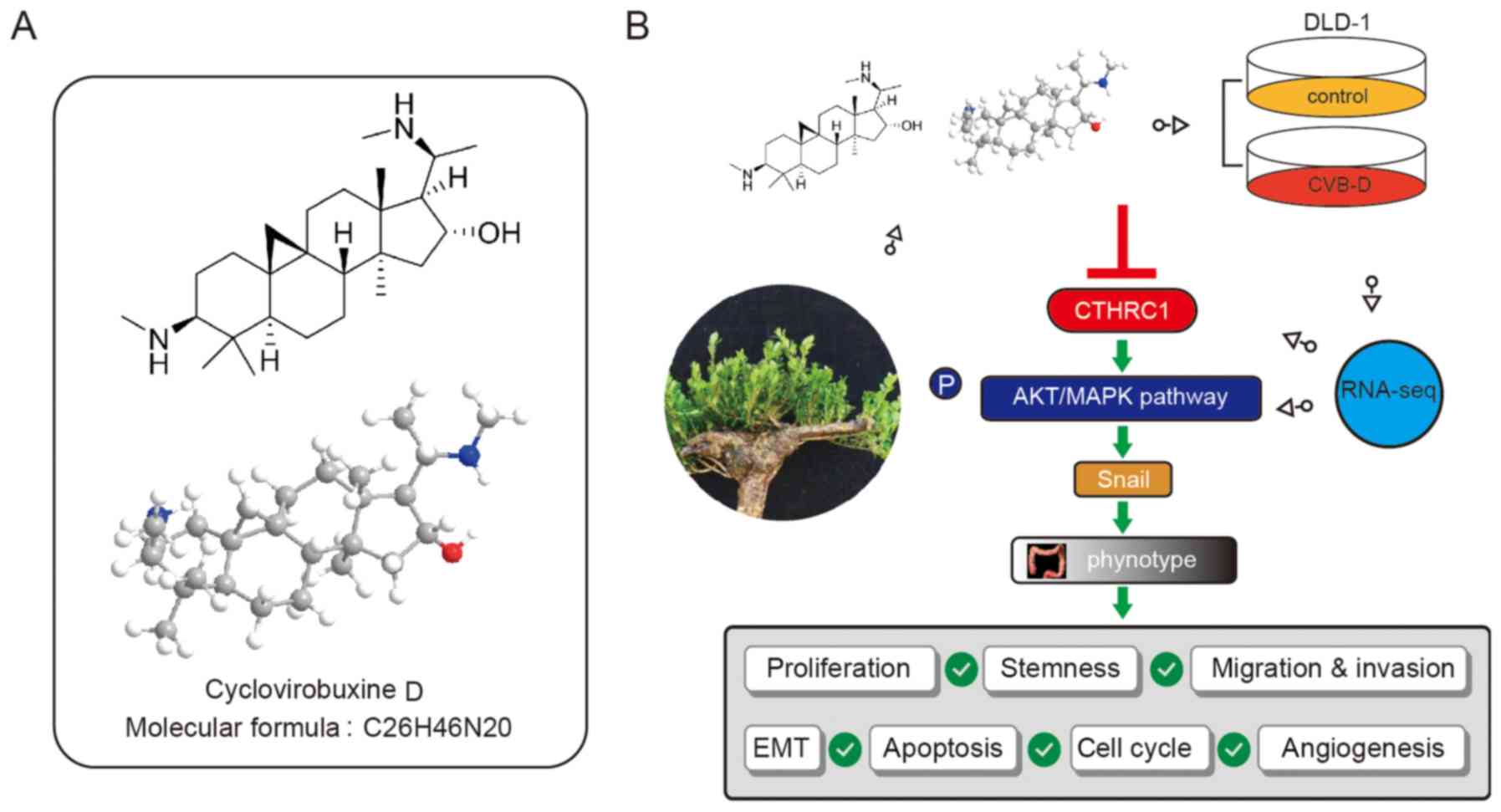
Introduction
Colorectal cancer (CRC), including colon and rectal
cancer, is one of the most common types of gastrointestinal
malignant tumors, ranking third in males and second in females, and
poses a serious threat to human health (1). Colon adenocarcinoma (COAD) is the
most common histopathological type of CRC, and COAD morbidity and
mortality rates are increasing. In 2018, the number of new cases of
COAD surpassed 1 million and >500,000 COAD patients succumbed to
the disease (2). Therefore,
studying the pathogenesis of and treatments for COAD are
particularly important (3).
Significant breakthroughs have been made in the early diagnosis and
treatment of CRC in recent decades (4). Unfortunately, the overall survival
(OS) rate of patients with CRC remains poor, especially in the
advanced stage, mainly due to recurrence and metastasis (5).
Accumulating evidence has shown that natural plant
compounds (NPCs), such as curcumin, resveratrol and dictamnine,
have significant clinical benefits in cancer, such as inhibiting
cell proliferation, affecting the cell cycle, and promoting
apoptosis and/or autophagy (6-8). In
addition, bioactive natural components extracted from plants have
received attention for their inhibition of epithelial-mesenchymal
transformation (EMT) through anti-inflammatory, anti-fibrosis or
anti-oxidant mechanisms (9). It
has been demonstrated that pathological EMT is inhibited by NPCs
via different cellular signal transduction pathways (10).
Cyclovirobuxine D (CVB-D; Chemical Abstracts Service
number: 860-79-7; C26H46N2O;
molecular weight: 402.66) is an alkaloid, which is mainly derived
from Buxus microphylla.
B. microphylla is a Traditional
Chinese Medicine with a long history of treating cardiovascular
diseases and was recorded hundreds of years ago in the Compendium
of Materia (11). Previous studies
have indicated that CVB-D can induce autophagy-related death in
human breast cancer cells via the AKT/mammalian target of rapamycin
(mTOR) pathway (12), and induce
mitochondria-mediated apoptosis, thereby inhibiting the
proliferation of gastric cancer cells (13). However, to the best of our
knowledge, there are no reports on the effects of CVB-D in CRC.
The metastasis of malignant tumors is the main
characteristic of the EMT. EMT is a complex biological process, in
which epithelial cells undergo multiple morphological and
biochemical changes and genetic rearrangements, leading to a
mesenchymal-like phenotype (14-16).
Enhanced mesenchymal phenotypes alter a series of biological
behaviors of cancer cells, such as proliferation, stemness,
anti-apoptosis, migration, invasion and radio-/chemo-sensitivity
(17). Therefore, targeting EMT is
a powerful approach for inhibiting the metastasis of malignant
tumors. A group of transcription factors, including Snail, Slug,
Twist, zinc finger E-box-binding homeobox (ZEB)1 and ZEB2, also
known as EMT regulators, directly or indirectly inhibit the
activity of the E-cadherin promoter and precisely orchestrate the
EMT process, leading to expression of the mesenchymal markers
N-cadherin and vimentin (18). In
addition, a number of oncogenes and tumor suppressor genes are
involved in the EMT process as upstream or downstream factors
(19). One of these proteins,
collagen triple helix repeat containing 1 (CTHRC1), was first found
to be involved in vascular remodeling and plays a key role in the
response of cells to arterial injury (20). CTHRC1 is overexpressed in numerous
solid tumors and plays an important role in tumorigenesis and
metastasis, particularly in CRC (21,22),
gastric cancer, melanoma, oral cancer, pancreatic cancer and
hepatocellular cancer (23-27).
CTHRC1 can activate multiple signaling pathways and promote
epithelial cell metastasis by inducing the EMT in CRC (21). This suggests that CTHRC1 may be a
potential therapeutic target for CRC.
Materials and methods
Cells and reagents
CRC cell lines DLD-1 and LoVo, and the human
intestinal epithelial cell line NCM460, were obtained from the
Fuheng Cell Center (Shanghai, China). NCM460 and DLD-1 cells were
cultured in Dulbecco's modified Eagle's medium (Thermo Fisher
Scientific, Inc.). LoVo cells were cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.). All media contained 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 100 mg/ml streptomycin. All cells were
cultured in a standard humidified incubator at 37°C with a 5%
CO2 atmosphere. Aladdin Industrial Corporation supplied
the CVB-D (cat. no. C117989). CVB-D was dissolved in methanol to
produce a 71 mmol/l stock solution.
MTT assay
CVB-D cytotoxicity was determined in CRC and NCM460
cells via MTT assay. DLD-1, LoVo and NCM460 cells were seeded into
96-well plates (5×103 per well) and cultured at 37°C for
24 h until the cells adhered to the plates. Different doses of
CVB-D (0, 10, 15, 20, 25, 30, 35, 40, 45 and 50 µM) were
added to the plates and cultured at 37°C for 24 or 48 h. MTT
solution (5 mg/ml; 20 µl/well; Sigma-Aldrich, Merck KGaA)
was then added and the plates were incubated at 37°C for 4 h. DMSO
(150 µl/well) was subsequently added to dissolve the
formazan crystals. Absorbance at a wavelength of 490 nm was
detected using a Microporous Plate automatic regulator (ELx808;
BioTek Instruments). The formula used for calculating the cell
proliferation inhibition rate was as follows: Inhibition rate
(%)=[1-A490 (test)/A490 (blank)] ×100%. Three biologically
independent experiments were conducted.
Plate colony-forming assay
CRC cells were seeded in a 6-cm diameter dish (1,000
cells/dish) and incubated for 24 h. CRC cells were then incubated
with different doses of CVB-D (0, 20, 30 and 40 µM) at 37°C
for 5 h. The CVB-D was then removed and replaced with normal
culture medium. After 14 days, the cells were fixed in methanol at
room temperature for 15 min and stained with 0.5% crystal violet at
room temperature for 10 min. Three biologically independent
experiments were performed.
Cell cycle assay
Adherent CRC cells were treated with CVB-D (30
µM) at 37°C for 48 h, then harvested and fixed in 75%
ethanol at 4°C overnight. The cells were digested and stained using
RNase and propidium iodide (4A Biotech), respectively. The cells
were stained at 37°C for 30 min in the dark. The cell cycle was
analyzed using a flow cytometer. ModFit LT V5.0 software (Verity
Software House, Inc.) was used for analysis. Three biologically
independent experiments were conducted.
