0.25, 0.5 and 1 µM). CSC2078
cells transfected with siRNAs for 24, 48 and 72 h. The Cell
Counting Kit-8 (CCK-8) solution (10 µl, MedChem Express) was
added to each well of the plate. Then, the plate was incubated at
37°C for 4 h. The absorbance was measured at 450 nm using a
microplate reader. The Cell Counting Kit-8 (cat. no. HY-K0301) and
JQ-1 (cat. no. HY-13030) was purchased from MedChem Express.
For the soft agar colony assay, 2,500 cells were
plated in the upper layer for each well in 6-well plate in
triplicate. The concentration of the lower layers of agar was 0.6%
and the upper layers were 0.4%. The agar was mixed with the
neurobasal medium or NeuroCult NS-A proliferation media at a ratio
of 1:1. The number of colonies in five random microscopic fields
were counted after 10-15 days of seeding GSCs and staining by 0.1%
crystal violet for 1 h at room temperature. Images were captured
using a light microscope at ×400 magnification in five random
microscopic fields (BX71 Olympus Corporation).
Self-renewal capacity was measured by neural sphere
formation assays. The cells were cultured into 6-well plates
(100/well) in triplicate and treated with JQ1 (0.1, 0.2, 0.25 and
0.5 µM) for 24 h and transfected with siRNAs for 48 h at
37°C. The neural spheres were cultured in NSC proliferation media
(HyClone; GE Healthcare Life Sciences) containing growth factors
(epidermal growth factor, 20 ng/ml; basic fibroblast growth factor,
10 ng/ml) for 10 days at 37°C. After 10 days, neurospheres
containing >50 cells were scored (22). The number of neural spheres in each
well was counted under an IX71 light microscope (Olympus
Corporation) at ×100 and 200 magnification. Data were presented as
the percentage of sphere-forming cells relative to control.
Cell-cycle distribution
Cells were seeded in 6-well plates (1×105
cells/well) and treated with JQ1 (0.05, 0.1, 0.125, 0.2, 0.25 and
0.5 µM) and/or linifanib (5 µM, cat. no. HY-50751,
MedChem Express) for 24 h. CSC2078 cells were transfected with
siRNAs for 48 h. We fixed cells with 1 ml 70% ethanol overnight at
4°C. The collected cells were detected using a propidium iodide
(PI)/RNase Staining Solution (cat. no. CY2001-P Sungene Biotech)
according to the manufacturer's protocols. The cell cycle was
analyzed using a flow cytometer (BD Biosciences) and ModFit LT 5.0
(Verity Software House).
Annexin V apoptosis assay, terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
Cells were seeded in 6-well plates overnight at a
density of 1×105 cells/well. Then, cells were treated
with JQ1 and/or linifanib for 24 h and were transfected with siRNAs
for 48 h. The vehicle-, JQ1/linifanib and JQ1 + Linifanib-treated
cells were stained with Annexin V-fluorescein isothiocyanate and PI
according to the instructions provided with the Apoptosis Detection
Kit (cat. no. WLA001, Wanleibio Co., Ltd.). The vehicle-treated
cells were treated with dimethyl sulfoxide (DMSO; maximum final
concentration of 0.01%). The apoptosis rate was calculated as the
sum of early apoptotic and late apoptotic cells Apoptosis was
analyzed using flow cytometry.
A TUNEL assay was conducted using the DeadEnd™
Colorimetric TUNEL System (Promega Coporation) and DeadEnd™
Fluorometric TUNEL System (Promega Corporation). The DeadEnd™
Colorimetric TUNEL System was used to detect the apoptosis of
CSC2078 treated with JQ1. Cells were treated with JQ1, or vehicle
(DMSO at a maximum final concentration of 0.01%) for 24 h. The
DeadEnd™ Fluorometric TUNEL System was used to detect the apoptosis
of tumors of CSC2078-subcutaneous xenograft mice as described
below. The TUNEL assay was performed according to the
manufacturer's instructions. Images of cells and tumor tissues were
captured using an Olympus BX53 microscope (light and fluorescence)
at ×400 magnification. For quantitation, the TUNEL-positive cells
were counted in five random area images for each sample using
ImageJ 1.46r (National Institutes of Health).
Hoechst 33342 staining
CSC1589 cells were treated with JQ1 for 24 h. Then,
the CSC1589 were fixed with 4% paraformal-dehyde for 5 min. Then,
CSC1589 were stained with 1 mg/ml Hoechst (Beyotime Biotechnology
Inc., Nantong, China) for 10 min at room temperature in the dark.
Finally, the cells were washed three times with PBS and imaged by
Olympus BX53 microscope (fluorescence) at ×400 magnification.
RNA isolation, reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA of CSC2078 and TS543 cells was extracted
using TRIzol (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Following isolation, total RNA was
reverse transcribed using the Revert Aid First Strand cDNA
Synthesis Kit (cat. no. K1622; Thermo Fisher Scientific, Inc.).
Briefly, RNA (2 µg) was incubated for 60 min at 42°C and for
5 sec at 70°C. qPCR was conducted using the SYBR® Premix
Ex Taq™ II (Tli RNaseH Plus, cat. no. RR820A, Takara Bio, Inc.)
according to the manufacturer's protocols. The thermocycling
conditions were as follows: 95°C initial denaturation for 30 sec;
followed by 40 cycles of denaturation at 95°C for 5 sec, and
annealing and extension at 60°C for 30 sec) using CFX96 (Bio-Rad
Laboratories, Inc.). After being normalized to the expression
levels of GADPH, the relative mRNA expression levels were
determined by the 2−ΔΔCq method (23). The primers used are described in
Table SII.
Protein extraction and western
blotting
GSCs were lysed in phenylmethylsulfonyl fluoride and
radioimmunopre-cipitation assay lysis buffer (1:100, Wuhan Boster
Biological Technology, Ltd.), and the collected lysates were
treated by sonication at 4°C, then centrifuged at 12,000 × g at 4°C
for 15 min. The supernatants were used as cell lysates. The protein
concentration was analyzed by a BCA protein assay kit (cat. no.
PP1001, Bioteke Corporation). Samples containing 30 µg of
protein were separated by SDS-PAGE (10, 12 and 15%) and transferred
onto polyvinylidene difluoride membranes which were probed with
antibodies. Prior, the membranes were blocked with 5% non-fat milk
in Tris-buffered saline containing 0.05% Tween-20 at room
temperature for 1 h. Subsequently, the membranes were incubated
overnight at 4°C with the following antibodies (1:1,000 dilutions)
against BRD4 antibody [EPR5150(2);
ab128874) from Abcam; phosphorylated (phosphor)-AKT (Ser473, cat
no. 4060) and phospho-retinoblastoma (Rb; Ser807/811, cat. no.
9308S) from Cell Signaling Technology, Inc.; total AKT (cat. no.
10176-2-AP), c-Myc (cat. no. 10828-1-AP), Bcl2-associated X protein
(BAX; cat. no. 50599-2-lg), BCL2 (cat. no. 12789-1-AP), caspase
3/p17/p19 (cat. no. 19677-1-AP), P27 (cat. no. 25614-1-AP), E2F1
(cat. no. 12171-1-AP), matrix metalloproteinase 2 (MMP2; cat. no.
10373-2-AP), MMP9 (cat. no. 10375-2-AP) and Actin antibody from
ProteinTech Group, Inc.; CCND1 (A11022) from ABclonal Biotech Co.,
Ltd.; proliferating cell nuclear antigen (PCNA; PC10, SC-56) from
Santa Cruz Biotechnology, Inc.; Rb (G3-245) from BD Pharmingen (BD
Biosciences); VEGF (cat. no. 05-443) from Upstate Biotechnology;
Histone H2A.X (D17A3) XP (H2AX, cat. no. 7631), phospho-Histone
H2A.X (Ser139, 20E3, γH2AX; cat. no. 9718), VEGFR2 (cat. no. 55B11)
and phospho-VEGF receptor 2 (Tyr1175, cat. no. 19A10) from Cell
Signaling Technology, Inc. AKT (phospho Thr308) from Immunoway
Biotechnology. The secondary antibodies, horseradish peroxidase
(HRP)-conjugated Affinipure Goat Anti-Mouse IgG (H+L) (cat. no.
