Introduction
The outgrowth of malignant tumour cells along
vascular routes facilitates their dissemination to distant organs,
leading to further colonization (1-3). An
early step of this multistep process is the destabilisation of the
endothelial barrier as a prerequisite for the cancer cell invasion
into and leakage out of lymphatic or blood vessels (intra- and
extravasation, respectively) (4).
The disintegration of the endothelium is induced by metabolites or
polypeptides (5,6), which are secreted by a variety of
tumour cell types. The present study focussed on a mechanism
triggered by 'S' stereo-isomer
12S-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid [12(S)-HETE], which
serves an important role in making the lymphatic wall accessible
(7). 12(S)-HETE causes loss of
vascular resilience due to the retraction of endothelial cells
(ECs) (8), resulting in the
formation of gaps in the endothelial layer, known as 'circular
chemorepellent-induced defects' (CCIDs), through which cancer cells
cross the endothelial wall and subsequently spread to the lymph
nodes (7). Under physiological
conditions, neutrophils traverse vessel barriers by similar or
lipoxin-induced mechanisms and reach sites of inflamed tissue
(9). Cancer cells seem to
re-activate processes, including 12(S)-HETE secretion, which enable
their movement through the vasculature and foreign tissues and are
otherwise used by neutrophils during the immune response (9).
12(S)-HETE triggers signalling through high- and
low-affinity receptors, including 12-HETER and leukotriene B4
receptor 2 (BLT2), respectively. Subsequently, phospholipase C,
Ca2+-release and CAM-kinase, as well as RHO, RAK and
myosin light chain kinase (MYLK), are actively phosphorylated and
induce the target of MYLK, myosin light chain 2 (MLC2). This causes
lymph EC (LEC) retraction and therefore the breakdown of the
endothelial barrier and CCID formation (10,11).
Furthermore, focal adhesion kinase (FAK), which is a non-receptor
tyrosine kinase that is required for cell-matrix contact, migration
and cell signalling, was demonstrated to contribute to this
phenomenon (12). However, how FAK
is associated in the signal transduction network triggering LEC
retraction, and whether FAK is regulated by 12(S)-HETE is yet to be
determined. There is a cross-talk between FAK and NF-κB (13,14),
and NF-κB has been reported to regulate the endothelial
lineage-determining transcription factor SRY-box transcription
factor 18 (SOX18), which supports endothelial cell differentiation
during embryonic development (15,16),
in human umbilical vein ECs (17)
and LECs (18). A previous study
demonstrated that SOX18, and its transcriptional target the lymph
endothelial transcription factor and marker protein prospero
homeobox 1 (PROX1), contributed to CCID formation in the
lymph-endothelial barrier (18).
The transcription factor NF-κB and its transcriptional target, the
intercellular adhesion molecule 1 (ICAM-1), were also revealed to
be required for CCID formation (6,19,20).
ICAM-1 is an effector necessary for cell-cell adhesion (20) and FAK is not only a signal
transducer but also an effector molecule regulating cell adherence
and migration (12), and both
ICAM-1 and FAK are obligatory for the retraction of LECs (12,20).
The retraction of LECs can be studied in a co-culture assay
consisting of LEC monolayers and tumour spheroids (5,7,12),
such as 12(S)-HETE-secreting HCT116 colon cancer cell spheroids,
which are placed on top of LECs. This assay monitors the formation
of CCIDs in the LEC monolayer underneath tumour emboli (7,19).
The elucidation of the mechanism that unlocks the
lymph-endothelial barrier, which is partly mediated by the
endothelial-specific transcription factors SOX18 and PROX1, may
allow the targeting and inhibition of an early metastatic step with
high accuracy and few side effects. Therefore, the present study
investigated whether v-rel avian reticuloendotheliosis viral
oncogene homolog A (RELA; the major component of the NF-κB
heterodimer), SOX18 and FAK are interconnected in a common signal
transduction pathway upon stimulation of LECs with 12(S)-HETE.
Materials and methods
Antibodies and reagents
Rabbit polyclonal antibodies against focal adhesion
kinase (FAK; 1:1,000; cat. no. 3285), phospho-Tyr397-FAK (pFAK;
1:1,000; cat. no. 3283) and mouse monoclonal anti-v-Rel avian
reticuloendotheliosis viral oncogene homolog A antibody (RELA/p65
clone L8F6; 1:1,000; cat. no. 6956) were purchased from Cell
Signalling Technology Inc. and mouse monoclonal anti-β-actin
antibody (clone AC-15; 1:5,000; cat. no. A3854) was purchased from
Sigma-Aldrich; Merck KGaA. Rabbit polyclonal anti-SRY-related
HMG-box 18 (SOX18; 1:600, cat. no. TA324592) was purchased OriGene
Technologies, Inc. and rabbit monoclonal anti-prospero homeobox 1
antibody (PROX1 clone EPR19273; 1:500; cat. no. Ab119359) and mouse
monoclonal anti-intercellular adhesion molecule 1 antibody (ICAM-1;
1:1,000; clone MEM111; cat. no. Ab2213) were purchased from Abcam.
HRP-conjugated swine anti-rabbit antibody (1:5,000; cat. no. P0217)
and HRP-conjugated rabbit anti-mouse (1:10,000; cat. no. P0260)
were purchased from Dako; Agilent Technologies, Inc.
siRNAs were used for transfection of LECs, which
were subsequently analyzed using western blot analysis, qPCR and in
CCID assay. siRNAs against RELA (siRELA; cat. no. s11914), PTK-2
(siFAK; cat. no. s11485), ICAM-1 (siICAM-1; cat. no. s7087) and
Silencer Select Negative Control SilencerR No. 1 si-RNA (n.t.Co;
cat. no. 4390843) were purchased from Ambion; Thermo Fisher
Scientific, Inc, and SOX18 (siSOX18; cat. no. L-019035-00-0005) and
PROX1 (siPROX1; cat. no. L-016913-005) were purchased from GE
Healthcare Dharmacon, Inc.
qPCR primers were used to quantify mRNA expression
in LECs upon transfection of siRNAs and treatment with 12(S)-HETE.
Q-PCR primers for SOX18 (cat. no. Hs00746079_ s1), RELA (cat. no.
Hs00153294_m1), ICAM-1 (cat. no. Hs00164932_m1), PROX1 (cat. no.
Hs00896294_m1) and β-actin (cat. no. Hs01060665_g1) were purchased
from TaqMan (Applied Biosystems; Thermo Fisher Scientific,
Inc.).
12(S)-HETE (CAS: 54397-83-0) was purchased from Enzo
Life Sciences (cat. no. BML-H012-0050), Bay11-7082 (cat. no.
196870) from EMD Millipore, parthenolide (cat. no. P0667),
arachidonic acid (cat. no. 10931), proadifen hydrochloride (cat.
no. P1061), guanfacine (cat. no. G1041), vinpocetine (cat. no.
V6382) and curcumin (cat. no. 08511) were purchased from
Sigma-Aldrich; Merck KGaA, and defactinib (cat. no. S7654; VS-6063
and PF-04554878) from Selleck Chemicals.