Apoptosis assay
CRC cells were attached to a six-well plate
(2.5×105 cells/well) and incubated overnight. Cells were
then treated with different doses of CVB-D (0, 20, 30 and 40
µM) at 37°C for 48 h, and then harvested and stained using
an Annexin V-FITC/PI Apoptosis Detection kit (cat. no. FXP018; 4A
Biotech) according to the manufacturer's protocol. A flow cytometer
was used to analyze apoptosis followed by analysis with FACS DiVa
6.1.3 software (BD Biosciences). Three biologically independent
experiments were conducted.
Wound healing assay
The 90% confluent monolayer of serum-starved CRC
cells was scraped with a 200-µl pipette tip and then treated
with CVB-D (0, 10 and 15 µM) at 37°C for 24 h. Subsequently,
photographs of the wound widths were obtained at different time
periods (0, 24 and 48 h) using a light microscope (magnification,
×100). The migration distances were analyzed by ImageJ V1.8.0
software (National Institutes of Health). Three biologically
independent experiments were conducted.
Transwell assay
Cell migration and invasion experiments were
conducted using Transwell chambers (BD Biosciences). CRC cells were
cultured in six-well plates with different doses of CVB-D (0, 10
and 15 µM) at 37°C for 24 h. Culture medium (500 µl)
containing 10% FBS was added to the lower chamber. Pretreated CRC
cells were resuspended in serum-free medium (10,000 cells/200
µl medium) and injected into the upper chamber. Unlike
migration analysis, 50 µl Matrigel (BD Biosciences) was used
to cover the upper side of the polycarbonate membrane for the
invasion assay. Following incubation for 48 h, migrated or invaded
cells were fixed in methanol at room temperature for 15 min. Cells
on the polycarbonate membrane were wiped off, and cells under the
polycarbonate membrane were stained with 0.5% crystal violet
solution at room temperature for 20 min. Under the light
microscope, five fields (magnification, ×100) were randomly
selected, and the mean number of cells was calculated
and used for statistical analysis. Three biologically independent
experiments were conducted.
Western blot analysis
CRC cells were treated with different concentrations
of CVB-D (0, 20, 30 and 40 µM) at 37°C for 48 h, and then
lysed with RIPA buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology) containing protease and phosphatase inhibitors.
Following 10 min of centrifugation at 12,000 x g and 4°C, the
supernatants were transferred to new test tubes. A Bradford Protein
assay kit (cat. no. P0012S) was used to measure the protein
concentration. Proteins (25 µg/lane) were separated by 10%
SDS-PAGE (Beyotime Institute of Biotechnology) and then
eletrotransferred onto polyvinylidene difluoride membranes (EMD
Millipore). Membranes were blocked with 5% non-fat
milk/TBS-Tween-20 for 1 h at room temperature and then incubated
with the corresponding primary antibodies overnight at 4°C. The
antibodies used were as follows: CTHRC1 rabbit polyclonal antibody
(1:1,000; cat. no. 16534-1-AP; Proteintech Group Inc.),
phosphorylated (p)-AKT (Ser473) rabbit polyclonal antibody
(1:1,000; cat. no. 4060S; Cell Signaling Technology, Inc.), p-ERK
(Thr202/Tyr204) rabbit polyclonal antibody (1:1,000; cat. no.
4370T; Cell Signaling Technology, Inc.), E-cadherin rabbit
polyclonal antibody (1:1,000; cat. no. 20874-1-AP; Proteintech
Group Inc.), N-cadherin rabbit polyclonal antibody (1:1,000; cat.
no. 22018-1-AP; Proteintech Group Inc.), vimentin rabbit polyclonal
antibody (1:1,000; cat. no. 10366-1-AP; Proteintech Group Inc.),
matrix metalloproteinase (MMP)2 rabbit polyclonal antibody
(1:1,000; cat. no. 10373-2-AP; Proteintech Group Inc.), MMP9 rabbit
polyclonal antibody (1:1,000; cat. no. 10375-2-AP; Proteintech
Group Inc.), CD133 rabbit polyclonal antibody (1:1,000; cat. no.
66666-1-Ig; Proteintech Group Inc.), cyclin D1 rabbit polyclonal
antibody (1:1,000; cat. no. 60186-1-Ig; Proteintech Group Inc.),
B-cell lymphoma 2 (Bcl-2)-associated X protein (BAX) rabbit
polyclonal antibody (1:1,000; cat. no. 50599-2-Ig; Proteintech
Group Inc.), Bcl-2 rabbit polyclonal antibody (1:1,000; cat. no.
12789-1-AP; Proteintech Group Inc.), ZEB1 rabbit polyclonal
antibody (1:1,000; cat. no. 21544-1-AP; Proteintech Group Inc.),
ZEB2 rabbit polyclonal antibody (1:1,000; cat. no. 14026-1-AP;
Proteintech Group Inc.), Snail rabbit polyclonal antibody (1:1,000;
cat. no. 13099-1-AP; Proteintech Group Inc.), Slug rabbit
polyclonal antibody (1:1,000; cat. no. 12129-1-AP; Proteintech
Group Inc.), AKT rabbit polyclonal antibody (1:1,000; cat. no.
10176-2-AP; Proteintech Group Inc.), ERK rabbit polyclonal antibody
(1:1,000; cat. no. 16443-1-AP; Proteintech Group Inc.), and β-actin
mouse polyclonal antibody (1:1,000; cat. no. TA-09; OriGene
Technologies, Inc.). The membranes were then incubated with
peroxidase-conjugated goat anti-rabbit secondary antibody
(1:10,000; cat. no. ZB-2301; ZSGB-BIO) or peroxidase-conjugated
goat anti-mouse secondary antibody (1:10,000; cat. no. ZB-2305;
ZSGB-BIO) for 1 h at room temperature. Protein signals were
visualized using enhanced chemiluminescence substrate (Thermo
Fisher Scientific, Inc.) with a Bio-Rad gel imaging system (Bio-Rad
Laboratories, Inc.). Image Lab version 2.0.1 (Bio-Rad Laboratories,
Inc.) was used for quantification of the western blot data. Three
biologically independent experiments were conducted.
RNA sequencing (RNA-seq) experiments
DLD-1 cells were inoculated into a 6-cm culture dish
during the exponential growth phase (2.5×105 cells/well)
and cultured overnight. After 48 h at 37°C of CVB-D (20 µM)
treatment, a cell suspension with <25% of the total cells was
selected as the experimental group. The cells were digested with
1.5 ml TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
then collected and transferred to −80°C for storage. This sample
collection step was repeated three times. The next steps including
RNA isolation, cDNA library preparation and RNA-seq were completed
by KangChen Bio-tech Inc. A NanoDrop was used to measure the
quantity and quality of RNA. The DNA library was constructed using
an Illumina kit, based on the KAPA stranded RNA-Seq Library, and
enrichment of RNA was performed with oligo(dT) magnetic beads. The
first-strand cDNA was generated by random primer inversion, and
dUTP was added for second-strand cDNA synthesis. The final library
was obtained by PCR amplification. Agilent 2100 was used to detect
the quality of the constructed library, and quantitative PCR was
used to quantify the library. After a series of strict quality
control steps, the trimmed data were compared to the reference
genome.
Bioinformatics prediction
GEPIA2 (http://gepia2.cancer-pku.cn/#index) analysis included
analyses of the differential expression of tumors/normal tissues,
the survival of patients and multiple gene comparisons. The human
protein atlas (http://www.proteinatlas.org) was employed to evaluate
CTHRC1 protein expression in COAD tissues. The Venny online tool
(https://bioinfogp.cnb.csic.es/tools/venny/) was used
to identify common data in different groups of data. Gene Ontology
(GO) (http://www.geneontology.org) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway (http://www.genome.jp/kegg) enrichment analyses were
performed based on the downregulated differentially expressed genes
(DEGs) obtained from RNA-seq experiments. The GO terms covers three
domains: Biological Process (BP), Cellular Component (CC) and
Molecular Function (MF). The targets of CVB-D should comply with
the following requirements: i) They were oncogenes; ii) they were
overexpressed in COAD; and iii) the overexpressed oncogenes were
accompanied by a decrease of OS and/or DFS.
Tumor xenograft experiments
A total of six female BALB/c nude mice (5-6-weeks
old; weight, 18 g) from Vital River Laboratory Animal Technology
Co., Ltd. were selected and reared in a pathogen-free environment.
Mice were housed at room temperature in a 12-h light/dark cycle and
had access to food and water ad libitum. The Medical Ethics
Committee of Harbin Medical University (Harbin, China) approved the
animal protocol. Following intraperitoneal injection of 1%
pentobarbital sodium into mice (50 mg/kg) (28), DLD-1 cells (0.1 ml;
1×107 cells/ml) were resuspended in culture medium and
subcutaneously implanted into the flanks of mice in equal volumes
to establish tumor xenografts. Mice were randomly divided into
treatment and control groups (three mice in each group) 7 days
after xenograft establishment. The experimental group and the
negative control group were intraperitoneally injected with 15
mg/kg CVB-D and PBS, respectively, once a day for 4 weeks.
Evaluation of tumor growth, angiogenesis, microangiogenesis and
tumor hardness was performed using B-ultrasound, color Doppler flow
imaging (CDFI), color power angiography (CPA), and ultrasonic
elastosonography (USE). Finally, mice were sacrificed and the
tumors, hearts, livers, spleens, lungs were collected for
pathological examination at the end of the experiment.
Immunohistochemistry (IHC), hematoxylin
and eosin (H&E) staining and TUNEL apoptosis detection
For IHC, briefly, formaldehyde-fixed (at room
temperature for 48 h) and paraffin-embedded tumour tissues were
sliced into 5-µm-thick sections. The sections were
deparaffinized by xylene, rehydrated in a graded series of ethanol
(100, 95, 85 and 75%), incubated with 3% H2O2
for 30 min, and blocked with 3% bovine serum albumin at room
temperature for 1 h. Sections were then immunostained with
anti-Ki-67 antibody (polyclonal, rabbit anti-human IgG; 1:200; cat.
no. WL01384; Wanleibio Co., Ltd.), CTHRC1 rabbit polyclonal
antibody (1:200; cat. no. 16534-1-AP; Proteintech Group Inc.),
Bcl-2 rabbit polyclonal antibody (1:100; cat. no. WL01556;
Wanleibio Co., Ltd.), N-cadherin rabbit polyclonal antibody (1:100;
cat. no. WL01047; Wanleibio Co., Ltd.), cyclin D1 rabbit polyclonal
antibody (1:200; cat. no. WL01435; Wanleibio Co., Ltd.) and HIF-1α
rabbit polyclonal antibody (1:500; cat. no. WL01607; Wanleibio Co.,
Ltd.) at 4°C overnight. The colour was developed with a
3,3′-diaminobenzidine Substrate kit (cat. no. DA1010; Beijing
Solarbio Science and Technology Co., Ltd.), and the sections were
counterstained with hematoxylin at room temperature for 3 min. Five
visual fields on each slide were randomly selected and photographed
under a light microscope (magnification, ×400).