SA00001-1; 1:2,000) and HRP-conjugated Affinipure Goat Anti-Rabbit
IgG (H+L) (cat. no. SA00001-2, 1:2,000) antibodies were purchased
from ProteinTech Group, Inc. After visualization using the enhanced
chemiluminescence (Thermo Fisher Scientific, Inc.), the density of
the bands was analyzed using image analysis software (Image J
1,46r).
Immunofluorescence
CSC1589 cells were cultured in 12-well plates at
density of 1×105 cells/well and treated with JQ1 for
0.125 or 0.25 µM. Single cell suspensions were seeded on
coverslips pre-coated with fibronectin (1 µg/ml, cat. no.
F1141-5MG, Sigma-Aldrich; Merck KGaA) and ornithine (50 mg/ml,
P3655-50MG, Sigma-Aldrich; Merck KGaA) overnight. CSC1589 cells
were fixed with 4% paraformaldehyde at room temperature for 10-15
min and permeabilized with 0.3% Triton X-100 (Sigma-Aldrich; Merck
KGaA) in PBS for 5 min. Then, potential non-specific binding sites
were blocked with antibody dilution buffer [10% fetal bovine serum
and (Sigma-Aldrich; Merck KGaA) and 1% bovine serum albumin
(Gentihold) in PBS]. Sections was incubated with an antibody
against Nestin (ab6320, 1:50; Abcam) overnight at 4°C. After
washing the sections with PBS for 5 min, secondary antibodies were
applied [Donkey anti-Mouse IgG (H+L) ReadyProbes™ Secondary
Antibody, Alexa Fluor® 488, R37114, 1:5,000; Invitrogen;
Thermo Fisher Scientific, Inc.] for 30 min at room temperature
before mounting in water soluble mounting medium (Wuhan Boster
Biological Technology, Ltd.) with DAPI (room temperature, 2 min).
After the final washing step by PBS, the glass coverslips were
mounted upside-down and visualized with an Olympus BX53 microscope
at ×400 magnification.
RNA-seq analysis
Total RNA of CSC2078 cells was isolated using the
TRIzol, after which the quality, concentration and integrity were
determined using a NanoDrop spectrophotometer (NanoDrop
Technologies; Thermo Scientific Fisher, Inc.). RNA (3 µg)
was used for the RNA sample preparations. Sequencing libraries were
generated using the TruSeq RNA Sample Preparation Kit (Illumina,
Inc.). Briefly, mRNA was purified from total RNA using poly-T
oligoattached magnetic beads. Fragmentation was carried out using
divalent cations under elevated temperature (94°C, 5 min) in an
Illumina proprietary fragmentation buffer. First strand cDNA was
synthesized using random oligonucleotides and SuperScript II (25°C
for 10 min; 42°C for 15 min; 70°C for 15 min, Illumine, Inc.)
reverse transcriptase. Then, the second strand cDNA synthesis was
performed by DNA Polymerase I and RNase H (16°C for 1 h, Illumine,
Inc.). Remaining overhangs were converted into blunt ends via
exonuclease/polymerase activities and the enzymes were removed.
After adenylation of the 3′ends of the DNA fragments, Illumina PE
adapter oligonucleotides were ligated to prepare for hybridization.
To select cDNA fragments of the preferred 200 bp in length, the
library fragments were purified using the AMPure XP system (Beckman
Coulter, Inc.). DNA fragments with ligated adaptor molecules on
both ends were selectively enriched using Illumina PCR Primer
Cocktail (Illumina, Inc.) in a 15 cycle PCR reaction (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 98°C initial denaturation for 30 sec;
followed 98°C denaturation for 10 sec, and 60°C annealing for 30
sec; and 72°C extension for 30 sec. Products were purified (AMPure
XP system) and quantified using the Agilent high sensitivity DNA
assay on a Bioanalyzer 2100 system (Agilent Technologies, Inc.).
The sequencing library was then sequenced on a Hiseq platform
(Illumina, Inc.) by Shanghai Personal Biotechnology Co. Ltd.
Gene ontology (GO) and Kyoto encyclopedia
of genes and genomes (KEGG) pathway analysis
We used HTSeq(0.9.1) statistics (https://htseq.readthedocs.io/en/release_0.11.1/history.html#version-0-9-1)
to compare the Read Count values on each gene as the original
expression of the gene, and then used fragments per kilobase of
transcript per million to standardize the expression. Then we used
DESeq (1.30.0) (24) to analyze
the genes of difference expression with screened conditions as
follows: Expression difference multiple |log2FoldChange| >1,
significance, P<0.05. In addition, we used R language Pheatmap
(1.0.8) software package (https://cran.r-project.org/web/packages/pheatmap/index.html)
to perform bi-directional clustering analysis of all different
genes of samples. We mapped all the genes to terms in the GO
database and calculated the numbers of differentially enriched
genes in each term. Based on the whole genome, terms with
significant enrichment of differentially enriched genes were
calculated by hypergeometric distribution. We counted the number of
differentially expressed genes following KEGG pathway enrichment
analysis (https://www.genome.jp/kegg/); the
metabolic pathways and signaling pathways that differentially
expressed genes mainly participate in were identified.
Animal studies
BALB/c male nude mice (age, 5-8 weeks) were obtained
from Beijing Vital River Laboratory Animal Technology. A total of
10 mice each weighing 18-22 g were used. Mice were housed under a
12 h light/dark cycle in an air-conditioned room at 22±2°C with
free access to food and water. In xenograft implantation
experiments, nude mice were injected subcutaneously in the right
flank with CSC2078 cells (1×107) suspended in 100
µl PBS. After 2 days, nude mice of the JQ1-treated group
were administered intraperitoneally with JQ1 (50 mg/kg/day) for 17
days. DMSO-containing JQ1 was diluted with physiological saline to
obtain a final solution of 5 mg/ml in JQ1-treated groups. Mice of
the control group were injected an equal amount of physiological
saline containing 5% DMSO per day. We measured the weight and tumor
size every day. Tumor size was measured with calipers. The tumor
volume was calculated with the following formula: V=(length) 2×
(width)/2. The humane endpoints were judged by mouse activity
assessment (hunching, stationary, poor grooming and ruffling) or
the mouse weight loss (>20% of total body weight). No animals
were sacrificed due to meeting these endpoints; all mice were
sacrificed via carbon dioxide after 17 days. The 10 mice were
anesthetized by an intraperitoneal injection of 10% chloral hydrate
at a dosage of 300 mg/kg body weight prior to carbon dioxide
euthanasia; 100% carbon dioxide was applied 10-30% volume per
minute and the mice were euthanized after exposure to carbon
dioxide for 5-6 min. Mice death was confirmed when no spontaneous
breathing for 2-3 min and no blinking reflex were observed. No
secondary method was applied to verify mice death. After the mice
were euthanized, tumor tissues were extracted were divided into two
parts: One part was frozen at -80°C for protein and RNA extraction,
and the other part was fixed in 4% paraformaldehyde (room
temperature, 24 h) for immunofluorescent staining. The splanchnic
tissues (heart, liver, spleen, lung and kidney) were excised for
hemoxylin and eosin (H&E) staining. The largest tumor in the
control group had a diameter of 13 mm and the JQ1-treated group had
a diameter of 7 mm. Animal experiments were approved by the Animal
Ethics Committee of Jilin University [approval no. 2018;(54)], and by the Institutional Committee
for the Care and Use of Laboratory Animals of the Experimental
Animal Center of Jilin University.