Cell lines
HCT 116 colon cancer cells (cat. no. 91091005) were
purchased from the European Collection of Authenticated Cell
Cultures (ECACC) by Public Health England and cultured at 37°C in
MEM medium (Gibco; Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% foetal calf serum (FCS; Gibco; Invitrogen;
Thermo Fisher Scientific, Inc.), 1% penicillin/streptomycin (PS)
and 1% non-essential amino acids (Gibco; Invitrogen; Thermo Fisher
Scientific, Inc.). Human micro-vessel endothelial cells were
purchased from Clonetics™ (Lonza Group, Ltd.). According to the
protocols of previous studies and following stable transfection
with telomerase, lymph endothelial cells (LECs) were isolated from
this immortalised mixture of human dermal endothelial cells
(21,22). LECs were grown at 37°C in EGM-2MV
(EBM2-based medium CC3156 and supplement CC4147; Lonza Group Ltd.).
Cells were kept at 37°C in a humidified atmosphere containing 5%
CO2 for subsequent experimentation.
SiRNA transfection
LECs were seeded in 6-well plates and grown at 37°C
to 80% confluence. siRELA, or siICAM-1, siSOX18, siPROX1, siFAK, or
Negative Control SilencerR No. 1 si-RNA (n.t.Co; 1.75 µg)
was mixed with 15 µl Hiperfect transfection reagent (cat.
no. 301705; Qiagen GmbH) in 100 µl serum-free EBM2-base
medium and left at room temperature to allow formation of
transfection complexes for 30 min. Old medium was exchanged for 1.4
ml pre-warmed EBM2-base medium, subsequently transfection-mix was
added dropwise to the cells and incubated at 37°C overnight. On the
next day the medium was exchanged for serum containing EGM2-MV
medium and cells were left to recover for 24 h.
For the CCID assay, LECs were seeded in 24-well
plates and grown at 37°C to 80% confluence. Subsequently, the
EGM2-MV medium was changed to serum free EBM2-base medium and the
transfection mix (0.75 µg siRNA; 6 µl Hiperfect
transfection reagent; 100 µl serum free medium) was added to
the cells and experiments were processed as aforementioned.
12(S)-HETE stimulation
Transfected LECs, which were allowed to recover
overnight, were starved in serum free EBM2-base medium at 37°C for
3 h. Then, LECs were stimulated at 37°C with 1 µM 12(S)-HETE
for 45 min to analyse gene and protein expression using reverse
transcription-quantitative (RT-q) PCR and western blot analysis,
respectively. Each experiment was performed with three biological
replicates.
Western blot analysis
LECs were seeded in 6-well plates and grown at 37°C
to 80% confluence, then 5, 10 and 15 µM Bay11-7082 was added
and cells were incubated for 4 h at 37°C. LECs were then lysed in
2X SDS lysis buffer containing 0.5 M Tris-HCl (pH 6.8), 20% SDS,
10% glycerol, 0.5 M Ethylenediaminetetraacetic acid, phosphatase
inhibitor cocktail and protease inhibitor cocktail and sonicated in
a pulsed manner using a Branson Sonifier 2000 (Emerson Electric
Co.). After centrifugation (12,000 × g; room temperature; 1 min),
the supernatant was mixed with 6X loading dye [0.6 M DTT, 24% (w/v)
SDS, 36% (v/v) glycine, bromophenol blue, 1.2 M Tris-HCl pH 6.8]
and heated for 5 min at 95°C. Equal amounts of protein (20
µg), as determined by Pierce BCA protein assay (Thermo
Fisher Scientific Inc.), were separated using 10% SDS-PAGE (80 V 10
min; 110 V 2 h; constant voltage) using Mini Protean Tetracell
(Bio-Rad Laboratories, Inc.) following the manufacturers protocol.
Subsequently, proteins were electro-blotted (20 V constant; on ice,
overnight) onto Immobilon FL PVDF membrane (0.45 µm pore
size; EMD Millipore) using transfer buffer containing 20 mM
Tris-base, 150 mM glycine, 20% (v/v) methanol, pH 8.5. Membranes
were stained using Ponceau S (Sigma-Aldrich; Merck KGaA) to control
transfer efficiency and equal loading. Membranes were blocked at
room temperature in 5% dried skim milk in TBS-T (1X Tris buffered
saline/0,1% Tween-20; pH 7.6) for 1 h and incubated with either
anti-pFAK, anti-FAK, anti-RELA, anti-ICAM-1, anti-SOX18, anti-PROX1
and anti-β-actin antibodies (specified in the subparagraph
'Antibodies and reagents'), agitated at 4°C, overnight. Then,
membranes were washed three times in TBS-T and incubated with
HRP-conjugated swine anti-rabbit antibody, or rabbit anti-mouse
antibody at room temperature for 1 h. Chemiluminescence was
measured using a Lumi-Imager F1 Workstation (Roche Diagnostics) and
densitometry was calculated using ImageJ software version 1.51
(National Institutes of Health) and Excel 2013 (Microsoft
Corporation). For repetitive analyses, membranes were stripped in
between antibody incubations with Restore Western Blot Stripping
Buffer (Thermo Fisher Scientific, Inc.) at 37°C for 5-10 min
followed by 3 washes in TBS-T (Sigma-Aldrich; Merck KGaA). Each
western blot analysis was performed using three biological
replicates.
RT-qPCR
RNA of transfected and stimulated LECs was extracted
using RNeasy-Mini-kit and Qia-shredder (both Qiagen GmbH). RNA
concentration was measured using a Nano-Drop fluoro-spectrometer
(Thermo Fisher Scientific, Inc.) and 2.5 µg RNA were
reverse-transcribed using RNA-to-cDNA-EcoDry™ Premix Random
Hexamers (Takara Bio Europe SAS) at 42°C for 60 min and the
reaction was stopped at 70°C for 10 min. Gene expression was
analysed using TaqMan Gene Expression Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.), TaqMan primers for
RELA, ICAM-1, SOX18, PROX1 and β-actin was used as a reference
gene, and the CFX 96 Real Time PCR Detection system (Bio-Rad
Laboratories, Inc.) and was quantified using the 2ΔΔCq
method (23). The thermocycling
conditions were as follows: First cycle 50°C for 2 min, then 1
cycle 95°C for 10 min, then 40 cycles at 95°C for 15 sec, 60°C for
30 sec, 72°C for 30 sec, and a last cycle at 72°C for 10 min.
Analysis was subsequently performed using three biological
replicates.
12(S)-HETE assay
HCT116 cells were seeded in 6-well plates and grown
at 37°C in complete MEM medium to 90% confluence and starved
overnight. Cells were then treated with 10 µM arachidonic
acid simultaneously with 40 µM proadifen, 40 µM
guanfacine, 40 µM vinpocetine or solvent (DMSO) in serum
free medium at 37°C for 4 h. The supernatant (1.5 ml) was
collected, butylated hydroxytoulene (0.001% final concentration;
cat. no. B1378; Sigma-Aldrich; Merck KGaA) was added to stabilise
the secreted eicosanoid, and centrifuged at 280 × g at 4°C for 5
min. The 12(S)-HETE concentration was determined as described
previously (24). Samples were
either flash frozen in liquid nitrogen and stored at -80°C until
further analysis or immediately passed through extraction
cartridges (Oasis™ HLB 1 cc; Warers Corporation; equilibrated with
2×1 ml methanol, 2×1 ml ddH2O immediately before use)
followed by washing of cartridges with 3×1 ml distilled
H2O. Bound 12(S)-HETE was eluted with 500 µl
methanol. After the evaporation of methanol with an Eppendorf
Speedvac Concentrator Plus (Eppendorf) at 30°C for 2 h, the samples
were reconstituted with 100 µl assay buffer of the
12(S)-HETE enzyme immunoassay kit (EIA; cat. no. ADI-900-050; Enzo
Life Sciences, Inc.) and subjected to 12(S)-HETE analysis according
to the manufacturers protocol (24). Absorbance at 405 nm was measured
with a Wallac 1420 Victor 2 multi-label plate reader (PerkinElmer,
Inc.). The concentration of 12(S)-HETE in the cellular supernatant
was normalised to cell number. The analysis was performed using
three biological replicates.