For H&E, briefly, tissues were fixed at room
temperature for 48 h and embedded in paraffin, sliced into
5-µm-thick sections, stained with hematoxylin at room
temperature for 5 min, treated with 1% acid ethanol for 3 sec,
washed with distilled water, dyed with eosin solution at room
temperature for 3 min, dehydrated with graded alcohol and cleaned
with xylene. After drying, five visual fields were randomly
selected on each slide and photographed using a light microscope
(magnification, ×200).
Apoptosis in pathological tissues was detected using
a TUNEL Apoptosis Detection kit (Wanlei Co., Ltd.; www.wanleibio.cn), according to the manufacturer's
protocol. Five visual fields were randomly selected on each slide
and photographed using a light microscope (magnification,
×400).
Transient transfection of small
interfering RNA (siRNA)
CTHRC1 expression was silenced by siRNA. CTHRC1
siRNA and negative control siRNA was synthesized by General
Biosystems, Inc. and 100 nM was transfected into DLD-1 and LoVo
cells using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.). The cells were incubated for 48 h and
then used in subsequent experiments. The siRNA sequences are
presented in Table SI.
Statistical analysis
Statistical analysis was conducted using SPSS 22.0
software (IBM Corp.). Data are presented as the mean ± standard
deviation. The difference of means between two groups was assessed
using Student's t-test. The difference of means between multiple
groups was assessed using one-way analysis of variance, and a
comparison between two groups was assessed using Bonferroni's
correction. P<0.05 was considered a statistically significant
difference.
Results
CVB-D suppresses CRC cell
proliferation
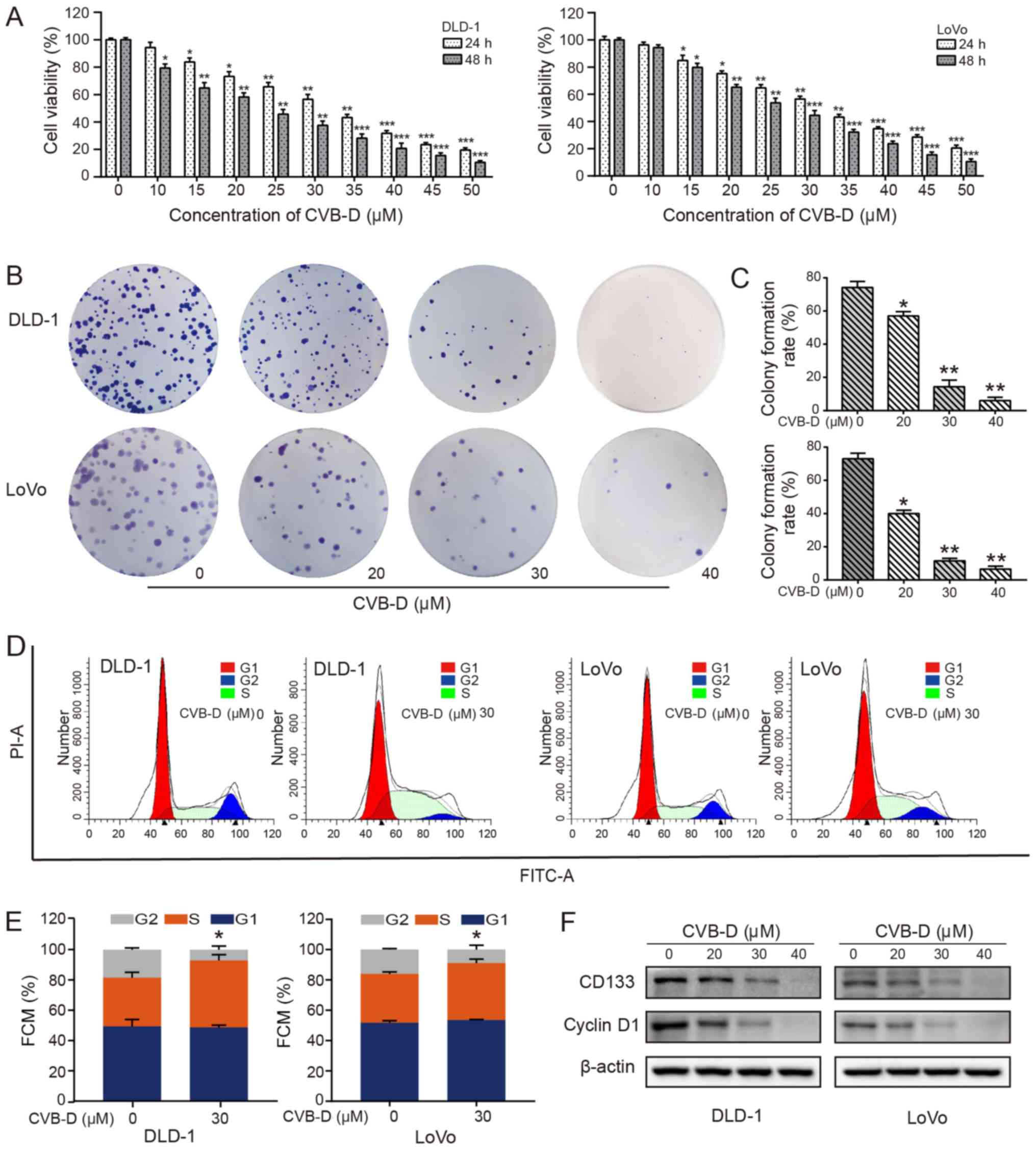
To evaluate the inhibitory effects of CVB-D on cell
viability, CRC cells were exposed to increasing concentrations
(0-50 µM) of CVB-D for 24 and 48 h. The results demonstrated
that CVB-D significantly inhibited the CRC cell viability in a
dose-dependent manner (Fig. 1A).
According to probit regression analysis, the IC50 of
CVB-D in DLD-1 and LoVo cells at 48 h was ~23.20 and ~26.12
µM, respectively. Significant dose-dependent reductions in
colony formation rate were also observed in DLD-1 and LoVo cells
treated with CVB-D, as determined by colony formation assay
(Fig. 1B), possibly due to a
decrease in stemness. CD133 is a transmembrane glycoprotein that is
considered to be a surface marker of tumor stem cells and is
overexpressed in CRC (29).
Patients with CRC with high CD133 expression are prone to
metastasis and recurrence (30,31).
The present results demonstrated that CD133 expression
significantly decreased with an increase in drug concentration
(Figs. 1F and S2), indicating that stemness may be
suppressed by CVB-D. In addition, CVB-D had an insignificant effect
on the viability of NCM460 cells (Fig. S1).
In cancer, control of the cell cycle is altered,
leading to uncontrolled cell proliferation, thereby making the cell
cycle an important target of cancer treatment (32). The overexpression of cyclin D1 in
patients with CRC indicates a poor prognosis (33). Therefore, enhancing the expression
of cyclin D1 and/or inhibiting its degradation is another important
mechanism by which oncogenic signaling pathways, including the
PI3K/AKT signaling pathway, regulate the proliferation and growth
of tumor cells (34,35). The regulation of cyclin D1
expression is a dynamic process in the cell. The expression levels
of cyclin D1 in the G1-S-G2 phases are high, low, and low,
respectively. The expression of cyclin D1 is low in the S phase;
however, it is not the decrease of cyclin D1 expression that leads
to the stagnation of S phase cells (36). The cell cycle experiments
demonstrated that CRC cells were arrested at the S phase following
CVB-D treatment, with a significantly higher percentage of cells in
this phase (Fig. 1D and E). The
expression of cyclin D1 in CRC cells was decreased following CVB-D
treatment, as shown by western blot analysis (Figs. 1F and S2).