Immunohistochemistry
Animals of the vehicle- and JQ1-treated CSC2078
xenograft model groups were sacrificed and the tumors were rinsed
in PBS, followed by fixation with 4% paraformaldehyde for 24 h at
room temperature. The tumors were cut into five-µm thick
sections and mounted on poly-D-lysine-coated slides. The sections
were then deparaffinized with xylene and rehydrated via alcohol,
followed by 100% alcohol for 5 min, 95% for alcohol for 5 min, 75%
alcohol for 5 min and 50% alcohol for 5 min. Then, the sections
were washed with PBS (1X) for 5 min twice at room temperature.
Antigen retrieval was performed by application of citrate buffer pH
6.00 for 20 min. The slides were incubated with primary antibodies
overnight at 4°C, against c-Myc (cat. no. 10828-1-AP, 1:100), BAX
(cat. no. 50599-2-lg, 1:100), BCL2 (cat. no. 12789-1-AP, 1:100),
MMP2 (cat. no. 10373-2-AP, 1:100) and MMP9 (cat. no. 10375-2-AP,
1:100) from ProteinTech Group, In. Subsequently, HRP secondary
antibodies were applied for 40 min at 37°C (SV0002; Wuhan Boster
Biological Technology, Ltd.) according to the manufacturer's
protocols. DAB (DA1010 Beijing Solarbio Science & Technology
Co., Ltd.) was applied at room temperature for 15 min and analysis
was performed using a BX53 microscope (Olympus Corporation).
Statistical analyses
Statistical analyses were performed with Microsoft
Excel (Microsoft Corporation) and GraphPad Prism 6 software
(GraphPad Software, Inc.). Comparison of the two sets of data was
using an unpaired Student's t-test. For comparison of more than two
sets, one-way ANOVA with a Newman-Keuls post-hoc test was
conducted. All quantifications were performed with at least three
independent experiments. Data are expressed as the mean ± standard
error of the mean. P<0.05 was considered to indicate a
statistically significant difference.
Results
Brd4 is a potential therapeutic target of
GBM
To evaluate the role of epigenetic genes in GSCs, we
acquired GSCs from hGFAP-Cre+ p53L/L PtenL/+
mice (20). We used siRNAs to
silence the expression of Brd4 to detect the effects of Brd4 on the
activity, proliferation and self-renewal ability of GSCs. Then,
western blotting was performed to detect the protein expression of
Brd4 in CSC2078 transfected with the Brd4-specific siRNAs. Compared
with the control group, we found that the expression of Brd4 was
significantly inhibited in the CSC2078 cells treated with siBrd4s,
especially siBrd4s-475 and siBrd4s-3820. This demonstrated the
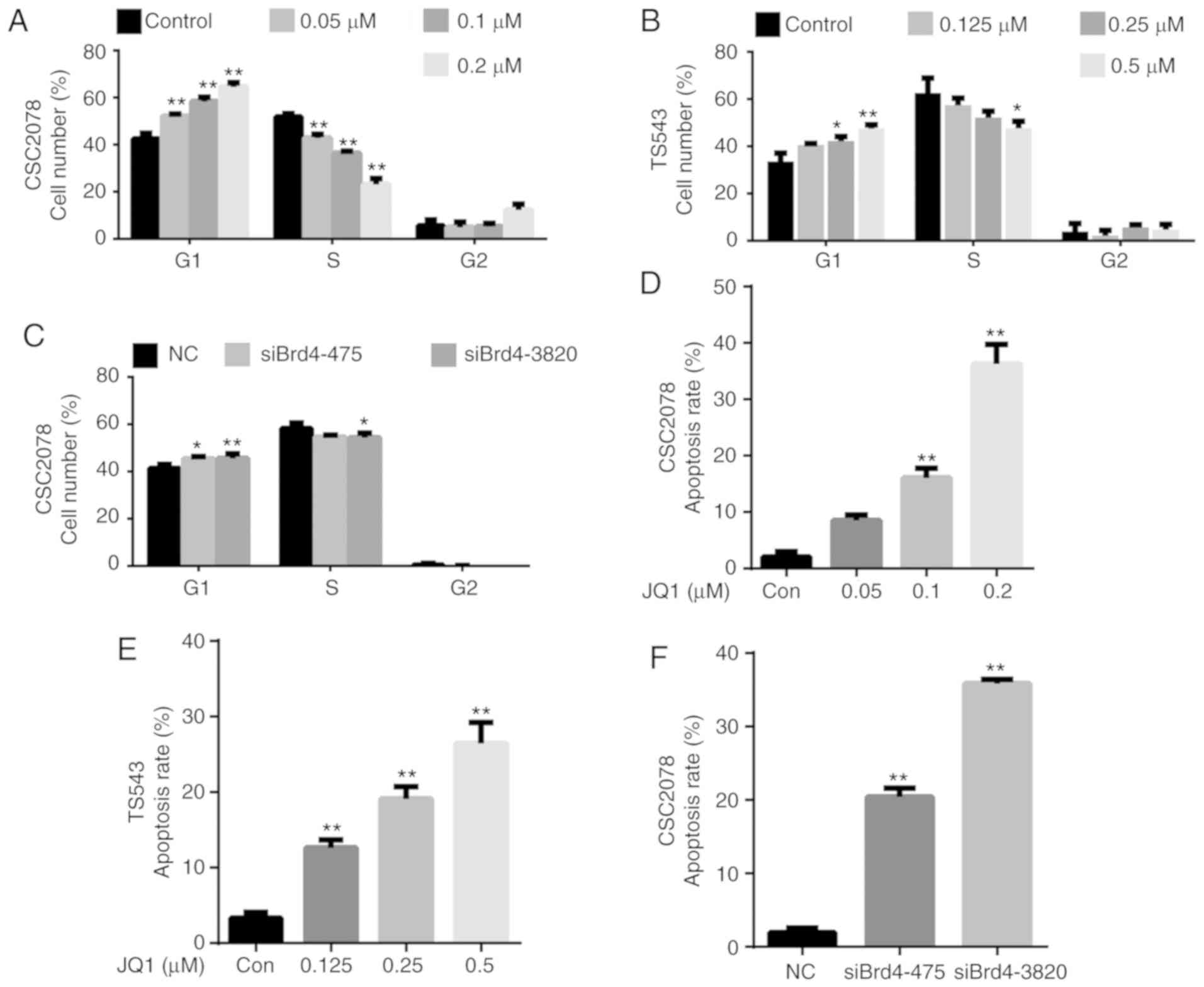
effectiveness of siRNAs targeting Brd4 (Fig. 1A and B). We next explored the
effects of silencing Brd4 on the proliferation and self-renewal of
GSCs. We detected the cell viability of CSC2078 transfected with
siBrd4s using a CCK-8 assay. The results showed that silencing of
Brd4 significantly reduced cell viability (Fig. 1C). A cell growth curve and a soft
agar colony formation assays demonstrated that the three
independent Brd4 siRNAs had significant anti-proliferative effects
on CSC2078 cells compared with the control (Figs. 1D and E, and S2A). Then, we conducted neural sphere
formation assays to detect the effects of the most effective siRNAs
on the self-renewal ability of CSC2078 cells. The results revealed
that the number of neural spheres in the siBrd4 groups were
significantly lower than the control group (Fig. 1F). The results illustrated that
siRNAs targeting Brd4 effectively inhibited the self-renewal
ability of CSC2078 cells (Figs. 1F
and S2E). Taken together, these
results suggested that Brd4 had important functions in mediating an
anti-tumor effect on GSCs, and that Brd4 could be a potential
therapeutic target of in ablating GSCs.