Spheroid formation
Per spheroid, 6000 HCT116 cells were added to MEM
medium containing 10% FCS and 0.3% methylcellulose (final
concentration) and 150 µl containing 6,000 cells were seeded
into each U-bottom shaped well of 96-well plates (Cellstar; cat.
no. 650185, Greiner Bio One International GmbH). After
centrifugation at 300 × g, room temperature for 15 min, spheroids
were grown at 37°C and 5% CO2 for two days.
CCID assay
LECs were seeded (20,000 cells/well) in 24-well
plates (Costar; cat. no. 3524, Sigma-Aldrich; Merck KGaA) and grown
at 37°C to ~100% confluence. To measure the size of the cell free
areas (circular chemorepellent-induced defects; CCIDs), which were
formed in the endothelial mono-layer directly underneath the tumour
spheroids, LECs were stained with CellTracker™ Green CMFDA (Thermo
Fisher Scientific, Inc.) at 37°C for 1 h. For the treatment with 2,
3 and 5 µM curcumin, 5 and 10 µM parthenolide, 2.5
and 5 µM Bay11-7082 and 2.5, 5, 10 and 15 µM
defactinib, HCT116 spheroids and LEC monolayers were washed with
PBS and pre-incubated with the different concentrations of these
compounds in EMB2-base medium at 37°C for 20 min. The EBM2-base
medium including the spheroids and inhibitors was transferred to
the LEC monolayers and co-incubated at 37°C for 4 h. CCID areas in
the LEC monolayers were imaged using an Axiovert fluorescence
microscope with a magnification of ×200 (Zeiss GmbH) using at least
15 fields of view; the size of the areas was calculated with Zen
Little 2012 software (Zeiss GmbH). For each condition, the CCIDs in
the LEC monolayer underneath at least 15 HCT116-spheroids, which
were between ~500-600 µM in diameter and surrounded by a
homogenously confluent LEC monolayer, were analysed. The CCID assay
was performed using three biological replicates of which at least 5
fields of view were analyzed.
Statistical analysis
Excel 2013 (Microsoft Corporation) software and
GraphPad Prism 6 (GraphPad Software, Inc.) were used for one-way
ANOVA and unpaired Student's t-test statistics. The data were
expressed as mean ± SEM. P<0.05 was considered to indicate a
statistically significant difference.
Results
12(S)-HETE upregulates RELA and SOX18,
which regulate each other in a positive feedback loop
When LECs were stimulated with 12(S)-HETE, the
expression of RELA, which is a constituent of the canonical NF-κB
transcription factor heterodimer, was induced (Fig. 1A). 12(S)-HETE also induced the
expression of SOX18 mRNA and protein in LECs (18) (Figs.
S1 and S2). Conversely, the specific inhibition of RELA by
siRNA (siRELA; the suppression of RELA at the mRNA and protein
levels by siRELA, as opposed to non-target RNA, is shown in
Fig. S3 and Fig. 1A, respectively) reduced the
expression of SOX18 mRNA (Fig.
1B), as well as that of PROX1, which is a transcriptional
target of SOX18 (Fig. 1C). This
demonstrated that the constitutive expression of SOX18 and PROX1
were positively regulated by RELA in LECs. It has recently been
demonstrated that RELA controls the 12(S)-HETE-stimulated
upregulation of SOX18 and PROX1 (18). Since ICAM-1 is an accepted target
of the NF-κB transcription factor in ECs (20,25),
and specifically of RELA (18),
the expression of ICAM-1 mRNA was examined, to control whether the
activity of RELA was increased upon 12(S)-HETE-treatment of LECs
(18) (Fig. S4). The inhibition of SOX18 by
siSOX18 abrogated the 12(S)-HETE-stimulated mRNA upregulation of
RELA (Fig. 1D) and ICAM-1
(Fig. 1E). To control whether the
SOX18 transcription factor was activated upon 12(S)-HETE treatment
of LECs the mRNA expression of its target, PROX1 expression was
analysed (Fig. S5; the
suppression of SOX18 mRNA- and protein levels by siSOX18, as
opposed to non-target RNA, is presented in Figs. S6 and S7, respectively). The data
indicated that RELA and SOX18 were positively feeding back to each
other in 12(S)-HETE-stimulated LECs (Fig. 1F).
 | Figure 112(S)-HETE stimulation was shown to
upregulate the expression of RELA and ICAM-1. LECs were stimulated
with 1 µM 12(S)-HETE for 45 min when (A) protein was
isolated for western blot analysis. Ponceau S staining (data not
shown) was used, and β-actin expression served as the loading
control. Relative protein expression was quantified using
densitometry. RELA was indicated to control the expression of SOX18
and PROX1. LECs were transiently transfected with siRNA targeting
RELA (siRELA) or non-targeting RNA (n.t.Co) and the mRNA expression
of (B) SOX18 and (C) PROX1 was determined using RT-qPCR. SOX18 was
shown to control the expression of RELA and ICAM-1. LECs were
transiently transfected with siRNA targeting SOX18 (siSOX18) or
non-targeting RNA (n.t.Co) and the mRNA expression of (D) RELA and
(E) ICAM-1 was determined using RT-qPCR. All experiments were
performed in triplicate. Error bars present the ± standard error
mean of at least 3 measurements. Statistical significance was
determined using ANOVA/Tukey's post hoc test (A, D and E) and
t-test (B and C). *P<0.05 vs. n.t.Co.
#P<0.05 vs. 12(S)-HETE stimulation. (F) Schematic
presentation of the regulation of SOX18, PROX1, RELA and ICAM-1
upon stimulation of LECs with 12(S)-HETE. 12(S)-HETE, 'S'
stereoisomer 12S-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid; RELA,
v-rel avian reticuloendotheliosis viral oncogene homolog A; LECs,
lymph endothelial cells; SOX18, prospero homeobox 1; PROX1,
prospero homeobox 1; ICAM-1, intercellular adhesion molecule 1;
RT-q, reverse transcription-quantitative; si, small interfering;
n.t.Co, non-targeting RNA. |
RELA and SOX18 regulate constitutive and
12(S)-HETE-induced FAK phosphorylation
Previous studies have reported that matrix
metalloproteinase-1 activated the protein activated receptor 1 and
Ca2+-release in LECs, inducing MLC2 and FAK, all of
which are prerequisites for their retraction and CCID formation
(6,12). In contrast, 12(S)-HETE has been
indicated to activate the receptors 12-HETER, BLT2 and
Ca2+-release (10,11),
as well as the transcription factors NF-κB and SOX18 in LECs
(18). NF-κB communicates with FAK
in macrophages (26) and,
conversely, NF-κB itself is controlled by FAK in endothelial and
other types of cells (13,14). Therefore, the current study
investigated whether 12(S)-HETE activated FAK in LECs and studied
the potential roles of RELA and SOX18 in such a signal transduction
pathway. The results revealed that 12(S)-HETE induced the
phosphorylation of FAK at tyrosine 397 (Fig. 2A). The transfection of siRELA
caused an increase in FAK phosphorylation under constitutive cell
culture conditions (Fig. 2A),
indicating that RELA suppressed constitutive FAK activity in LECs
(Fig. S8). Additionally, 15
µM Bay11-7082 was indicated to induce hyperphosphorylation
of FAK and an increase in FAK protein expression, whereas lower
Bay11-7082 concentrations (5 and 10 µM) caused a gradual
decrease in FAK phosphorylation (Fig.