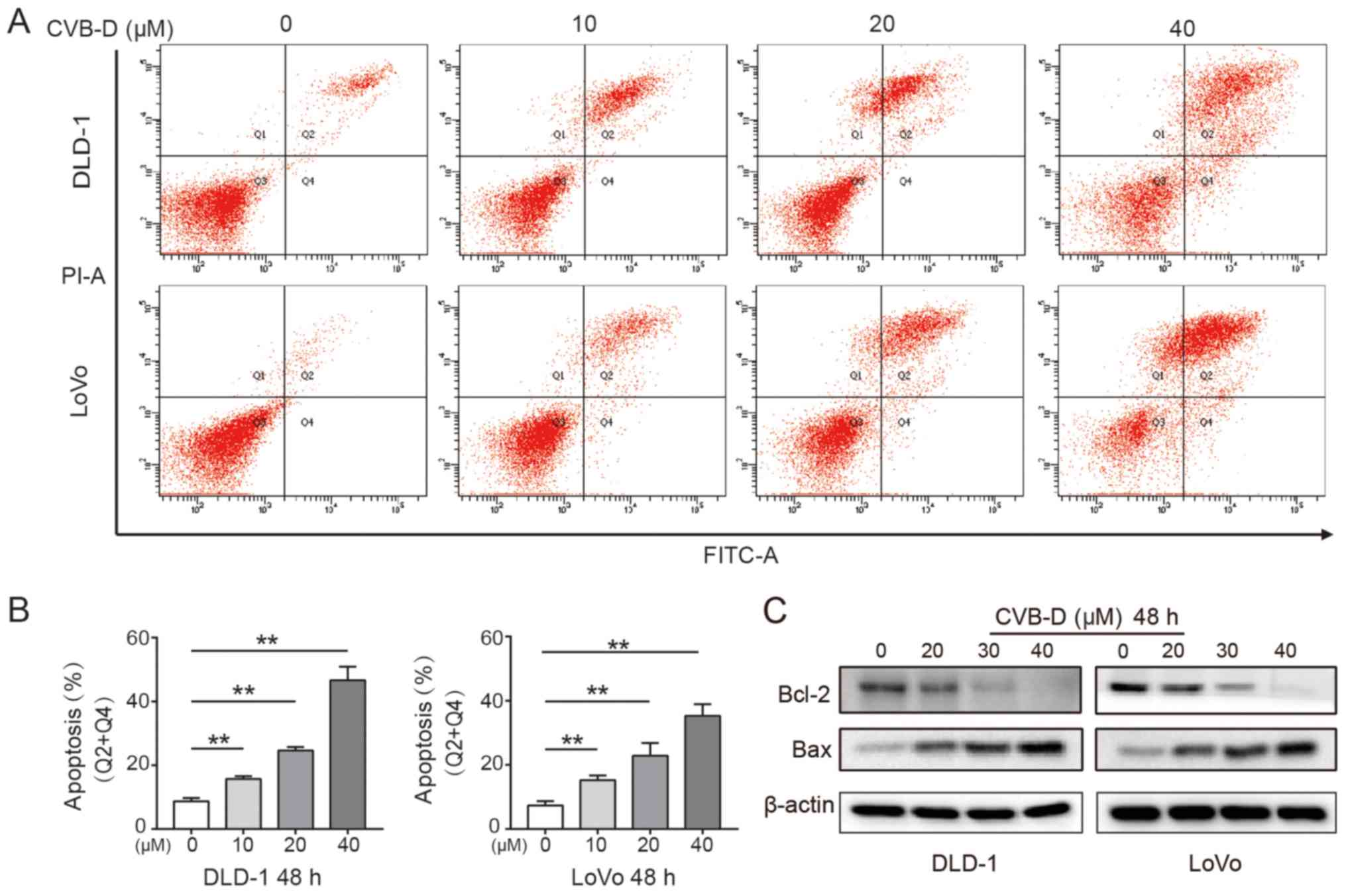
CVB-D induces apoptosis of CRC cells
Apoptosis is the key steady-state mechanism that
balances cell division and cell death to restrain the uncontrolled
proliferation of cancer cells; thus, induction of apoptosis is an
important mechanism of anticancer drugs (37). The present study found that CVB-D
significantly induced apoptosis of CRC cells in a dose-dependent
manner (Fig. 2A and B). Western
blot analysis demonstrated that Bcl-2 expression significantly
decreased and Bax expression significantly increased in CRC cells
following treatment, indicating that CVB-D induced apoptosis
(Figs. 2C and S3).
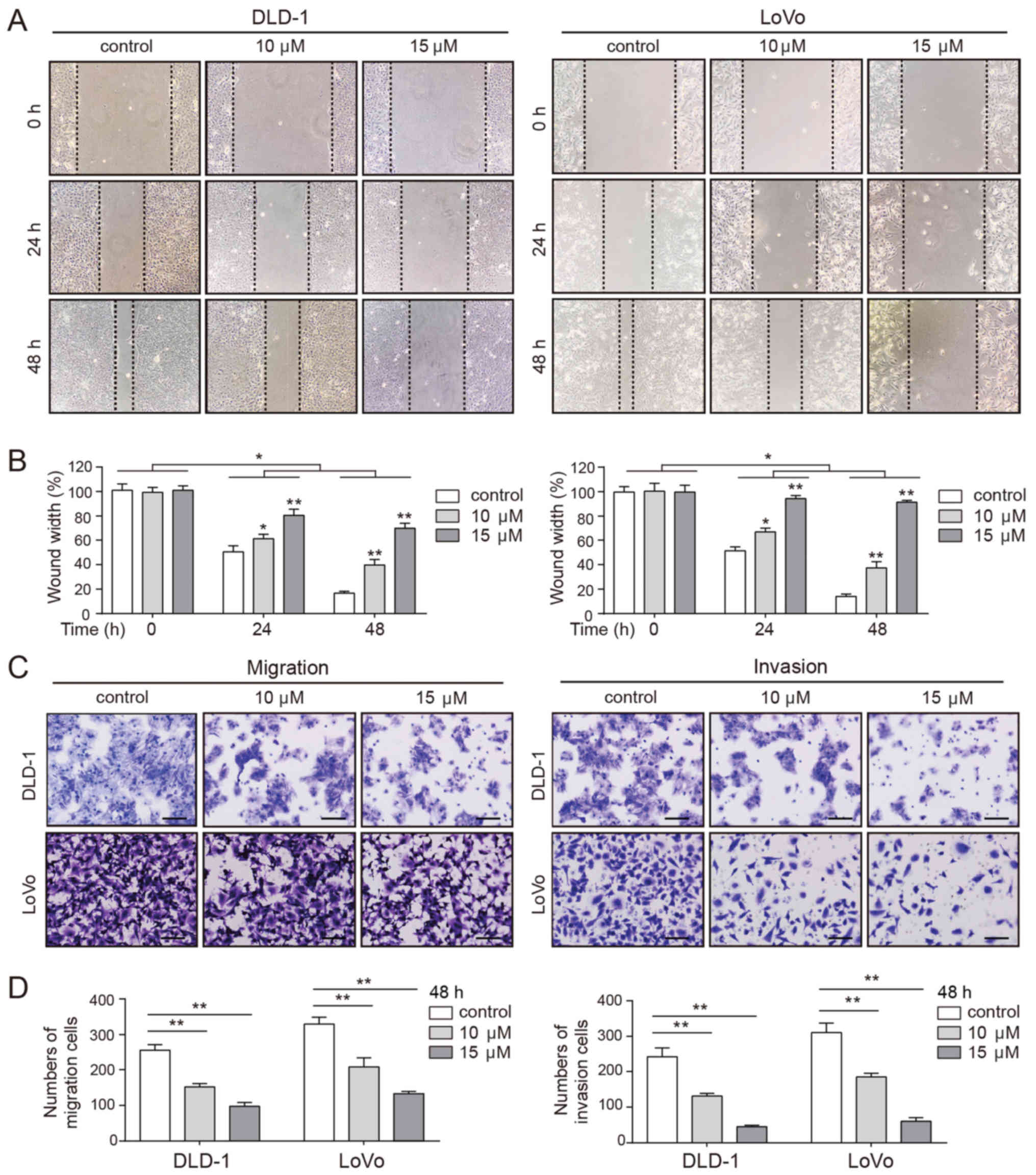
CVB-D decreases the mobility of CRC
cells
Pro-apoptotic chemotherapeutic agents decrease the
mobility of tumor cells (38).
Thus, the effects of CVB-D on cell migration and invasion were
evaluated by wound healing and Transwell assays. As presented in
Fig. 3A and B, the wound in the
untreated cells healed rapidly, while the wound of the
CVB-D-treated cells healed more slowly in a dose-dependent manner.
Consistent with these observations, Transwell assays demonstrated a
significantly dose-dependent decrease in the number of migrating
and invading cells following CVB-D treatment (Fig. 3C and D). These results confirmed
that CVB-D inhibited the mobility of CRC cells in a dose-dependent
manner.
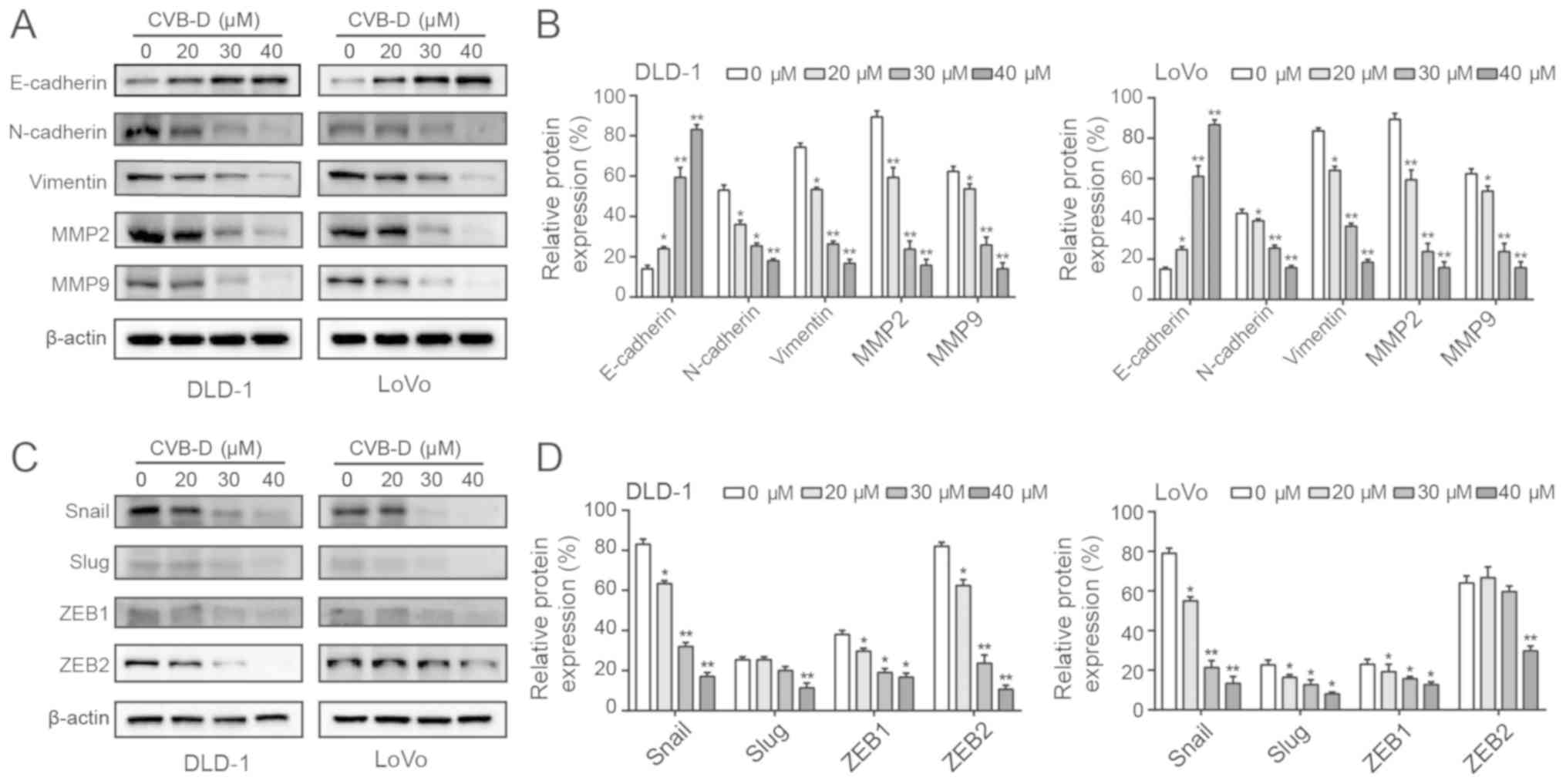
CVB-D affects the EMT through Snail
E-cadherin, vimentin, and N-cadherin are main
biomarkers of the EMT (39).