JQ1 suppresses the proliferation and
self-renewal of GSCs
To explore the effects of JQ1 on tumorigenic
properties, we assessed cell proliferation and self-renewal in
TS543, CSC2078 and CSC1589. CCK-8 assays revealed that cell
viability was significantly reduced compared with the control after
24 and 48 h of treatment with JQ1 in a concentration-dependent
manner (Figs. 2A and B, and
S1A). Next, we analyzed cell
proliferation using cell growth curves following 3 days of
treatment of CSC2078, TS543 and CSC1589 with JQ1. JQ1 significantly
inhibited the proliferation of all three GSC lines compared with
the control (Figs. 2C and D, and
S1B). In addition, JQ1
significantly reduced the clone formation capacity of GSCs compared
with the control. Specifically, the number of cell clones was
significantly lower after 10 days of treatment with JQ1. This
further supported that JQ1 effectively inhibited GSCs proliferation
(Figs. 2E and F, S1C and D, and S2A). In addition, neural
sphere formation assays revealed that neural sphere numbers were
significantly decreased by treatment with various concentrations of
JQ1 for 10 days compared with the control (Figs. 2G and H, and S2B-D).
These results demonstrated that the treatment of
GSCs with JQ1 has similar effects as silencing of Brd4. The
anti-neoplastic effects of JQ1 effectively inhibited the
proliferation and self-renewal of GSCs.
JQ1 and siBrd4 inhibits cell
proliferation by inducing G1 arrest and apoptosis of GSCs
To further explore the mechanisms by which JQ1 and
siBrd4 inhibit cell proliferation, we evaluated their effects on
the cell cycle and apoptosis of GSCs. We analyzed the cell cycle
via flow cytometry of CSC2078 and TS543 cells treated with JQ1 for
24 h, and CSC2078 transfected with siBrd4 for 48 h. We found that
JQ1 significantly increased the percentage of cells in G1 and
reduced that in S phase compared with the control; notable effects
on the percentage of cells in G2/M phase were observed (Figs. 3A and B, and S3). We observed a similar G1/S cell
cycle arrest effect in CSC2078 transfected with siBrd4 (Figs. 3C and S3). Collectively, these data suggested
that JQ1 impeded cell cycle progression.
Treatment with JQ1 and siBrd4s also induced
apoptosis in GSCs. We assessed the apoptosis of CSC2078 and TS543
cells via flow cytometry using Annexin V/PI staining. The number of
apoptotic cells was significantly increased in both cells lines
treated with JQ1 compared with the control (Figs. 3D and E, S4, S7 and S8A-D). Similarly, silencing
of Brd4 using siRNAs significantly induced the apoptosis of CSC2078
cells compared with the control (Figs.
3F, S4, and S8E-G).
Consistent with the aforementioned results, Hoechst 33342 staining
illustrated that the number of apoptotic CSC1589 cells was
significantly with JQ1 treatment than in the control group
(Fig. S5A and B). The results of
TUNEL staining also confirmed the ability of JQ1 to promote the
apoptosis of CSC2078 cells (Fig. S5C
and D). These data suggested that JQ1 reduces the viability and
inhibits the proliferation of GSCs by inducing cell cycle arrest
and apoptosis.
Downregulation of Brd4 by JQ1 can
potentially promote GSC differentiation
GO enrichment analysis revealed that treatment of
CSC2078 with JQ1 induced multiple changes of biological activities,
among which 'cell differentiation' was particularly prominent
(Fig. S6A). RNA-seq analysis of
differentially expressed genes (DEGs) illustrated that the neural
stem cell (NSC) markers Nestin and ciliary neurotrophic factor were
significantly downregulated in the JQ1-treated groups (Fig. S6B). We detected the expression of
Nestin via immuno-fluorescence staining of CSC1589 cells. The
results indicated that protein expression levels of Nestin were
significantly downregulated in JQ1-treated cells (Fig. S6C and D). Then, we detected the
mRNA expression of Nestin by RT-qPCR in CSC2078. The results
indicated that the mRNA expression levels of Nestin were
significantly downregulated after treatment with JQ1 for 24 h
(Fig. S6E). In addition, the
astrocyte marker S-100β was upregulated, whereas the
oligodendrocyte marker platelet-derived growth factor α (PDGFα) was
strongly downregulated (Fig.
S6B). The neuronal precursor marker class III b-tubulin was
markedly upregulated in JQ1-treated cells. The results revealed
that JQ1 can inhibit the expression of NSC markers and promote GSC
differentiation to astrocytes under proliferative conditions
(Fig. S6B).
PI3K/AKT signaling pathway plays an
important role in mediating the anti-tumor effect of JQ1
To further explore the mechanism by which
Brd4 regulates the growth of GSCs, we performed RNA-seq comparisons
of the CSC2078 cells treated with JQ1 for 24 h. The results
revealed that 1,327 and 1,484 genes were upregulated and
downregulated, respectively (Fig. 4A
and B). KEGG is a collection of databases containing genomes,
biological pathways, diseases. Compared with the whole genomic
background, these databases can identify pathways of significant
enrichment between candidate target genes (25). The top 20 pathways of candidate
target gene enrichment were shown in the enrich scatter diagram,
and enrichment factors, q-value and gene number were used to report
KEGG enrichment degree. Our results showed that PI3K/AKT signaling
pathway was significantly enriched and the q-value of this pathway
was close to zero; the DEG numbers were higher (Fig. 5). Therefore, we used western
blotting to investigate the relationship between Brd4 and the
PI3K/AKT signal pathway. The results demonstrated that inhibition
of Brd4 using siRNAs or JQ1 inhibited PI3K and phospho-AKT (Ser473
or Thr308); thus, we speculated that Brd4 regulates GSC growth
through PI3K/AKT signaling (Fig.
4C-H).
JQ1 exerts anti-tumor effects in GSCs
through the VEGF/PI3K/AKT signaling pathway
It has been reported that PI3K signaling is
activated by various growth factors, including VEGF (26). We mainly analyzed the downregulated
growth factors in CSC2078 cells treated with JQ1 from the results
of RNA-seq analysis It was found that VEGF was inhibited to the
highest degree (Fig. 6A). VEGF is
released by cancer cells to induce tumor angiogenesis and can be
secreted from the extracellular matrix (ECM) by MMP9 to promotes
tumor growth (27). Thus, we used
western blotting to detect the expression of VEGF and MMP in
CSC2078 cells treated with JQ1. The results demonstrated that the
expression of VEGF, MMP2 and MMP9 were significantly inhibited by
JQ1 (Fig. 6B and C). Therefore, we
preliminarily speculated that JQ1 exhibits anti-tumor effects via
the VEGF/PI3K/AKT signaling pathway. In addition, we proposed that
MMP2 and MMP9 proteins downstream of VEGF can be downregulated by
JQ1, controlling the angiogenesis and metastasis of GBM and
inhibiting tumor development.
 | Figure 6JQ1 induces cycle arrest and
apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling
pathway. (A) The mRNA expression of genes related growth factors in
CSC2078 expressed as fold change. (B and C) Western blotting
analysis of VEGF, MMP9 and MMP2 expression in CSC2078 treated with
JQ1 (error bars represent standard error of the mean). (D and E)
CSC2078 cells were treated with JQ1 and or linifanib for 24 h.
JQ1=100 nM, linifanib=5 µM. The cell cycle was analyzed by
flow cytometry. Quantitation of the cell cycle. Data are
representative of three independent experiments (error bars
represent standard error of the mean). (F and G) CSC2078 cells were
treated with JQ1 and or linifanib for 24 h. JQ1=100 nM, linifanib=5
µM. Effects of CSC2078 apoptosis were detected by flow
cytometry. Quantitation of the apoptosis rate of CSC2078. Data are
representative of three independent experiments. (Error bars
represent standard error of the mean). (H and I) Western blotting
analysis of p-VEGFR2, VEGFR2, PI3K, Akt, p-Akt (Ser473), CyclinB1,
Bcl-2 and Bax in control, JQ1 and/or linifanib treated groups.