2B). Bay11-7082 inhibited RELA and the degradation of IκBα, and
therefore inhibited the entire panel of NF-κB transcription factors
(and possibly also other cellular activities; Fig. 2B). This may have been the reason
for the increase in FAK protein expression and for the
dose-independent phosphorylation of FAK (Fig. 2B). When RELA was inhibited by
siRELA, simultaneous stimulation with 12(S)-HETE did not
significantly change the phosphorylation level of FAK, although an
additive over-phosphorylation of FAK was hypothetically expected
(Fig. 2A). Therefore, it remained
unresolved which condition (siRELA-transfection or
12(S)-HETE-stimulation) caused the increase of FAK phosphorylation,
as the interpretation of the observed effect was not able to be
performed.
 | Figure 2RELA and SOX18 signalling is
associated with FAK. (A) LECs were transiently transfected with
non-targeting RNA (n.t.Co) or siRNA targeting RELA (siRELA)
followed by stimulation with 1 µM 12(S)-HETE for 45 min. (B)
LECs were treated with the solvent (DMSO or Co) or with 5-15
µM Bay11-7082 for 4 h. (C) LECs were transiently transfected
with non-targeting RNA (n.t.Co) or siRNA targeting SOX18 (siSOX18)
followed by stimulation with 1 µM 12(S)-HETE for 45 min.
(A-C) Next, the expression of FAK and its phosphorylation at
tyrosine 397 was analysed using western blotting. Ponceau S
staining (data not shown) was used, and β-actin expression served
as the loading control. Relative protein expression was quantified
using densitometry. pFAK expression was normalised to respective
FAK protein levels. All experiments were performed in triplicate.
Error bars present the ± standard error mean of at least 3
measurements. Statistical significance was determined using
ANOVA/Tukey's post hoc test (A and C) and ANOVA/Dunnett's post hoc
test (B). *P<0.05 vs. n.t.Co. #P<0.05
vs. 12(S)-HETE stimulation. (D) Schematic presentation of SOX18
positively regulating 12(S)-HETE-stimulated phosphorylation of FAK
in LECs. RELA, v-rel avian reticuloendotheliosis viral oncogene
homolog A; 12(S)-HETE, 'S' stereoisomer
12S-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid; SOX18, prospero
homeobox 1; FAK, focal adhesion kinase; LECs, lymph endothelial
cells; si, small interfering; p, phosphorylated; n.t.Co,
non-targeting RNA. |
Similar to siRELA, siSOX18 also induced the
phosphorylation of FAK under constitutive conditions (Fig. 2C), demonstrating an inhibitory
effect of SOX18 on FAK activity (Fig.
S8). However, in contrast to siRELA, siSOX18 prevented the
12(S)-HETE-triggered phosphorylation of FAK (Fig. 2C). Hence, the RELA/SOX18 circuit
suppressed the phosphorylation of FAK under constitutive
conditions, whereas the suppressive function of SOX18 on FAK became
an activating one when LECs were stimulated with 12(S)-HETE
(Fig. 2D).
FAK feeds back to RELA and SOX18
When FAK was inhibited by siFAK transfection (FAK
protein suppression is presented in Fig. S9), the 12(S)-HETE-triggered
upregulation of RELA and ICAM-1 mRNAs was prevented (Fig. 3A and B, respectively). Therefore,
it can be suggested that FAK positively regulated RELA when LECs
were stimulated with 12(S)-HETE (Fig.
3C).
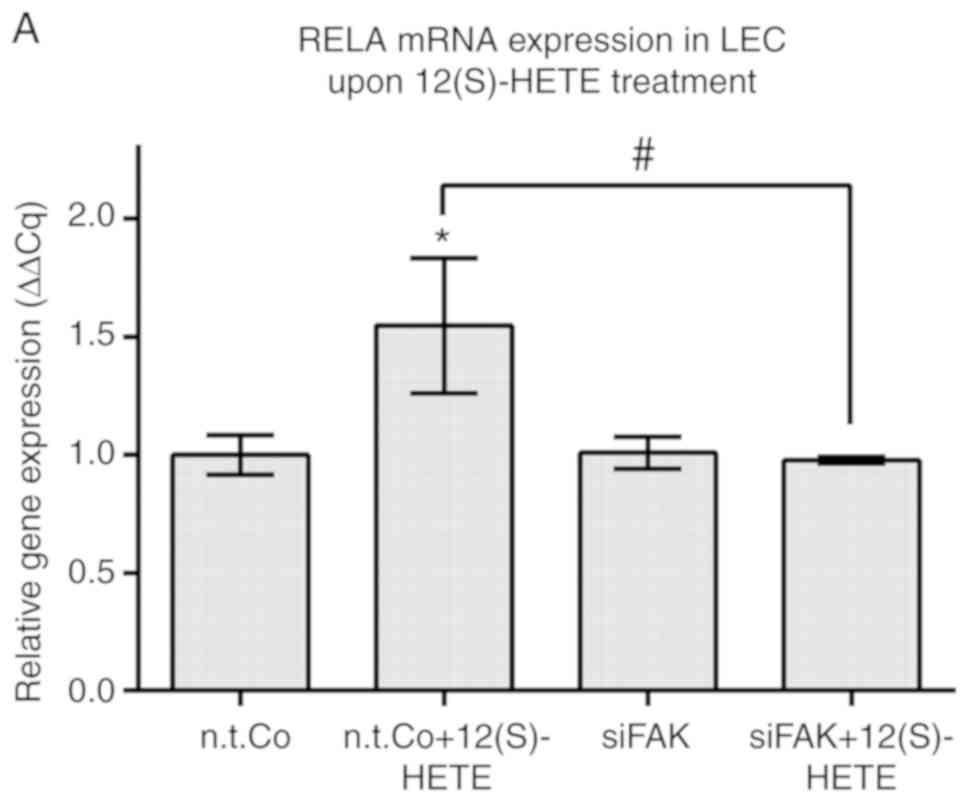 | Figure 3FAK was shown to regulate RELA,
ICAM-1 and SOX18 in 12(S)-HETE stimulated cells. LECs were
transiently transfected with siRNA targeting FAK (siFAK) or with
non-targeting RNA (n.t.Co), stimulated with 1 µM 12(S)-HETE
for 45 min and then the mRNA expression of (A) RELA, (B) ICAM-1,
(D) SOX18 and (E) PROX1 was analysed by RT-qPCR. All experiments
were performed in triplicate. Error bars present the ± standard
error mean of at least 3 measurements. Statistical significance was
determined by ANOVA/Tukey's post hoc test. *P<0.05
vs. n.t.Co. P<0.05 vs. 12(S)-HETE stimulation. (C and F)
Schematic presentations of (C) FAK positively regulating RELA when
LECs were stimulated with 12(S)-HETE, and (F) 12(S)-HETE inducing
the RELA/SOX18 circuit, which upregulated PROX1 and activated FAK.