Western blot analysis revealed that CVB-D treatment significantly
increased the expression of E-cadherin and significantly decreased
the expression of vimentin and N-cadherin (Fig. 4A and B). Degradation of the
extracellular matrix (ECM) promotes the ability of malignant tumor
cells to invade adjacent tissues, blood and lymphatic vessels in an
MMP-dependent manner (40). In
particular, MMP2 and MMP9 have been shown to promote cancer cell
invasion and lymph node metastasis in gastrointestinal tumors
(41). In the present study, MMP2
and MMP9 expression levels were decreased following treatment with
CVB-D (Fig. 4A and B). In
addition, western blotting demonstrated that CVB-D significantly
decreased the expression of Snail in CRC cells in a dose-dependent
manner. In DLD-1 cells, the expression of Slug was significantly
inhibited when the concentration reached 40 µM, and the
expression levels of ZEB1 and ZEB2 were reduced by CVB-D in a dose
dependent manner. In LoVo cells, the expression levels of Slug and
ZEB1 were decreased in a dose-dependent manner, and the expression
of ZEB2 was significantly decreased at 40 µM. Therefore,
compared with Slug, ZEB1 and ZEB2, only the expression of Snail was
significantly decreased both in DLD-1 and LoVo cells in a
dose-dependent manner (Fig. 4C and
D). In view of these results, the present study mainly focused
on Snail. The GEPIA database confirmed that Snail expression was
significantly higher in COAD compared with adjacent tissue
(Fig. S4A). Furthermore, a higher
expression of Snail was significantly associated with a reduced OS
rate of patients with COAD (Fig.
S4B). These data suggested that inhibition of the EMT by CVB-D
was regulated by Snail.
RNA-seq indicates that CVB-D exerts
anticancer effects on CRC cells mainly by inhibiting the AKT/ERK
signaling pathway
RNA-seq was used to investigate the mechanisms
underlying the effects of CVB-D on CRC. The experimental flowchart
is presented in Fig. 5A.
Hierarchical clustering and volcano plots demonstrated that 1,932
DEGs were upregulated and 2,559 DEGs were downregulated, and 7,255
DEGs were unchanged in RNA-seq data (Fig. 5B and C). According to the
functional annotation in the GO database, the downregulated DEGs
were mostly enriched in the following BP terms 'metabolic process'
and 'heterocycle metabolic process'; the CC terms 'intracellular'
and 'mitochondrion'; and the MF terms 'catalytic activity' and 'RNA
binding' (Fig. 5D). As cellular
metabolism is the foundation of all biological activities, studying
the mechanism of the metabolic process may enhance cancer treatment
methods (42). The classical MAPK,
PI3K-AKT and cell cycle pathway pathways were associated with the
group of downregulated DEGs in the RNA-seq dataset (Fig. 5E). Western blot analysis verified
that the protein expression levels of p-AKT, p-ERK1/2 and PI3K were
inhibited by CVB-D in a dose-dependent manner, while, the levels of
total AKT and ERK1/2 demonstrated no difference, indicating that
CVB-D exerts anticancer effects on CRC by inhibiting the AKT/ERK
signaling pathway (Figs. 5F and
S5).
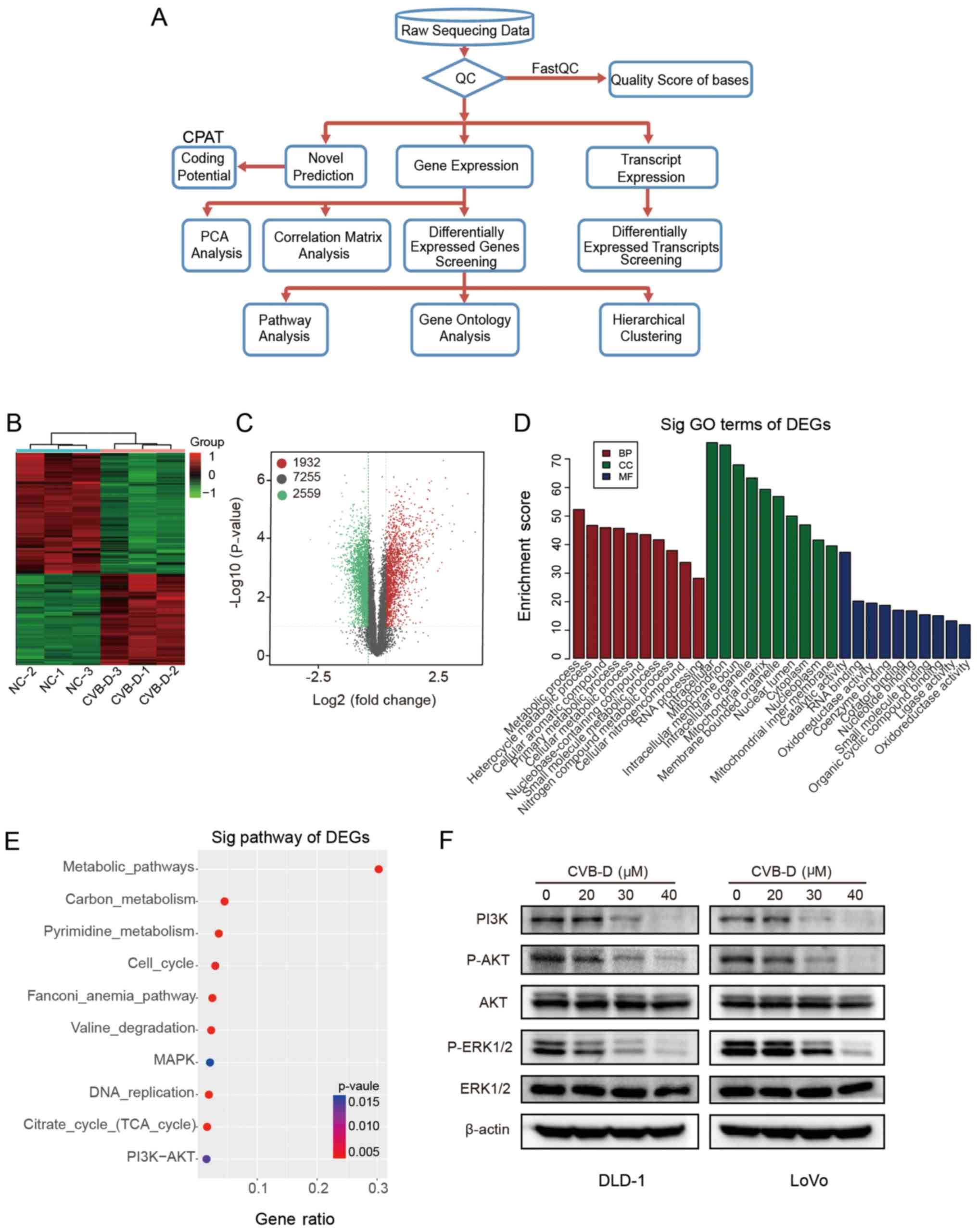 | Figure 5Analysis of RNA-seq data obtained
from DLD-1 cells treated with CVB-D. (A) Experimental flowchart of
RNA-seq. (B) Correlation of gene expression levels among samples.
(C) A heat map shows the hierarchical clustering of DEGs in DLD-1
cells following treatment with CVB-D. Upregulated and
downregu-lated genes are represented by red and green,
respectively. (D) A volcano plot shows the number of upregulated,
downregulated and non-differentially expressed genes. (E) GO
analysis obtained information regarding BP, CC and MF terms
associated with the downregulated DEGs (F) Western blot analysis
verified the expression of major proteins of the AKT/ERK signaling
pathway in CRC cells treated with CVB-D (0-40 µM, 48 h).
Data are presented as mean ± standard deviation (n=3). BP,
biological process; CC, cellular component; MF, molecular function;
RNA-seq, RNA sequencing; CVB-D, cyclovirobuxine D; DEG,
differentially expressed gene; GO, Gene Ontology; NC, negative
control; p-, phosphorylated; sig, significant. |
CTHRC1 is a therapeutic target of CRC
cells
By mining the GEPIA2 database, a total of 5,331 DEGs
related to COAD and 926 most differentially expressed survival
genes associated with the OS and/or disease-free survival (DFS)
rates of patients with COAD were identified. Using the Venny online
tool, 24 overlapping genes were found between the two
aforementioned groups of data and the downregulated genes revealed
by RNA-seq (Fig. 6A). Then,
potential targets were selected according to three conditions: i)
They were oncogenes; ii) they were overexpressed in COAD (Fig. S6); and iii) the overexpressed
oncogenes were accompanied by a decrease in OS and/or DFS (Figs. S7 and S8).