JQ1=100 nM, linifanib=5 µM. Densitometry was performed and
fold change of protein expression was presented (error bars
represent standard error of the mean). *P<0.05,
**P<0.01 vs. Con. Bax, Bcl2-associated X protein;
Con, control; J/C, JQ1/Con; MMP, matrix metalloproteinase; p,
phosphorylated; VEGFR2, vascular endothelial growth factor; VEGFR2,
receptor 2. |
In order to further verify that JQ1 plays an
anti-tumor role in GSCs through the VEGF/PI3K/AKT signaling
pathway, we selected the VEGR2 inhibitor linifanib for further
experiments. Linifanib is a novel tyrosine-kinase inhibitor, which
selectively targets VEGFR and PDGFR, and has low off-target
inhibitory activity (28). We
analyzed the cell cycle via flow cytometry of CSC2078 treated with
JQ1 and/or linifanib for 24 h. We found that JQ1, linifanib alone
or in combination induced the cell cycle arrest of CSC2078
(Fig. 6D and E). In addition,
treatment with JQ1 and/or Linifanib significantly induced CSC2078
cell apoptosis. Importantly, the apoptosis rate in JQ1-treated
group was not significant different compared with that
linifanib-treated group or the combination group (Figs. 6F and G, and S9). Consistent with our observations,
there was a reduction in the levels of phospho-VEGFR2, PI3K,
phospho-AKT (Ser473), Cyclin B1 and Bcl-2 in CSC2078 treatment with
JQ1, Linifanib or in combination for 24 h. However, the expression
of Bax was increased and VEGFR2 and AKT was not significantly
altered (Fig. 6H and I). Compared
with treatment with each agent alone, co-treatment with JQ1 and
linifanib did not cause greater attenuation of PI3K/AKT activity
and further induce cell cycle arrest/apoptosis. These results
further indicated that JQ1 exerts anti-tumor effects on GSCs
through the VEGF/PI3K/AKT signaling pathway.
JQ1 and siBrd4 induces cycle arrest and
apoptosis of GSCs through the VEGF/PI3K/AKT signaling pathway
The cell cycle was significantly enriched in KEGG
pathway enrichment analysis. To further investigate the mechanism
by which Brd4 regulates cell cycle progression in GSCs, we used
RT-qPCR to detect cell cycle-related genes, namely c-Myc, cyclin
D1, p21, and p27, in CSC2078 and TS543 cells treated with JQ1. We
found that the mRNA expression levels of the c-Myc and CyclinD1
were significantly inhibited, while P21 and P27 were significantly
upregulated (Figs. 7A and 8A). Then, we detected the protein levels
of cell cycle proteins including Cyclin D1, CDK inhibitor (CDKI)
p27, Rb, phospho-Rb, E2F1 and c-Myc after treatment CSC2078 with
JQ1, or siBrd4 by western blotting. We found that the protein
expression of the c-Myc and CyclinD1 was significantly inhibited,
while P27 was significantly upregulated. JQ1 or siBrd4 reduced the
expression of Rb protein, but the expression level of p-Rb protein
did not significantly change; thus, Rb protein was not activated.
Therefore, E2F1 cannot be released from the RB/E2F1 complex to
promote transcription of downstream genes. The Rb/E2F1 complex was
deactivated resulting in a cycle arrest in GSCs treated with JQ1 or
siRNAs (Fig. 7B-D).
 | Figure 7Inhibition of the vascular
endothelial growth factor/PI3K/AKT signaling pathway induces cell
cycle arrest in glioma stem cells. (A) Reverse
transcription-quantitative polymerase chain reaction of c-Myc,
cyclinD1, P21, P27, Bim, BAX, Bak, Bcl-2 and Bcl-xl in CSC2078
treated with JQ1 for 24 h (error bars represent standard error of
the mean). (B) Western blotting analysis of cyclinD1, P27, Rb,
p-Rb, E2F1 and c-Myc in CSC2078 treated by JQ1 or siBrd4. (C and D)
Quantification (error bars represent standard error of the mean).
*P<0.05, **P<0.01 vs. Control. p,
phosphorylated Rb, retinoblastoma; siBrd4, small interfering RNA
against bromodomain-containing protein 4. |
 | Figure 8Inhibition of VEGF/PI3K/AKT signaling
pathway induces apoptosis in GSCs. (A) Reverse
transcription-quantitative polymerase chain reaction of c-Myc,
cyclinD1, P21, P27, Bim, BAX, Bak and Bcl-2 in TS543 treated with
JQ1 for 24 h (error bars represent standard error of the mean). (B)
Western blotting analysis of Bcl-2, BAX, cleaved-caspase 3, γH2AX
and H2AX in GSCs treated with JQ1 or siBrd4. (C-E) Quantification
(error bars represent standard error of the mean). (F) Proposed
schematic model of VEGF/PI3K/AKT signaling pathway in GSCs.
*P<0.05, **P<0.01 vs. Con. BAX,
Bcl-2-associated X protein; Bak, Bcl2 antagonist/killer 1; Con,
control; GSCs, glioma stem cells; H2AX, H2A histone family member
X; siBrd4, small interfering RNA against bromodomain-containing
protein 4; VEGF, vascular endothelial growth factor. |
As AKT can activate apoptosis in many types of
cancer, we investigated the effect of Brd4 on apoptotic genes in
GSCs. We used RT-qPCR to verify the effects of JQ1 and siBrd4 on
important apoptotic genes in PI3K/AKT signaling, including Bim,
BAX, Bak, Bcl-2, and Bcl-xl in CSC2078 and/or TS543 cells (Figs. 7A and 8A). We also investigated the effects of
JQ1 or siBrd4 on apoptotic proteins, including BAX, Bcl-2, and
cleaved caspase 3 in GSCs via western blotting. We found that the
protein expression of cleaved caspase 3 and Bax were significantly
upregulated, while Bcl-2 was inhibited. In addition, we found that
the protein expression of γH2AX were significantly upregulated. JQ1
or siBrd4 caused DNA damage as indicated by the ratio of
γH2AX/H2AX, which further promoted the apoptosis of GSCs. From our
results, the trend was consistent between JQ1- and siBrd4s-treated
cells. The results indicated that siBrd4 or JQ1 could exhibit
pro-apoptotic effects through the VEGF/PI3K/AKT signal axis in GSCs
(Fig. 8B-E).
JQ1 has anti-tumor activity in a
glioblastoma tumor xenograft model
In addition, we assessed the therapeutic effects of
JQ1 on a glioblastoma tumor xenograft model. Nude mice bearing
CSC2078 subcutaneous tumors were treated with 50 mg/kg of JQ1 via
an intraperitoneal injection once daily and sacrificed on the 17th
day (Fig. 9A). The results
demonstrated that the tumor volume was significantly smaller in the
JQ1-treated group than that in the control group (Fig. 9B and C). Apoptosis in tumor tissues
obtained from control and JQ1-treated mice was detected using TUNEL
staining. The number of apoptotic cells was markedly increased in
JQ1-treated tumors (Fig. 9D). The
results of H&E staining indicated that tumor tissues displayed
obvious nuclear shrinkage in JQ1-treatment group (Fig. 9E). The results were consistent with
the in vitro findings. Immunohistochemical staining revealed
that c-Myc, PCNA, Bcl-2, MMP2 and MMP9 expression was decreased in
tumor tissues from JQ1-treated mice, whereas BAX expression was
elevated (Fig. 9F). To detect the
toxicity of JQ1 in mice, H&E staining was performed in the
organs of mice. There were no obvious histopathological findings in
the JQ1-treated mice, as shown in Fig. 10A. Compared with the findings in
control tumors, the protein expression levels of Brd4 and AKT were
markedly unchanged in JQ1-treated tumors, whereas PI3K and
phosphor-AKT (Ser473) was downregulated. The results were
consistent with the in vitro findings (Fig. 10B and C). We observed significant
reductions in c-Myc, Cyclin D1, and Bcl-2 levels in JQ1-treated
mice compared with the control. By contrast, BAX and γH2AX
expression was significantly increased (Fig. 10D and E). Together, these data
suggested that JQ1 could effectively inhibit tumorigenesis and the
development of GBM. The therapeutic effects of JQ1 may warrant a
clinical trial.
 | Figure 10JQ1 has notable anti-tumor effects on
CSC2078 subcutaneous xenograft mice with low toxicity. (A) H&E
staining of the heart, liver, spleen, lung and kidney tissues.