FAK positively signalled back to RELA, ICAM-1 and SOX18. RELA,
v-rel avian reticuloendotheliosis viral oncogene homolog A; ICAM-1,
intercellular adhesion molecule 1; SOX18, prospero homeobox 1;
12(S)-HETE, 'S' stereoisomer
12S-hydroxy-5Z,8Z,10E,14Z-eicosatetraenoic acid; LECs, lymph
endothelial cells; FAK, focal adhesion kinase; PROX1, prospero
homeobox 1; si, small interfering; n.t.Co, non-targeting RNA. |
The transfection of siFAK into LECs reduced the
constitutive expression of SOX18 mRNA (unlike RELA mRNA) and
abolished the 12(S)-HETE-triggered upregulation of SOX18 mRNA
(Fig. 3D), such as that of RELA
mRNA (Fig. 3A).
Therefore, FAK positively controlled SOX18
expression in 12(S)-HETE-treated and untreated LECs. siFAK
inhibited the target of SOX18, PROX1 under constitutive conditions,
whereas upon stimulation with 12(S)-HETE, PROX1 was independent of
FAK (Fig. 3E). Although SOX18 mRNA
was downregulated by siFAK, the SOX18 protein may have been
activated by 12(S)-HETE-treatment, thereby inducing the
transcription of PROX1. This could explain why PROX1 mRNA (unlike
ICAM-1 mRNA) escaped the siFAK-induced down-regulation. In
conclusion, when LECs were stimulated with 12(S)-HETE, the
expression of RELA/SOX18 was induced and positively regulated.
Furthermore, the RELA/SOX18 circuit activated FAK, which itself was
positively feeding back to SOX18/RELA (Fig. 3F).
Inhibition of lymph endothelial barrier
breaching by molecular silencing and pharmacological drugs
The destabilization of the lymph endothelial barrier
due to LEC-retraction is a prerequisite for cancer intra- and
extravasation (7), and is a
hallmark for the formation of the pre-metastatic niche (27). A trigger of LEC-retraction is
12(S)-HETE, which is secreted by a number of cancer types (7,32).
In HCT116 cells, 12(S)-HETE is metabolised by cytochrome P450 1A1
(CYP1A1) from arachidonic acid (28,29)
as evidenced by the compromised 12(S)-HETE synthesis through the
CYP1A1 inhibitors proadifen, guanfacine and vinpocetine (29-32;
Fig. S10). RELA, as the major
monomer of the heterodimeric transcription factor NF-κB, regulates
the expression of a large number of genes; SOX18 is also known to
control the expression of several genes. In order to monitor the
prointravasative effect of these transcription factors in the CCID
assay more specifically, the current study did not knock down RELA
and SOX18. Instead, their targets, ICAM-1 and PROX1, were assessed,
respectively. These targets were silenced using siRNAs, as they
were both reported to attenuate CCID formation, similar to RELA and
SOX18 (6,20,18).
The specific inhibition of PROX1 or FAK reduced CCID formation by
~25% and the inhibition of ICAM-1 by ~12%. When PROX1 and ICAM-1,
or FAK and ICAM-1 were simultaneously inhibited, CCIDs were also
reduced, if only by ~25%, similar to the inhibition of PROX1 or FAK
alone (Fig. 4A). This suggested
that PROX1 and ICAM-1, as well as FAK and ICAM-1, resided in common
pathways involving RELA. The suppression of the protein levels of
ICAM-1 by siICAM-1 and of PROX1 by siPROX1, as opposed to
non-target RNA, is presented in Figs.
S11 and S12. The suppression of the respective ICAM-1
and PROX1 mRNA levels has been demonstrated in previous
studies (6,18). The simultaneous inhibition of PROX1
and FAK demonstrated an additive effect and suppressed CCID
formation by ~50%, which suggested that PROX1 and FAK were
residents of distinct cross-talking pathways. The cross-talking
link may have been SOX18, mediating separate signals to FAK and
PROX1 (Fig. 3F). SOX18 could
therefore have caused endothelial barrier destabilisation via two
distinct mechanisms.
 | Figure 4Molecular inhibition of LEC barrier
breaching as measured using CCID formation. (A) LECs were
transiently transfected with either siRNA alone targeting FAK
(siFAK), ICAM-1 (siICAM-1), PROX1 (siPROX1) and non-targeting RNA
(n.t.Co), or in the following combinations: siFAK and siICAM-1,
siFAK and siPROX1, siICAM-1 and siPROX1. Curcumin, parthenolide,
Bay11-7082 and defactinib were shown to inhibit CCID formation.
(B-D) LECs were pre-treated with the solvent (DMSO or Co) or the
indicated concentrations of (B) curcumin and parthenolide,
naturally occurring NF-κB inhibitors, for 20 min, (C and D)
defactinib, a FAK inhibitor currently at phase II clinical trial,
or (D) the indicated concentrations of Bay 11-7082 alone or in
combination with defactinib for 15 min. Experiments were conducted
in triplicate analysing at least five spheroids per replicate.
Error bars present the ± standard error mean of ≥15 measurements.
Statistical significance was determined by ANOVA/Tukey's post hoc
test (A and D) and ANOVA/Dunnett's post hoc test (B and C).
*P<0.05 vs. n.t.Co or Co. #P<0.05. LEC,
lymph endothelial cell; CCID, circular chemorepellent-induced
defect; FAK, focal adhesion kinase; ICAM-1, intercellular adhesion
molecule 1; PROX1, prospero homeobox 1; si, small interfering; Co,
control treatment using DMSO (solvent); n.t.Co, non-targeting
RNA. |
siRNA-based approaches are not a treatment option
currently. However, natural NF-κB inhibitors are available.
Therefore, the HCT116/LEC model was treated with curcumin and
parthenolide to inhibit NF-κB (Fig.
4B), or with defactinib to inhibit FAK (Fig. 4C), all of which attenuated CCID
formation. As curcumin and parthenolide are likely to inhibit a
broad spectrum of molecules in addition to NF-κB, the HCT116/LEC
model was also treated with the synthetic and considerably more
specific NF-κB inhibitor Bay11-7082 (Fig. 4D). The combination of Bay11-7082
and defactinib attenuated CCID formation additively (Fig. 4D). This suggested that Bay11-7082
may have also inhibited SOX18/PROX1, as a consequence of the
RELA/SOX18 circuit, or that defactinib inhibited additional
molecules beyond FAK.
Discussion
The aim of the present study was to investigate
whether interference with the NF-κB/SOX18/FAK feedback loops may
have affected the resilience of the lymph endothelial barrier by
measuring the spheroid-induced destabilisation of the LEC
monolayer.