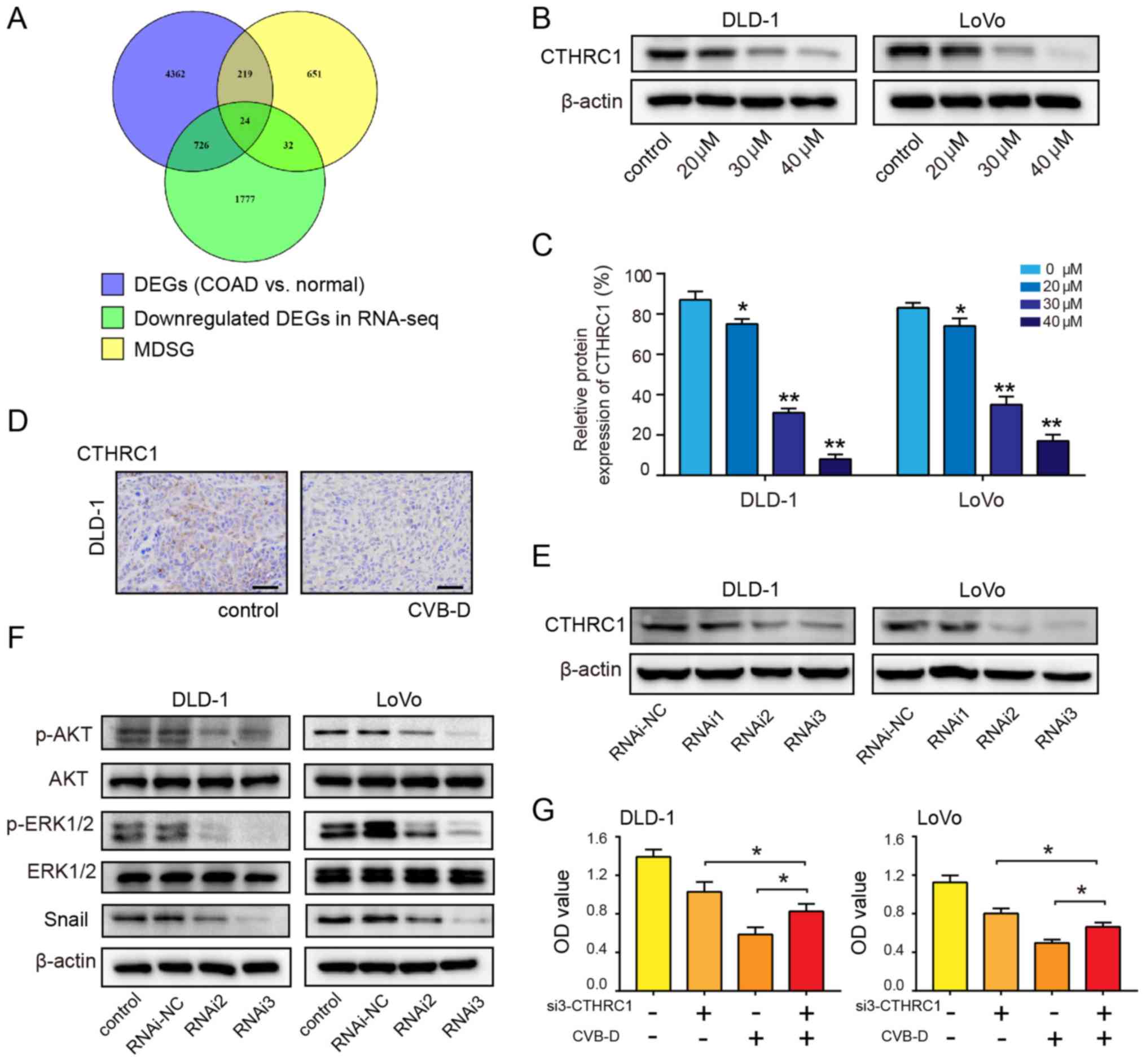 | Figure 6CTHRC1 as a therapeutic target of
CRC. (A) Venn diagram showing the number of potential targets in
three sets of data. (B and C) Western blot analysis was performed
to evaluate the expression of CTHRC1 in CRC cells following
treatment with CVB-D (0-40 µM) for 48 h. (D)
Immunohistochemical analysis of CTHRC1 expression after treatment
with CVB-D (15 mg/kg, once a day for a total of 4 weeks) in the
DLD-1 xenograft tumors (magnification, ×400; scale bar, 100
µM). (E) siRNA-mediated knockdown of CTHRC1 in CRC cells was
evaluated by western blot assay. (F) siRNA-mediated knockdown of
CTHRC1 in CRC cells resulted in reduced levels of p-AKT, p-ERK and
Snail. (G) CVB-D significantly reversed the effect of
CTHRC1-knockdown on the viability of CRC cell. Data are presented
as mean ± standard deviation (n=3). *P<0.05,
**P<0.01 vs. untreated control group. CVB-D,
cyclovirobuxine D; DEG, differentially expressed gene; CTHRC1,
collagen triple helix repeat containing 1; p-, phosphorylated; OD,
optical density; si, small interfering; CRC, colorectal cancer; NC,
negative control; MDSG, most differential survival genes that are
associated with the overall survival and/or disease-free survival
rates of patients. |
The remaining four genes, CTHRC1, C2orf70, NIFK and
COMT, were considered the most likely potential targets for CVB-D.
GEPIA2 was used to analyze the correlation between the four genes
(CTHRC1, COMT, C2orf70 and NIFK) and transcription factors. The
correlations were low between Snail and COMT (r=0.35,
P=1.1×10-10), Snail and C2orf70 (r=0.26,
P=2.4×10-6), and Snail and NIFK (r=0.38, P=1.6×10
-12). However, there was a significantly high
correlation between Snail and CTHRC1 (r=0.69;
P=3.1×10-45) (Fig.
S9A). In addition, the correlation between other transcription
factors and the four potential targets was also analyzed. COMT,
C2orf70 and NIFK had no or low correlation with Slug, ZEB1 and
ZEB2. Whereas, CTHRC1 had a certain degree of correlation with
other three transcription factors (CTHRC1 and Slug (r=0.57,
P=4.5×10-29); CTHRC1 and ZEB1 (r=0.43,
P=1×10-15); CTHRC1 and ZEB2 (r=0.37, P=8.7×10
-12) (Fig. S9). The
protein and mRNA expression analysis using the online databases
demonstrated that the expression of CTHRC1 was significantly higher
in COAD tissues compared with adjacent normal tissues (Fig. S10A and B). The CTHRC1 mRNA
expression in COAD ranked 13th highest among 31 types of tumors
(Fig. S10C). Patient survival
analysis revealed that overexpression of CTHRC1 predicted poor OS
and DFS rates in COAD (Fig.
S10D). Western blotting reveled that the expression of CTHRC1
in DLD-1 and LoVo cells decreased in a dose-dependent manner
following treatment with CVB-D (Fig.
6B and C). IHC results demonstrated that CTHRC1 expression in
the CVB-D group was lower compared with the control group (Fig. 6D). RNAi2 and RNAi3 effectively
downregulated the expression of CTHRC1 (Figs. 6E and S11). Western blot analysis demonstrated
that the ratios of p-AKT/AKT and p-ERK/ERK significantly decreased
after siRNA-mediated CTHRC1-knockdown; the expression level of
Snail was also decreased after siRNA-mediated CTHRC1-knockdown
(Figs. 6F and S12), indicating that CTHRC1 may be
located upstream of p-AKT, p-ERK and Snail. In the siRNA3 rescue
experiment, siRNA3-mediated suppression of CTHRC1 in CRC cells
resulted in a partial reversal of CVB-D-induced growth inhibition
(Fig. 6G). These results suggest
that the growth of CRC cells inhibited by CVB-D is mediated by a
CTHRC1-dependent mechanism.
CVB-D inhibition on the growth of CRC
xenograft tumors in nude mice
The DLD-1 xenograft model was established in BALB/C
nude mice to evaluate the antitumor ability of CVB-D. After 4
weeks, tumor weight and size in the CVB-D treatment group were
significantly smaller compared with the control group (Fig. 7A-C). The expression levels of Ki67,
cyclin D1, Bcl-2, HIF1A and N-cadherin were decreased in the CVB-D
treatment compared with the control group, as detected by IHC
(Fig. 7E).
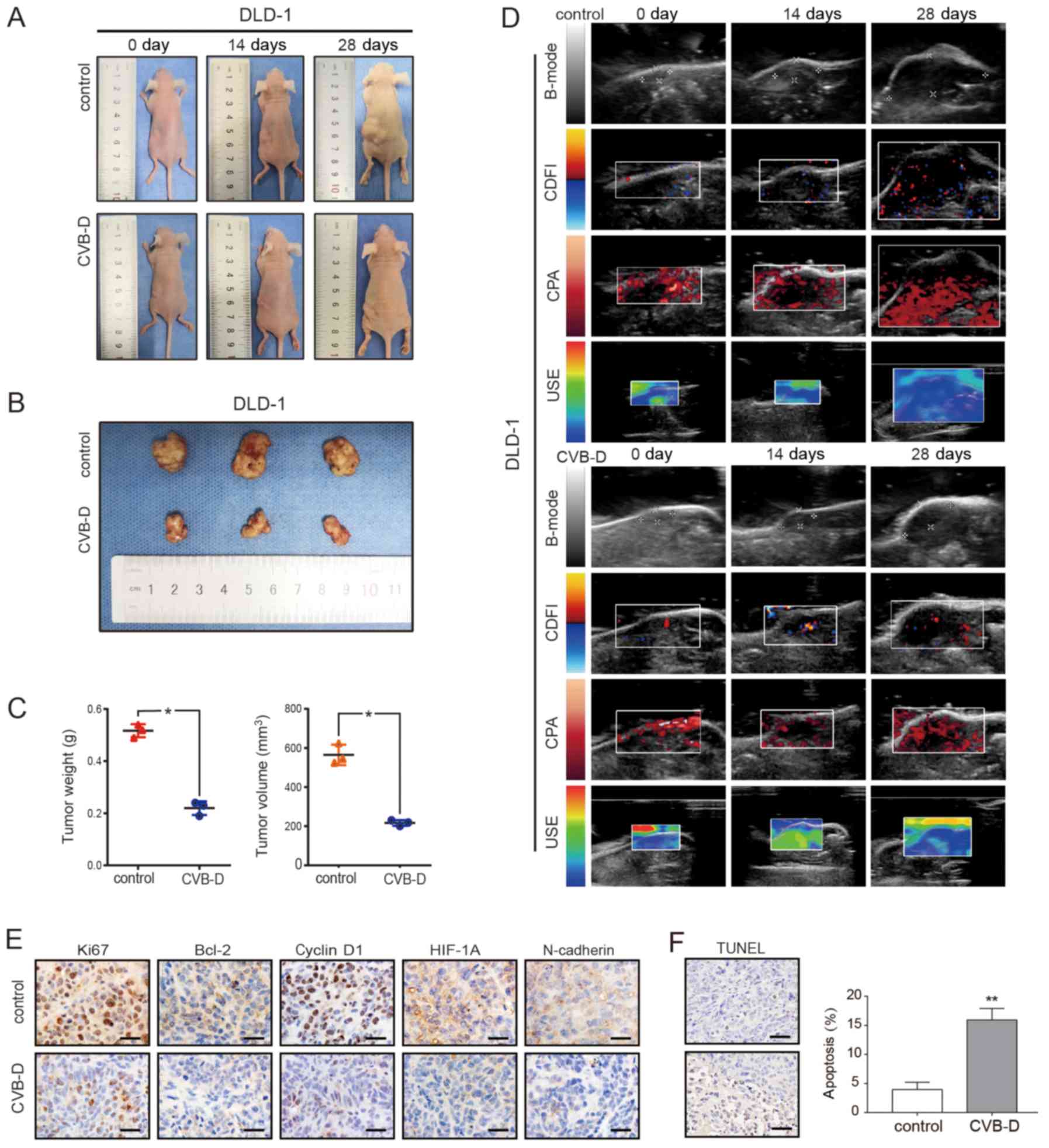 | Figure 7Inhibitory effect of CVB-D on DLD-1
xenografts in BALB/c nude mice. (A-C) The xenograft tumor model of
DLD-1 cells in nude mice was established and then treated with
CVB-D (15 mg/kg/day for 4 weeks). All mice were observed, and the
tumor weight and volume were measured. (D) B-mode, CDFI, CPA and
USE of ultrasonography were used to evaluate xenografts in the
treatment and control groups. (E) The expressions of Ki67, Bcl-2,
cyclin D1, HIF-1α and N-cadherin were detected by
immunohistochemical staining (original magnification, ×400; scale
bar, 100 µM), (F) A TUNEL assay was used to detect apoptosis
in pathological tissues. Data are presented as mean ± standard
deviation (n=3). *P<0.05 and **P<0.01.