Scale bar=20 µm. (B and C) Western blotting analysis
proteins expression of Brd4, PI3K, AKT and P-AKT (Ser473), in tumor
tissues (Error bars represent standard error of the mean). (D and
E) Western blotting analysis of c-Myc, Cyclin D1, BAX, Bcl-2, γH2AX
and H2AX expression in tumor tissues (error bars represent standard
error of the mean). *P<0.05, **P<0.01
vs. Con. Con, control; p, phosphorylated; BAX, Bcl-2-associated X
protein; Brd4, bromodomain-containing protein 4; H2AX, H2A histone
family member X. |
Discussion
Previous reports and the current study have
demonstrated that Brd4 is of great value as a therapeutic target
for GBM (22,29,30).
Therefore, therapies targeting Brd4 may aid the development of more
effective treatment options for improving quality of life and
prolonging the survival of patients with GBM (31).
Previous studies illustrated that epigenetic
abnormalities were widespread in glioma; thus, epigenetic analysis
might be critical for developing more effective treatment
strategies for GBM (32,33). The epigenetic reader Brd4 has
emerged as a therapeutic target for many cancers. Brd4 is an
important therapeutic target for NUT midline cancer and
hematopoietic diseases, and encouraging results have been obtained
(11,34,35).
Research on Brd4 as a drug target for hepatocarcinoma, breast
cancer, and pancreatic cancer has become more extensive in past
decade (14,36,37).
To date, few studies have explored the role of Brd4 as a drug
target for glioma cells, especially GSCs. GBM is a highly
heterogeneous tumor; this heterogeneity is dominated by the
presence of GSCs (7). Most
importantly, the reason to study GSCs is that they have shown to be
highly tumorigenic in vivo, and exhibited marked resistance
to conventional chemotherapy and radiotherapy (38,39).
In addition, GSCs are present throughout the tumor and can migrate
along white matter pathways, often evading even gross-total
resection, which provides a possibility for the recurrence of GBM
(40). Over the past decade, the
body of research regarding GSCs has indicated that highly resistant
and tumorigenic sub-populations are maintained in specific
microenvironmental niches, including the vascular niche (41). GBM is one of the tumors with the
highest degree of vascularization in solid tumors (42). Microvascular hyperplasia has been
considered as an important feature of the initiation and
development of GBM (42). GSCs
highly promotes angiogen-esis and the expression of VEGF,
attracting endothelial cells to the tumor and driving neovascular
growth (41,43). Therefore, using GSCs to perform
experiments may increase the value of our results.
JQ1 and Brd4 have co-crystal structures, and thus,
JQ1 has high affinity and specificity for Brd4 (44). In our experiments, we reported that
the anti-tumor effects of JQ1 are consistent with those of siBrd4.
Importantly, the results of RNA-seq experiments indicated that JQ1
selected a few closely related targets and affected only a few
genes. JQ1 exhibits a small number of off-target effects, improving
its efficacy as an epigenetic therapy (45). In addition, JQ1 has excellent
pharmacokinetic properties including 49% oral bioavailability and a
strong ability to cross the blood-brain barrier, which provides a
basis for its use in treating GBM (46). Previous experiments focused on the
inhibitory effect of JQ1 on the proliferation of tumor cells, and
JQ1 also has a certain potential to promote cell differentiation
(47). In our study, we found that
JQ1 can inhibit the expression of NSC markers Nestin and ciliary
neurotrophic factor and promote GSC differentiation to astrocytes
under proliferative conditions. The role of JQ1 in promoting GSCs
needs further study.
As a drug target for tumor treatment, Brd4 is
involved in many signaling pathways, including the Wnt, Hedgehog,
and PI3K-AKT pathways (48-50).
There are two reasons why we selected the PI3K/AKT pathway for
further research in our study. Firstly, the PI3K/AKT signaling
pathway plays an important role in the regulation of signal
transduction (51). The PI3K/AKT
signaling pathway mediates various biological processes such as
cell proliferation, apoptosis, metabolism and angiogenesis in GBM
(52,53). Over the last few decades, it has
been recognized that this signaling pathway is frequently activated
by genetic and epigenetic alterations in malignant brain tumors,
including GBM. Hyper-activation of the PI3K/AKT signaling pathway
is closely related to rapid growth, tumor progression and multidrug
resistance of GBM cells (52).
Secondly, in addition to the PI3K/AKT signaling pathway, there are
the Rap1, P53 and FoxO signaling pathways. The enrichment factors
and false discovery rate values of these signaling pathways are not
significantly superior to the PI3K/AKT signaling pathway. However,
the PI3K/AKT signaling pathway has the most significant DEG numbers
in these signaling pathways. In conclusion, the PI3K/AKT signaling
pathway was selected for further analysis in our study. AKT mainly
has two important phosphorylation sites, Thr308 and Ser473; serine
473 phosphorylation is necessary for the full activation of AKT
(54,55). It has been reported that
phosphorylation of T308 only partially activates AKT, but it is not
yet clear whether it is necessary prior to phosphorylation of S473
(55). Secondly, it has reported
that elevated AKT activation in human cancers can result from
enhanced activation phosphorylation of AKT on the Ser473 site
(56). Therefore, in the present
study, we chose phospho-AKT (Ser473) as the PI3K substrate for
research. AKT is an important mediator of VEGF. PI3K/AKT signaling
mediates the VEGF-induced angiogenic stimulation of endothelial
cells (57). As important
components of the PI3K/AKT signaling pathway, AKT isoforms play key
roles in several cellular processes including anti-apoptosis,
proliferation, growth, DNA repair, transport, metabolism,
angiogenesis, and stem cell self-renewal (58,59).
In this article, we reported the novel VEGF/PI3K/AKT signaling axis
that serves an important role in GSCs. The experimental results of
this study indicated that JQ1 inhibits VEGF and phospho-VEGFR2
expression, thereby reducing the expression of PI3K downstream of
VEGF and inhibiting the activity of phosphor-AKT (Ser473 or
Thr308). In addition, although the mechanism is unclear, MMP
protein expression was significantly decreased by JQ1, preventing
VEGF secreting from the ECM to promote angiogenesis and tumor
growth (27). Additionally,
treatment with JQ1 or siBrd4 caused corresponding changes in
apoptosis- and cell cycle-related genes downstream of AKT. The Rb
protein binds to and regulates members of the E2F family. E2F1 is a
transcriptional activator of E2F family members that positively
regulates the transition from G1 phase to S phase (60-62).
Active cyclin D/CDK complexes phosphorylate the Rb protein, and
phosphorylated Rb is unable to interact with E2F. Therefore, E2F is
activated, permitting it to promote the transcription of genes
necessary for entry into S phase (63). By contrast, in our study, because
AKT phosphorylation at Ser473 was inhibited, the expression of the
downstream target gene cyclin D1 was decreased, and that of CDKIs
(P21 and P27) was increased. Thus, the Rb protein was not
phosphorylated, and E2F1 cannot be released from the Rb/E2F1
complex to facilitate transcription of downstream genes including
c-Myc, leading to cell cycle arrest. In addition, after Brd4
silencing, the expression of apoptosis-related genes downstream of
AKT, such as cleaved caspase 3, BAX, and Bcl-2, was affected.