FAK is widely associated with the outgrowth,
dissemination and colonisation of various tumours at distant sites
(33). Cancer cells often travel
through blood and lymphatic vessels and reach premetastatic niches,
which require the destabilisation and crossing of endothelial walls
(4,7,27).
The endothelial barrier-destabilising contribution of FAK, which
supports the mobility of LECs, was demonstrated in a previous study
(12). In the present study, a
formerly unknown signalling network activating FAK upon stimulation
of LECs with 12(S)-HETE (an arachidonic acid metabolite), which
triggers LEC retraction (8) and
CCID formation, is reported, and this was indicated to be a
prerequisite for tumour cell intra- and extravasation (7). Due to its involvement in cancer
dissemination, FAK has been attracting increasing attention in
anti-metastasis research. Currently the effect of the FAK inhibitor
defactinib has been tested in 15 clinical trials against a variety
of malignancies. Of which, a total of 15 trials were terminated and
five were completed, of which one was already at phase II (Clinical
Trials. gov Identifier: NCT01951690). However, no results have been
published so far. A total of 5 other trials are still recruiting
patients currently.
The present data indicated that the activation of
FAK upon stimulation of LECs with 12(S)-HETE was regulated by SOX18
and RELA. RELA and SOX18 were positively feeding-back to each other
and also influenced the expression of the partner targets (RELA
influenced SOX18 and PROX1 and SOX18 influenced RELA and ICAM-1).
As has been previously reported, SOX18 and NF-κB are associated
with eachother (17,18) and NF-κB was demonstrated to
regulate the expression of FAK (26). These observations were consistent
with the data of the current study.
FAK was also indicated to feed back to RELA, ICAM-1
and SOX18 but not to PROX1, suggesting that the signal triggered by
12(S)-HETE arrived at RELA before it activated the other
components. RELA and SOX18 are transcription factors without a
known kinase function. Therefore, the phos-phorylation of FAK must
be associated with tyrosine kinases or phosphatases when cells were
treated with 12(S)-HETE under constitutive conditions,
respectively. FAK tyrosine 397 becomes cis- or
trans-autophosphorylated (34)
when FAK is associated with integrin-beta subunits. The
upregulation of integrin αvβ3 was demonstrated to depend on NF-κB,
leading to the activation of FAK and increased the motility of
human lung cancer cells (35).
Integrins also become activated when in contact with
transcriptional targets of NF-κB, including vimentin (36). Alternatively, NF-κB activates FAK
in LECs through the upregulation of interleukins (IL; including
IL-6), which causes the recruitment of Src at the Src Homology 2
(SH-2) and SH-3 domains of FAK in LECs (37). In turn, Src itself and other
Src-family kinases, including Fyn and Yes have been indicated to
phosphorylate tyrosine 397 (38,39).
Conversely, this residue is dephosphorylated by the SH2 domain
containing protein-tyrosine phosphatase 2 (SHP-2) in ECs, which
stabilises vascular permeability (40), and SHP-2 is regulated by NF-κB
(41). Hence, SHP-2, Src, vimentin
and integrins might have been involved in NF-κB-dependent
constitutive- or 12(S)-HETE-induced phosphorylation of FAK tyrosine
397.
SOX18, which inhibited FAK activity under
constitutive conditions, became an activator of FAK upon
stimulation with 12(S)-HETE. Physiologically, 12(S)-HETE and other
molecules are secreted by neutrophils and macrophages, which enable
their passage through lymph endothelial barriers and into the
adjacent stroma (9). In this
scenario, the regulatory function of NF-κB/RELA helps combat
infections. Pathologically, tumour-secreted 12(S)-HETE unlocks the
lymph endothelial cell wall, and in this scenario, RELA supports
malignant dissemination. Therefore, an immune cell-specific
mechanism, which is required to reach various types of body
tissues, becomes re-activated in cancer cells. It has previously
been demonstrated that 12(S)-HETE causes the disintegration of
endothelial barriers by inducing endothelial-to-mesenchymal
transition (endo-MT) (19,42). In molecular terms, 12(S)-HETE has
been revealed to upregulate S100A4, ZEB1 and the mobility proteins
MCL2 and myosin phosphatase target, and to transiently downregulate
vascular endothelial-cadherin in ECs (7,11,20).
These polypeptides, which are tightly linked to endo-MT and cause
endothelial retraction, also facilitate the formation of
pre-metastatic niche (PMN) (43,44).
Other authors have provided evidence that activated FAK contributes
to the formation of PMN at distant sites in pancreatic (45) and breast (46) cancer, which is based on the reduced
endothelial barrier function adjacent to the pre-metastatic organ.
This is not only a hallmark of PMN (27), but also indicative for endo-MT
(12,19). In support of this observation, the
siRNA-mediated suppression of FAK was shown to inhibit the
haptotaxis/haptokinesis (directional migration) of pancreatic
carcinoma cells (47), as well as
directional endothelial cell migration that was triggered by breast
cancer cell spheroids (12).
Therefore, 12(S)-HETE fulfils the criteria of an endo-MT trigger
factor (19,27) and might therefore contribute to the
formation of the PMN at a very early step of metastatic
dissemination. Targeting crucial signalling pathways might
attenuate the metastatic process. The current study demonstrated
that RELA, SOX18 and FAK were integrated in a signalling network
when LECs were stimulated with 12(S)-HETE. As both NF-κB and FAK
are common signal transducers for various cell functions in several
cell types, the specificity is marginal and the toxic side effects
are probably substantial. However, as this pro-metastatic mechanism
also involved SOX18 and its target PROX1 (18), both transcription factors are
specific for endothelial development and lymph endothelial
maintenance (15,16), therefore, identifying specific
inhibitors that target these polypeptides is required. Inhibitors
of SOX18, and eventually PROX1, may reduce non-specific toxicity
and exert improved anti-metastatic effects by strengthening the
resilience of the lymphatic barrier and attenuating the voyage of
cancer cells through the lymphatic route.
Supplementary Data
Funding
JH was supported by a State Scholarship Fund of
China Scholarship Council, the National Natural Science Foundation
of China (grant no. 81202853), the Natural Science Foundation of
Jiangsu Province (grant no. BK2012444) and Jiangsu Planned Projects
for Postdoctoral Research Funds. AF was supported by a DIKTI-OeAD
fellowship, and CHN by a technology grant (TSA Doktorat) financed
by the Austria Federal Ministry of Science and Research (BMFW) in
frame of Asea Uninet. GK was supported by the Austrian Science Fund
(FWF)/Herzfelder'sche Familienstiftung (project no. P 30572-B28 and
ZB-H) by a grant of the Medical Scientific Funds of the Mayor of
the City of Vienna (grant no. 15116).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GK, RDM, WJ, ZBH conceptualized the current study.
GK, WJ, RDM, DM and SK designed the current study. SE, DM, KK, SB,
JH and NR acquired the data. GK, NH, JR and CHN performed quality
control of the data and algorithms. SE, WJ, RDM and GK analyzed and
interpreted the data. SE, DM and KK performed statistical analysis.