B-mode, B-ultrasound; CDFI, color Doppler flow imaging; CPA, color
power angiography; USE, ultrasonic elastosonography; CVB-D,
cyclovirobuxine D; HIF-1α, human inducible factor-1α. |
In addition to dynamically observing the growth of
the transplanted tumor and comparing the size of the excised tumor
after execution, ultrasonic imaging (USI) was employed to
dynamically detect the tumor indicators. USI can continuously and
dynamically scan the lesions, and can show stereoscopic changes,
which are not limited by image stratification, and does not damage
the nude mice.
USI can also be evaluated systematically with
multi-parameters such as Doppler and elastography (43). The ultrasound scan mode in this
experiment included B-mode, CDFI, CPA and USE. The results
demonstrated that tumor growth slowed after CVB-D treatment;
angiogenesis decreased, especially microvascular; the elasticity
score, which is highly related to tumor malignancy, decreased; and
hardness of tumor decreased (Fig.
7D). The H&E staining result revealed that the number of
blood vessels in the experimental group was decreased after CVB-D
treatment (Fig. S13). H&E
staining demonstrated that there were no obvious morphological
changes in the heart, liver, spleen and lung (Fig. S14). The apoptosis rate of
xenograft tumor cells, as detected by the TUNEL assay, was 15.91%
in the CVB-D treatment group and 3.96% in the control group,
demonstrating the antitumor effects of CVB-D (Fig. 7F).
CVB-D chemical structure and the
inhibitory mechanism of action in CRC
The chemical and 3D structure of CVB-D is presented
in Fig. 8A. A schematic
presentation of the mechanism underlying CVB-D-induced inhibition
of CRC and the relevant molecular mechanism associated with the
AKT/ERK-Snail signaling pathway is presented in Fig. 8B.
Discussion
It has been reported that NPCs exhibit anticancer,
anti-inflammatory and anti-fibrotic effects, which inhibits
pathological EMT through different cell signal transduction
pathways (10). In the present
study, the expression of epithelial marker E-cadherin increased
following CVB-D treatment, while the mesenchymal markers N-cadherin
and vimentin decreased. Therefore, it was speculated that CVB-D can
prevent EMT. In addition, the expression levels of the stemness
marker CD133, invasion and metastasis related markers MMP2 and
MMP9, and following CVB-D treatment as detected by western
blotting. Subsequently, the expression of transcription factors,
which have been demonstrated to be involved in the upstream
regulation of EMT, was investigated. The consistent and significant
inhibitory effect of CVB-D on Snail expression in both types of CRC
cells suggests that Snail may be the optimum candidate among EMT
regulatory factors. Notably, database analysis identified that the
expression of Snail is higher in COAD tissues compared with
paracancerous tissues, further supporting Snail as the optimum EMT
regulator candidate. Additionally, high Snail expression is
associated with shorter survival time and promotes the occurrence
and development of CRC (44).
These results suggest that CVB-D may suppress EMT by regulating the
expression of Snail.
RNA-seq techniques have been used for the study of
numerous diseases, such as bladder, gastric, liver, cervical, colon
and breast cancers (45-50). RNA-seq is an effective tool for
sequencing transcripts using high-throughput sequencing technology.
The complete mechanism of drug action may be obtained by analyzing
RNA-seq data, such as the potential targets of drug action and
oncogenic signaling pathways (51). Generally, antitumor agents
typically inhibit oncogenic signaling pathways; therefore, the
present study focused on the downregulated signaling pathways
revealed by RNA-seq. According to the RNA-seq data, each pathway
corresponded to a statistical P-value to indicate significance. The
smaller the P-value, the more significant the association between
the differential genome and the signaling pathway. The P-value of
the metabolic signaling pathway was lowest in the downregulated
signaling pathways; however, the purpose of this experiment was to
determine the association between drugs and oncogenic signaling
pathways; therefore, metabolic signaling pathways were removed. The
oncogenic signaling pathways ranked high, i.e. classical MAPK and
PI3K-AKT pathways, were given primary attention in the
downregulated signaling pathways revealed by RNA-seq, which were
demonstrated to be important signaling pathways in CRC (52). Activation of the PI3K-AKT signaling
pathway regulates multiple biological processes, such as promoting
invasion and metastasis of CRC (53). Abnormal activation of the
RAS/Raf/MRK/ERK signaling pathway promotes the progression of colon
cancer and has been identified as a novel target for tumor therapy
(54). Zhao et al (55) demonstrated that CAPS1 accelerated
CRC metastasis via the EMT process mediated by the PI3K/AKT/Snail
signaling pathway, and also confirmed that Snail silencing can
attenuate the CASP1 overexpression-induced migration and invasion
of SW480 cells. Zhu et al (56) demonstrated that ZC3H13 inhibits
proliferation and invasion of CRC via the Ras-ERK-Snail signaling
pathway. These studies indicate that these two signaling pathways
play an important role in CRC and can serve a role upstream of
Snail. Western blotting confirmed that CVB-D plays an anticancer
role by downregulating the AKT/ERK pathway.
It was hypothesized that drug-target genes may be
differentially expressed in COAD. By mining the GEPIA2 database, a
total of 5,331 DEGs in COAD and 926 MDSG were identified. Using the
Venny online tool, 24 overlapping genes were found between the two
aforementioned groups of data and the downregulated genes revealed
by RNA-seq. Four oncogenes, including CTHRC1, C2orf70, NIFK and
COMT, were further selected because they were overexpressed in COAD
and associated with poor OS and/or DFS, which were considered the
most likely potential targets for CVB-D. At the start of the
present study, it was identified that of the EMT regulatory factors
analyzed, Snail decreased most notably following CVB-D treatment;
therefore, correlations among the four genes (CTHRC1, COMT, C2orf70
and NIFK) and Snail were analyzed in GEPIA2. Accordingly, a high
correlation between Snail and CTHRC1 was identified. Therefore,
CTHRC1 was selected as the potential therapeutic target of CVB-D
for further experimental study.
CTHRC1 has been reported to be an oncogene in
previous studies (21-27), and it can promote tumorigenesis by
multiple mechanisms. It has been confirmed that CTHRC1 can activate
the SRC and ERK signal cascades and upregulate the expression of
MMP9, thus promoting the invasion of CRC cells (22). Overexpression of CTHRC1 has been
found to promote the occurrence and development of cervical cancer
by activating the Wnt/PCP oncogenic signaling pathway (57). CTHRC1 induces EMT changes and MMP
expression through the PI3K/AKT/ERK/CREB signaling pathway to
promote the invasiveness and metastasis of hepatocellular carcinoma
(58). In addition, there is a
close relationship between CTHRC1 and EMT. Ni et al
(21) demonstrated that CTHRC1 can
promote epithelial cell metastasis by inducing the EMT in CRC
cells. In a study by Kim et al (22), CTHRC1-knockdown prevented EMT and
inhibited the proliferation of renal cell carcinoma. Liu et
al (59) reported that
CTHRC1-knockdown inhibits EMT and cell migration in glioblastoma
cells. Notably, CTHRC1 can be used as a prognostic indicator for
numerous tumors. Gu et al (60) found CTHRC1 overexpression in a
large proportion of patients with gastric cancer, and the high
expression was associated with gastric cancer progression and poor
prognosis. Kaplan-Meier analysis by Hou et al (61) revealed that patients with
epithelial ovarian cancer with high CTHRC1 expression had
significantly shorter OS times. Multivariate cox analysis
demonstrated that for patients with CRC, CTHRC1 could be used as an
independent prognostic factor for OS (21).
In the present study, the inhibitory effect of CVB-D
on the expression of CTHRC1 protein in CRC was confirmed by western
blotting with cell lines and IHC of the subcutaneous transplanted
tumors in nude mice. Then, CTHRC1-knockdown experiments were
performed. Following siRNA-mediated knockdown, the levels of p-AKT
and p-ERK were decreased, indicating that CTHRC1 is located
upstream of the AKT/ERK pathway. Finally, CTHRC1-knockdown cell
lines were treated with CVB-D for rescue experiments. CVB-D alone
was superior to si-CTHRC1 alone in inhibiting cell viability, and
the addition of CVB-D to CRC cells pretreated with CTHRC1 siRNA did
not cause further inhibition. Therefore, the present experiments
demonstrated that the anticancer effect of CVB-D was predominantly
achieved by inhibiting the CTHRC1-AKT/ERK-Snail signal pathway.
In the past, oral or intravenous administration of
traditional chemotherapy has struggled to increase local drug
concentration in tumors, leading to low drug utilization and poor
therapeutic effectiveness (62).