Inhibition of Brd4 by siRNAs or JQ1 treatment resulted in increased
DNA damage, which further promoted apoptosis in GSCs.
The role of Brd4 in GBM has attracted our attention,
but there were some limitations in our study. In the future,
further experiments are needed to silence or overexpress the genes
involved in the pathway to confirm the results and obtain further
insight. JQ1 also has great potential for promoting the
differentiation of GSCs, making further research valuable. Despite
its limitations, this study identified the epigenetic protein Brd4
as a promising target for the treatment of GBM and proposed the
feasibility of treating GBM using JQ1. In summary, our experiments
identified promising prospects of epigenetic-based targeted
therapeutic approaches for GBM, and these findings will may
facilitate the rapid clinical evaluation of this new strategy.
Supplementary Data
Abbreviations:
|
GBM
|
glioblastoma multiforme
|
|
Brd4
|
bromodomain-containing protein 4
|
|
BET
|
bromodomain and extraterminal
domain
|
|
GSCs
|
glioma stem cells
|
|
VEGF
|
vascular endothelial growth factor
|
|
MMP
|
matrix metalloproteinase protein
|
|
ECM
|
extracellular matrix
|
Acknowledgments
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81773217 and 81472344) and
Jilin Provincial Education Department (grant no. Jijiaokehezi
[2016]455) and Jilin University Bethune Plan B Projects (grant no.
2015220) and Jilin Provincial Research Foundation for the
Development of Science and Technology Projects (grant no.
20190701065GH) and Project funded by China Postdoctoral Science
Foundation (grant no. 2018M631885).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NW, BG, LZ and YL contributed to the conception and
design, acquisition of data, analysis and interpretation of data.
HZ contributed to the conception and revised the manuscript
critically. LX, QW and DW performed molecular analyses and
statistical tests. HL, XC and SZ contributed to analysis and
interpretation of data. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the Animal
Ethics Committee of Jilin University [approval no. 2018;(54)], and by the Institutional Committee
for the Care and Use of Laboratory Animals of the Experimental
Animal Center of Jilin University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Appin CL and Brat DJ: Molecular genetics
of gliomas. Cancer J. 20:66–72. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aldape K, Zadeh G, Mansouri S,
Reifenberger G and von Deimling A: Glioblastoma: Pathology,
molecular mechanisms and markers. Acta Neuropathol. 129:829–848.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Auffinger B, Spencer D, Pytel P, Ahmed AU
and Lesniak MS: The role of glioma stem cells in chemotherapy
resistance and glioblastoma multiforme recurrence. Expert Rev
Neurother. 15:741–752. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Balça-Silva J, Matias D, Carmo AD,
Sarmento-Ribeiro AB, Lopes MC and Moura-Neto V: Cellular and
molecular mechanisms of glioblastoma malignancy: Implications in
resistance and therapeutic strategies. Semin Cancer Biol. Sep
25–2018. View Article : Google Scholar : Epub ahead of
print. PubMed/NCBI
|
|
6
|
Ahmed AU, Auffinger B and Lesniak MS:
Understanding glioma stem cells: Rationale, clinical relevance and
therapeutic strategies. Expert Rev Neurother. 13:545–555. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang
J, Aggarwal A and Rosenfeld MG: Brd4 and JMJD6-associated
anti-pause enhancers in regulation of transcriptional pause
release. Cell. 155:1581–1595. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu T, Kamikawa YF and Donohoe ME: Brd4's
bromodomains mediate histone H3 acetylation and chromatin
remodeling in pluripotent cells through P300 and Brg1. Cell Rep.
25:1756–1771. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Donati B, Lorenzini E and Ciarrocchi A:
BRD4 and cancer: Going beyond transcriptional regulation. Mol
Cancer. 17:1642018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
French CA, Ramirez CL, Kolmakova J,
Hickman TT, Cameron MJ, Thyne ME, Kutok JL, Toretsky JA, Tadavarthy
AK, Kees UR, et al: BRD-NUT oncoproteins: A family of closely
related nuclear proteins that block epithelial differentiation and
maintain the growth of carcinoma cells. Oncogene. 27:2237–2242.
2008. View Article : Google Scholar
|
|
11
|
Zuber J, Shi J, Wang E, Rappaport AR,
Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al:
RNAi screen identifies Brd4 as a therapeutic target in acute
myeloid leukaemia. Nature. 478:524–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang
Q, Lin Y, Li J, Kang T, Tao M, et al: Disrupting the interaction of
BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like
breast cancer. Cancer Cell. 25:210–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Asangani IA, Dommeti VL, Wang X, Malik R,
Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S,
Engelke C, et al: Therapeutic targeting of BET bromodomain proteins
in castration-resistant prostate cancer. Nature. 510:278–282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou J, Li W, Guo J, Li G, Chen F and Zhou
J: Downregulation of miR-329 promotes cell invasion by regulating
BRD4 and predicts poor prognosis in hepatocellular carcinoma.
Tumour Biol. 37:3561–3569. 2016. View Article : Google Scholar
|
|
15
|
Leal AS, Williams CR, Royce DB, Pioli PA,
Sporn MB and Liby KT: Bromodomain inhibitors, JQ1 and I-BET 762, as
potential therapies for pancreatic cancer. Cancer Lett. 394:76–87.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB,
Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et
al: BET bromodomain inhibition as a therapeutic strategy to target
c-Myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bandopadhayay P, Bergthold G, Nguyen B,
Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R,
Masoud S, et al: BET bromodomain inhibition of MYC-amplified
medulloblastoma. Clin Cancer Res. 20:912–925. 2014. View Article : Google Scholar
|
|
18
|
Das A, Chai JC, Yang CS, Lee YS, Das ND,
Jung KH and Chai YG: Dual transcriptome sequencing reveals
resistance of TLR4 ligand-activated bone marrow-derived macrophages
to inflammation mediated by the BET inhibitor JQ1. Sci Rep.
5:169322015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shao Q, Kannan A, Lin Z, Stack BC Jr, Suen
JY and Gao L: BET protein inhibitor JQ1 attenuates Myc-amplified
MCC tumor growth in vivo. Cancer Res. 74:7090–7102. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng H, Ying H, Yan H, Kimmelman AC,
Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al: p53
and Pten control neural and glioma stem/progenitor cell renewal and
differentiation. Nature. 455:1129–1133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang R, Vider J, Kovar JL, Olive DM,
Mellinghoff IK, Mayer-Kuckuk P, Kircher MF and Blasberg RG:
Integrin avβ3-targeted IRDye 800CW near-infrared imaging of
glioblastoma. Clin Cancer Res. 18:5731–5740. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce
LA, Thompson RC, Muller S, Knapp S and Wang J: Inhibition of BET
bromodomain targets genetically diverse glioblastoma. Clin Cancer
Res. 19:1748–1759. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11:R1062010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanehisa M, Goto S, Sato Y, Kawashima M,
Furumichi M and Tanabe M: Data, information, knowledge and
principle: Back to metabolism in KEGG. Nucleic Acids Res.
42(Database Issue): D199–D205. 2014. View Article : Google Scholar :
|
|
26
|
Ola R, Dubrac A, Han J, Zhang F, Fang JS,
Larrivée B, Lee M, Urarte AA, Kraehling JR, Genet G, et al: PI3
kinase inhibition improves vascular malformations in mouse models
of hereditary haemorrhagic telangiectasia. Nat Commun. 7:136502016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee SH, Jeong D, Han YS and Baek MJ:
Pivotal role of vascular endothelial growth factor pathway in tumor
angiogenesis. Ann Surg Treat Res. 89:1–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aversa C, Leone F, Zucchini G, Serini G,
Geuna E, Milani A, Valdembri D, Martinello R and Montemurro F:
Linifanib: Current status and future potential in cancer therapy.