GK, SE, RDM and WJ prepared the manuscript. SE, ZBH, SK, GK, RDM
and WJ edited the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
12(S)-HETE
|
12S-hydroxy-5Z,8Z,10E,14Z-
eicosatetraenoic acid, 'S' stereoisomer
|
|
12-HETER
|
12(S)-HETE receptor (syn. GPR31)
|
|
BLT2
|
Leukotriene B4 receptor 2 (syn. BLT2
receptor, BLT2R, low affinity LTB4 receptor)
|
|
CCID
|
Circular chemorepellent induced
defect
|
|
CYP1A1
|
Cytochrome P450 1A1
|
|
FAK
|
Focal adhesion kinase
|
|
ICAM-1
|
Intercellular adhesion molecule 1
|
|
IKK1
|
IκB kinase 1 (also IKK-α)
|
|
PMN
|
pre-metastatic niche
|
|
PROX1
|
Prospero homeobox protein 1
|
|
RELA
|
V-Rel avian reticuloendotheliosis
viral oncogene homolog A
|
|
SOX18
|
SRY-related HMG-box 18
|
Acknowledgments
The authors would like to thank Dr Adryan
Fristiohady (Faculty of Pharmacy, Halu Oleo University, Kendari,
Indonesia) for helping to train the masters student Stefanie
Engleitner, and the authors would also like to thank Mr. Toni
Jäger, Department of Pathology, Medical University of Vienna, for
preparing the figures.
References
|
1
|
Paget S: The distribution of secondary
growth in cancer of the breast. Lancet. 1:99–101. 1889.
|
|
2
|
Fidler IJ: The pathogenesis of cancer
metastasis: The 'seed and soil' hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nguyen DX, Bos PD and Massagué J:
Metastasis: From dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reymond N, d'Água BB and Ridley AJ:
Crossing the endothelial barrier during metastasis. Nat Rev Cancer.
13:858–870. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uchide K, Sakon M, Ariyoshi H, Nakamori S,
Tokunaga M and Monden M: Cancer cells cause vascular endothelial
cell (vEC) retraction via 12(S)HETE secretion; the possible role of
cancer cell derived microparticle. Ann Surg Oncol. 14:862–868.
2007. View Article : Google Scholar
|
|
6
|
Nguyen CH, Senfter D, Basilio J, Holzner
S, Stadler S, Krieger S, Huttary N, Milovanovic D, Viola K,
Simonitsch-Klupp I, et al: NF-κB contributes to MMP1 expression in
breast cancer spheroids causing paracrine PAR1 activation and
disintegrations in the lymph endothelial barrier in vitro.
Oncotarget. 6:39262–39275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kerjaschki D, Bago-Horvath Z, Rudas M,
Sexl V, Schneckenleithner C, Wolbank S, Bartel G, Krieger S, Kalt
R, Hantusch B, et al: Lipoxygenase mediates invasion of
intrameta-static lymphatic vessels and propagates lymph node
metastasis of human mammary carcinoma xenografts in mouse. J Clin
Invest. 121:2000–2012. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Honn KV, Tang DG, Grossi I, Duniec ZM,
Timar J, Renaud C, Leithauser M, Blair I, Johnson CR, Diglio CA, et
al: Tumor cell-derived 12(S)-hydroxyeicosatetraenoic acid induces
micro-vascular endothelial cell retraction. Cancer Res. 54:565–574.
1994.PubMed/NCBI
|
|
9
|
Rigby DA, Ferguson DJ, Johnson LA and
Jackson DG: Neutrophils rapidly transit inflamed lymphatic vessel
endothe-lium via integrin-dependent proteolysis and lipoxin-induced
junctional retraction. J Leukoc Biol. 98:897–912. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nguyen CH, Brenner S, Huttary N, Li Y,
Atanasov AG, Dirsch VM, Holzner S, Stadler S, Riha J, Krieger S, et
al: 12(S)-HETE increases intracellular Ca(2+) in lymph-endothelial
cells disrupting their barrier function in vitro; stabilization by
clinical drugs impairing calcium supply. Cancer Lett. 380:174–183.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nguyen CH, Stadler S, Brenner S, Huttary
N, Krieger S, Jäger W, Dolznig H and Krupitza G: Cancer
cell-derived 12(S)-HETE signals via 12-HETE receptor, RHO, ROCK and
MLC2 to induce lymph endothelial barrier breaching. Br J Cancer.
115:364–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong J, Fristiohady A, Nguyen CH,
Milovanovic D, Huttary N, Krieger S, Hong J, Geleff S, Birner P,
Jäger W, et al: Apigenin and luteolin attenuate the breaching of
MDA-MB231 breast cancer spheroids through the lymph endothelial
barrier in vitro. Front Pharmacol. 9:2202018. View Article : Google Scholar :
|
|
13
|
Dwyer SF, Gao L and Gelman IH:
Identification of novel focal adhesion kinase substrates: Role for
FAK in NFB signaling. Int J Biol Sci. 11:404–410. 2015. View Article : Google Scholar :
|
|
14
|
Tavora B, Reynolds LE, Batista S,
Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong PP,
Alexopoulou A, Elia G, et al: Endothelial-cell FAK targeting
sensitizes tumours to DNA-damaging therapy. Nature. 514:112–116.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanki Y, Nakaki R, Shimamura T, Matsunaga
T, Yamamizu K, Katayama S, Suehiro JI, Osawa T, Aburatani H, Kodama
T, et al: Dynamically and epigenetically coordinated GATA/ETS/SOX
transcription factor expression is indispensable for endothelial
cell differentiation. Nucleic Acids Res. 45:4344–4358. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
François M, Caprini A, Hosking B, Orsenigo
F, Wilhelm D, Browne C, Paavonen K, Karnezis T, Shayan R, Downes M,
et al: Sox18 induces development of the lymphatic vasculature in
mice. Nature. 456:643–647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Basilio J, Hoeth M, Holper-Schichl YM,
Resch U, Mayer H, Hofer-Warbinek R and de Martin R: TNFα-induced
down-regulation of Sox18 in endothelial cells is dependent on
NF-κB. Biochem Biophys Res Commun. 442:221–226. 2013. View Article : Google Scholar
|
|
18
|
Fristiohady A, Milovanovic D, Krieger S,
Basilio J, Jäger W, de Martin R and Krupitza G: 12(S)-HETE induces
lymphendo-thelial cell retraction in vitro through contribution of
SOX18 and PROX1. Int J Oncol. 50:307–316. 2018.
|
|
19
|
Vonach C, Viola K, Giessrigl B, Huttary N,
Raab I, Kalt R, Krieger S, Vo TP, Madlener S, Bauer S, et al: NF-κB
mediates the 12(S)-HETE-induced endothelial to mesenchymal
transition of lymphendothelial cells during the intravasation of
breast carcinoma cells. Br J Cancer. 105:263–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Viola K, Kopf S, Huttary N, Vonach C,
Kretschy N, Teichmann M, Giessrigl B, Raab I, Stary S, Krieger S,
et al: Bay11-7082 inhibits the disintegration of the
lymphendothelial barrier triggered by MCF-7 breast cancer
spheroids; The role of ICAM-1 and adhesion. Br J Cancer.
108:564–569. 2013. View Article : Google Scholar :
|
|
21
|
Yang J, Chang E, Cherry AM, Bangs CD, Oei
Y, Bodnar A, Bronstein A, Chiu CP and Herron GS: Human endothelial
cell life extension by telomerase expression. J Biol Chem.