Expert Rev Anticancer Ther. 15:677–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pastori C, Daniel M, Penas C, Volmar CH,
Johnstone AL, Brothers SP, Graham RM, Allen B, Sarkaria JN, Komotar
RJ, et al: BET bromodomain proteins are required for glioblastoma
cell proliferation. Epigenetics. 9:611–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ishida CT, Zhang Y, Bianchetti E, Shu C,
Nguyen TTT, Kleiner G, Sanchez-Quintero MJ, Quinzii CM, Westhoff
MA, Karpel-Massler G, et al: Metabolic reprogramming by Dual
AKT/ERK inhibition through imipridones elicits unique
vulnerabilities in glioblastoma. Clin Cancer Res. 24:5392–5406.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Segatto M, Fittipaldi R, Pin F, Sartori R,
Dae Ko K, Zare H, Fenizia C, Zanchettin G, Pierobon ES, Hatakeyama
S, et al: Epigenetic targeting of bromodomain protein BRD4
counteracts cancer cachexia and prolongs survival. Nat Commun.
8:17072017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dunn GP, Rinne ML, Wykosky J, Genovese G,
Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et
al: Emerging insights into the molecular and cellular basis of
glio-blastoma. Genes Dev. 26:756–784. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kondo Y, Katsushima K, Ohka F, Natsume A
and Shinjo K: Epigenetic dysregulation in glioma. Cancer Sci.
105:363–369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
French CA, Miyoshi I, Kubonishi I, Grier
HE, Perez-Atayde AR and Fletcher JA: BRD4-NUT fusion oncogene: A
novel mechanism in aggressive carcinoma. Cancer Res. 63:304–307.
2003.PubMed/NCBI
|
|
35
|
Roe JS and Vakoc CR: The essential
transcriptional function of BRD4 in acute myeloid leukemia. Cold
Spring Harb Symp Quant Biol. 81:61–66. 2016. View Article : Google Scholar
|
|
36
|
Andrieu G, Tran AH, Strissel KJ and Denis
GV: BRD4 regulates breast cancer dissemination through
Jagged1/Notch1 signaling. Cancer Res. 76:6555–6557. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sahai V, Kumar K, Knab LM, Chow CR, Raza
SS, Bentrem DJ, Ebine K and Munshi HG: BET bromodomain inhibitors
block growth of pancreatic cancer cells in three-dimensional
collagen. Mol Cancer Ther. 13:1907–1917. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bayin NS, Modrek AS and Placantonakis DG:
Glioblastoma stem cells: Molecular characteristics and therapeutic
implications. World J Stem Cells. 6:230–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aum DJ, Kim DH, Beaumont TL, Leuthardt EC,
Dunn GP and Kim AH: Molecular and cellular heterogeneity: The
hallmark of glioblastoma. Neurosurg Focus. 37:E112014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ropolo M, Daga A, Griffero F, Foresta M,
Casartelli G, Zunino A, Poggi A, Cappelli E, Zona G, Spaziante R,
et al: Comparative analysis of DNA repair in stem and nonstem
glioma cell cultures. Mol Cancer Res. 7:383–392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thomas TM and Yu JS: Metabolic regulation
of glioma stem-like cells in the tumor micro-environment. Cancer
Lett. 408:174–181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jain RK, di Tomaso E, Duda DG, Loeffler
JS, Sorensen AG and Batchelor TT: Angiogenesis in brain tumours.
Nat Rev Neurosci. 8:610–622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shi J and Vakoc CR: The mechanisms behind
the therapeutic activity of BET bromodomain inhibition. Mol Cell.
54:728–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dawson MA, Kouzarides T and Huntly BJ:
Targeting epigenetic readers in cancer. N Engl J Med. 367:647–657.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Filippakopoulos P, Qi J, Picaud S, Shen Y,
Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et
al: Selective inhibition of BET bromodomains. Nature.
468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee S, Rellinger EJ, Kim KW, Craig BT,
Romain CV, Qiao J and Chung DH: Bromodomain and extraterminal
inhibition blocks tumor progression and promotes differentiation in
neuroblastoma. Surgery. 158:819–826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Engelke CG and Chinnaiyan AM: aBETting
therapeutic resistance by Wnt signaling. Cell Res. 25:1187–1188.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tang Y, Gholamin S, Schubert S, Willardson
MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N,
et al: Epigenetic targeting of Hedgehog pathway transcriptional
output through BET bromodomain inhibition. Nat Med. 20:732–740.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chautard E, Ouédraogo ZG, Biau J and
Verrelle P: Role of Akt in human malignant glioma: From oncogenesis
to tumor aggressiveness. J Neurooncol. 117:205–215. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S,
Malaponte G, et al: Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade
inhibitors: How mutations can result in therapy resistance and how
to overcome resistance. Oncotarget. 3:1068–1111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao HF, Wang J, Shao W, Wu CP, Chen ZP,
To ST and Li WP: Recent advances in the use of PI3K inhibitors for
glioblastoma multiforme: Current preclinical and clinical
development. Mol Cancer. 16:1002017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lv D, Jia F, Hou Y, Sang Y, Alvarez AA,
Zhang W, Gao WQ, Hu B, Cheng SY, Ge J, et al: Histone
acetyltransferase KAT6A upregulates PI3K/AKT signaling through
TRIM24 binding. Cancer Res. 77:6190–6201. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Freudlsperger C, Horn D, Weißfuß S,
Weichert W, Weber KJ, Saure D, Sharma S, Dyckhoff G, Grabe N,
Plinkert P, et al: Phosphorylation of AKT(Ser473) serves as an
independent prognostic marker for radiosensitivity in advanced head
and neck squamous cell carcinoma. Int J Cancer. 136:2775–2785.
2015. View Article : Google Scholar
|
|
55
|
Yung HW, Charnock-Jones DS and Burton GJ:
Regulation of AKT phosphorylation at Ser473 and Thr308 by
endoplasmic reticulum stress modulates substrate specificity in a
severity dependent manner. PLoS One. 6:e178942011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liao Y and Hung MC: Physiological
regulation of Akt activity and stability. Am J Transl Res. 2:19–42.
2010.PubMed/NCBI
|
|
57
|
Li F, Sawada J and Komatsu M: R-Ras-Akt
axis induces endothelial lumenogenesis and regulates the patency of
regenerating vasculature. Nat Commun. 8:17202017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu Q, Turner KM, Alfred Yung WK, Chen K
and Zhang W: Role of AKT signaling in DNA repair and clinical
response to cancer therapy. Neuro Oncol. 16:1313–1323. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Khan KH, Yap TA, Yan L and Cunningham D:
Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J
Cancer. 32:253–265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sun H, Chang Y, Schweers B, Dyer MA, Zhang
X, Hayward SW and Goodrich DW: An E2F binding-deficient Rb1 protein
partially rescues developmental defects associated with Rb1
nullizygosity. Mol Cell Biol. 26:1527–1537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jiang H, Martin V, Gomez-Manzano C,
Johnson DG, Alonso M, White E, Xu J, McDonnell TJ, Shinojima N and
Fueyo J: The RB-E2F1 pathway regulates autophagy. Cancer Res.
70:7882–7893. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chang MM, Lai MS, Hong SY, Pan BS, Huang
H, Yang SH, Wu CC, Sun HS, Chuang JI, Wang CY and Huang BM:
FGF9/FGFR2 increase cell proliferation by activating ERK1/2,
Rb/E2F1, and cell cycle pathways in mouse Leydig tumor cells.
Cancer Sci. 109:3503–3518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sammons SL, Topping DL and Blackwell KL:
HR+, HER2-advanced breast cancer and CDK4/6 inhibitors: Mode of
action, clinical activity, and safety profiles. Curr Cancer Drug
Targets. 17:637–649. 2017. View Article : Google Scholar :
|