274:26141–26148. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schoppmann SF, Soleiman A, Kalt R, Okubo
Y, Benisch C, Nagavarapu U, Herron GS and Geleff S:
Telomerase-immortalized lymphatic and blood vessel endothelial
cells are functionally stable and retain their lineage specificity.
Microcirculation. 11:261–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Stadler S, Nguyen CH, Schachner H,
Milovanovic D, Holzner S, Brenner S, Eichsteininger J, Stadler M,
Senfter D, Krenn L, et al: Colon cancer cell-derived 12(S)-HETE
induces the retraction of cancer-associated fibroblast via MLC2,
RHO/ROCK and Ca2+ signalling. Cell Mol Life Sci.
74:1907–1921. 2017. View Article : Google Scholar
|
|
25
|
Zhong L, Simard MJ and Huot J: Endothelial
microRNAs regulating the NF-κB pathway and cell adhesion molecules
during inflammation. FASEB J. 32:4070–4084. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Wang X, Li X, Fan Y, Li G, Guo C,
Zhu F, Zhang L and Shi Y: CD68(+)HLA-DR(+) M1-like macrophages
promote motility of HCC cells via NF-κB/FAK pathway. Cancer Lett.
345:91–99. 2014. View Article : Google Scholar
|
|
27
|
Peinado H, Zhang H, Matei IR, Costa-Silva
B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang
Y, et al: Pre-metastatic niches: Organ-specific homes for
metastases. Nat Rev Cancer. 17:302–317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zapletal O, Tylichová Z, Neča J, Kohoutek
J, Machala M, Milcová A, Pokorná M, Topinka J, Moyer MP, Hofmanová
J, et al: Butyrate alters expression of cytochrome P450 1A1 and
metabolism of benzo[a]pyrene via its histone deacetylase activity
in colon epithelial cell models. Arch Toxicol. 91:2135–2150. 2017.
View Article : Google Scholar
|
|
29
|
Nguyen CH, Brenner S, Huttary N, Atanasov
AG, Dirsch VM, Chatuphonprasert W, Holzner S, Stadler S, Riha J,
Krieger S, et al: AHR/CYP1A1 interplay triggers lymphatic barrier
breaching in breast cancer spheroids by inducing 12(S)-HETE
synthesis. Hum Mol Genet. 25:5006–5016. 2016.
|
|
30
|
Taira Z, Yamase D and Ueda Y: A new
technique for assaying cytochrome P450 enzyme activity in a single
cell. Cell Biol Toxicol. 23:143–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teichmann M, Kretschy N, Kopf S,
Jarukamjorn K, Atanasov AG, Viola K, Giessrigl B, Saiko P, Szekeres
T, Mikulits W, et al: Inhibition of tumour spheroid-induced
prometastatic intravasation gates in the lymph endothelial cell
barrier by carbamazepine: Drug testing in a 3D model. Arch Toxicol.
88:691–699. 2014.
|
|
32
|
Holzner S, Brenner S, Atanasov AG, Senfter
D, Stadler S, Nguyen CH, Fristiohady A, Milovanovic D, Huttary N,
Krieger S, et al: Intravasation of SW620 colon cancer cell
spheroids through the blood endothelial barrier is inhibited by
clinical drugs and flavonoids in vitro. Food Chem Toxicol.
111:114–124. 2018. View Article : Google Scholar
|
|
33
|
Cooper J and Giancotti FG: Integrin
signaling in cancer: Mechanotransduction, stemness, epithelial
plasticity, and therapeutic resistance. Cancer Cell. 35:347–367.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hanks SK, Ryzhova L, Shin NY and Brábek J:
Focal adhesion kinase signaling activities and their implications
in the control of cell survival and motility. Front Biosci.
8:d982–d996. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu J, Luo J, Li Y, Jia M, Wang Y, Huang Y
and Ke S: HMGB1 induces human non-small cell lung cancer cell
motility by activating integrin αvβ3/FAK through TLR4/NF-κB
signaling pathway. Biochem Biophys Res Commun. 480:522–527. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lilienbaum A, Duc Dodon M, Alexandre C,
Gazzolo L and Paulin D: Effect of human T-cell leukemia virus type
I tax protein on activation of the human vimentin gene. J Virol.
64:256–263. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang YH, Yang HY, Huang SW, Ou G, Hsu YF
and Hsu MJ: Interleukin-6 induces vascular endothelial growth
factor-C expression via Src-FAK-STAT3 signaling in lymphatic
endothelial cells. PLoS One. 11:e01588392016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Calalb MB, Polte TR and Hanks SK: Tyrosine
phosphorylation of focal adhesion kinase at sites in the catalytic
domain regulates kinase activity: A role for Src family kinases.
Mol Cell Biol. 15:954–963. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Klinghoffer RA, Sachsenmaier C, Cooper JA
and Soriano P: Src family kinases are required for integrin but not
PDGFR signal transduction. EMBO J. 18:2459–2471. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chichger H, Braza J, Duong H and
Harrington EO: SH2 domain-containing protein tyrosine phosphatase 2
and focal adhesion kinase protein interactions regulate pulmonary
endo-thelium barrier function. Am J Respir Cell Mol Biol.
52:695–707. 2015. View Article : Google Scholar :
|
|
41
|
Kang HJ, Chung DH, Sung CO, Yoo SH, Yu E,
Kim N, Lee SH, Song JY, Kim CJ and Choi J: SHP2 is induced by the
HBx-NF-κB pathway and contributes to fibrosis during human early
hepatocellular carcinoma development. Oncotarget. 8:27263–27276.
2017.PubMed/NCBI
|
|
42
|
Senfter D, Holzner S, Kalipciyan M,
Staribacher A, Walzl A, Huttary N, Krieger S, Brenner S, Jäger W,
Krupitza G, et al: Loss of miR-200 family in 5-fluorouracil
resistant colon cancer drives lymphendothelial invasiveness in
vitro. Hum Mol Genet. 24:3689–3698. 2015.PubMed/NCBI
|
|
43
|
Kaplan RN, Riba RD, Zacharoulis S, Bramley
AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et
al: VEGFR1-positive haematopoietic bone marrow progenitors initiate
the pre-metastatic niche. Nature. 438:820–827. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kaplan RN, Psaila B and Lyden D: Bone
marrow cells in the 'pre-metastatic niche': Within bone and beyond.
Cancer Metastasis Rev. 25:521–529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Houg DS and Bijlsma MF: The hepatic
pre-metastatic niche in pancreatic ductal adenocarcinoma. Mol
Cancer. 17:952018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hiratsuka S, Goel S, Kamoun WS, Maru Y,
Fukumura D, Duda DG and Jain RK: Endothelial focal adhesion kinase
mediates cancer cell homing to discrete regions of the lungs via
E-selectin upregulation. Proc Natl Acad Sci U S A. 108:3725–3730.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lu J, Zhou S, Siech M, Habisch H,
Seufferlein T and Bachem MG: Pancreatic stellate cells promote
hapto-migration of cancer cells through collagen I-mediated
signalling pathway. Br J Cancer. 110:409–420. 2014. View Article : Google Scholar :
|