The rapid development of scientific progress has resulted in the
development of precise and complex nanoscale devices, which can
actively drive drugs to the target at the molecular level (63). This will greatly increase the local
blood concentration, further reduce toxicity and side effects, and
effectively amplify multi-target anticancer mechanisms of
drugs.
There were several limitations of the present study.
First, due to ethical limitations, the sample size of the animal
experiment was small; however, the results were consistent. Second,
there were differences in CTHRC1-knockdown efficiency between the
two siRNAs, which may have been due to off-target effects.
In conclusion, as an alkaloid chemotherapeutic drug,
CVB-D was shown to inhibit the viability, migration, invasion and
stemness of CRC cells, and induce apoptosis and cell cycle arrest.
Through RNA-seq analysis, it was further confirmed that CVB-D can
inhibit the tumorigenesis of CRC via the CTHRC1-AKT/ERK-Snail
signaling pathway.
Supplementary Data
Funding
This work was supported by the Scientific Planning
Project of Heilongjiang Province (grant. no. 201713) and the
Innovation Project of Harbin Medical University (grant. no.
YJSKYCX2018-37HYD).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FJ, YC and DP conceived and designed the study. FJ,
SR and YC performed the experiments. YZ, KS and YX performed the
pathology experiments. ZL performed ultrasonic imaging experiments.
FJ wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal studies were approved by the Ethics
Committee of the First Affiliated Hospital of Harbin Medical
University (Heilongjiang, China) and were conducted according to
the national regulations in China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
CVB-D
|
cyclovirobuxine D
|
|
CTHRC1
|
collagen triple helix repeat
containing 1
|
|
COAD
|
colon adenocarcinoma
|
|
CRC
|
colorectal cancer
|
|
NPCs
|
natural plant compounds
|
|
EMT
|
epithelial-mesenchymal transition
|
|
DEGs
|
differentially expressed genes
|
|
OS
|
overall survival
|
|
DFS
|
disease-free survival
|
|
CPA
|
color power angiography
|
|
CFI
|
color Doppler flow imaging
|
|
USE
|
ultrasonic elastosonography
|
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Liu Z, Bai Y, Xie F, Miao F and Du F:
Comprehensive analysis for identifying diagnostic and prognostic
biomarkers in colon adeno-carcinoma. DNA Cell Biol. Feb 7–2020.Epub
ahead of print. View Article : Google Scholar
|
|
3
|
Rasool S, Kadla SA, Rasool V and Ganai BA:
A comparative overview of general risk factors associated with the
incidence of colorectal cancer. Tumour Biol. 34:2469–2476. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van Cutsem E and Nordlinger B: Advanced
colorectal cancer: ESMO clinical practice guidelines for treatment.
Ann Oncol. 21(Suppl 5): v93–v97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen C, Xu ZQ, Zong YP, Ou BC, Shen XH,
Feng H, Zheng MH, Zhao JK and Lu AG: CXCL5 induces tumor
angiogenesis via enhancing the expression of FOXD1 mediated by the
AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 10:1782019.
View Article : Google Scholar
|
|
6
|
Tomeh MA, Hadianamrei R and Zhao X: A
review of curcumin and its derivatives as anticancer agents. Int J
Mol Sci. 20:E10332019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee SR, Jin H, Kim WT, Kim WJ, Kim SZ,
Leem SH and Kim SM: Tristetraprolin activation by resveratrol
inhibits the proliferation and metastasis of colorectal cancer
cells. Int J Oncol. 53:1269–1278. 2018.PubMed/NCBI
|
|
8
|
Wang JY, Wang Z, Li MY, Zhang Z, Mi C, Zuo
HX, Xing Y, Wu YL, Lian LH, Xu GH, et al: Dictamnine promotes
apoptosis and inhibits epithelial-mesenchymal transition,
migration, invasion and proliferation by downregulating the HIF-1α
and Slug signaling pathways. Chem Biol Interact. 296:134–144. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boldbaatar A, Lee S, Han S, Jeong AL, Ka
HI, Buyanravjikh S, Lee JH, Lim JS, Lee M and Yang Y: Eupatolide
inhibits the TGF-β1-induced migration of breast cancer cells via
downregulation of SMAD3 phosphorylation and transcriptional
repression of ALK5. Oncol Lett. 14:6031–6039. 2017.PubMed/NCBI
|
|
10
|
Avila-Carrasco L, Majano P, Sánchez-Toméro
JA, Selgas R, López-Cabrera M, Aguilera A and González Mateo G:
Natural plants compounds as modulators of epithelial-to-mesenchymal
transition. Front Pharmacol. 10:7152019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu B, Fang TH, Lü GH, Xu HQ and Lu JF:
Beneficial effect of Cyclovirobuxine D on heart failure rats
following myocardial infarction. Fitoterapia. 82:868–877. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu J, Sun D, Gao S, Gao Y, Ye J and Liu P:
Cyclovirobuxine D induces autophagy-associated cell death via the
Akt/mTOR pathway in MCF-7 human breast cancer cells. J Pharmacol
Sci. 125:74–82. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu J, Tan Z, Chen J and Dong C:
Cyclovirobuxine D inhibits cell proliferation and induces
mitochondria-mediated apoptosis in human gastric cancer cells.
Molecules. 20:20659–20668. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21(Suppl 7): vii89–vii92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kong D, Zhang F, Shao J, Wu L, Zhang X,
Chen L, Lu Y and Zheng S: Curcumin inhibits cobalt chloride-induced
epithelial-to-mesenchymal transition associated with interference
with TGF-β/Smad signaling in hepatocytes. Lab Invest. 95:1234–1245.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Z, Chen Y, An T, Liu P, Zhu J, Yang H,
Zhang W, Dong T, Jiang J, Zhang Y, et al: Nuciferine inhibits the
progression of glioblastoma by suppressing the SOX2-AKT/STAT3-Slug
signaling pathway. J Exp Clin Cancer Res. 38:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao H, Xu E, Liu H, Wan L and Lai M:
Epithelial-mesenchymal transition in colorectal cancer metastasis:
A system review. Pathol Res Pract. 211:557–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barr S, Thomson S, Buck E, Russo S, Petti
F, Sujka-Kwok I, Eyzaguirre A, Rosenfeld-Franklin M, Gibson NW,
Miglarese M, et al: Bypassing cellular EGF receptor dependence
through epithelial-to-mesenchymal-like transitions. Clin Exp
Metastasis. 25:685–693. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pyagay P, Heroult M, Wang Q, Lehnert W,
Belden J, Liaw L, Friesel RE and Lindner V: Collagen triple helix
repeat containing 1, a novel secreted protein in injured and
diseased arteries, inhibits collagen expression and promotes cell
migration. Circ Res. 96:261–268. 2005. View Article : Google Scholar
|
|
21
|
Ni S, Ren F, Xu M, Tan C, Weng W, Huang Z,
Sheng W and Huang D: CTHRC1 overexpression predicts poor survival
and enhances epithelial-mesenchymal transition in colorectal
cancer. Cancer Med. 7:5643–5654. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim HC, Kim YS, Oh HW, Kim K, Oh SS, Kim
JT, Kim BY, Lee SJ, Choe YK, Kim DH, et al: Collagen triple helix
repeat containing 1 (CTHRC1) acts via ERK-dependent induction of
MMP9 to promote invasion of colorectal cancer cells. Oncotarget.
5:519–529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang L, Dai DL, Su M, Martinka M, Li G and
Zhou Y: Aberrant expression of collagen triple helix repeat
containing 1 in human solid cancers. Clin Cancer Res. 12:3716–3722.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang G and Li G: Novel multiple markers
to distinguish melanoma from dysplastic nevi. PLoS One.
7:e450372012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu G, Sengupta PK, Jamal B, Yang HY,
Bouchie MP, Lindner V, Varelas X and Kukuruzinska MA:
N-glycosylation induces the CTHRC1 protein and drives oral cancer
cell migration. J Biol Chem. 288:20217–20227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park EH, Kim S, Jo JY, Kim SJ, Hwang Y,
Kim JM, Song SY, Lee DK and Koh SS: Collagen triple helix repeat
containing-1 promotes pancreatic cancer progression by regulating
migration and adhesion of tumor cells. Carcinogenesis. 34:694–702.
2013. View Article : Google Scholar
|
|
27
|
Chen YL, Wang TH, Hsu HC, Yuan RH and Jeng
YM: Overexpression of CTHRC1 in hepatocellular carcinoma promotes
tumor invasion and predicts poor prognosis. PLoS One. 8:e703242013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Erhardt W, Hebestedt A, Aschenbrenner G,
Pichotka B and Blümel G: A comparative study with various
anesthetics in mice (Pentobarbitone, Ketamine-Xylazine,
Carfentanyl-Etomidate). Res Exp Med (Berl). 184:159–169. 1984.
View Article : Google Scholar
|
|
29
|
Aghajani M, Mansoori B, Mohammadi A,
Asadzadeh Z and Baradaran B: New emerging roles of CD133 in cancer
stem cell: Signaling pathway and miRNA regulation. J Cell Physiol.
234:21642–21661. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lim SH, Jang J, Park JO, Kim KM, Kim ST,
Park YS, Lee J and Kim HC: CD133-positive tumor cell content is a
predictor of early recurrence in colorectal cancer. J Gastrointest
Oncol. 5:447–456. 2014.PubMed/NCBI
|
|
31
|
Kang M, Kim S and Ko J: Roles of CD133 in
microvesicle formation and oncoprotein trafficking in colon cancer.
FASEB J. 33:4248–4260. 2019. View Article : Google Scholar
|
|
32
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in
cancer. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McKay JA, Douglas JJ, Ross VG, Curran S,
Murray GI, Cassidy J and McLeod HL: Cyclin D1 protein expression
and gene polymorphism in colorectal cancer. Aberdeen colorectal
initiative. Int J Cancer. 88:77–81. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qie S and Diehl JA: Cyclin D1, cancer
progression, and opportunities in cancer treatment. J Mol Med
(Berl). 94:1313–1326. 2016. View Article : Google Scholar
|
|
35
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Y, Zhuang Z, Meng Q, Jiao Y, Xu J
and Fan S: Polydatin inhibits growth of lung cancer cells by
inducing apoptosis and causing cell cycle arrest. Oncol Lett.
7:295–301. 2014. View Article : Google Scholar
|
|
37
|
Plati J, Bucur O and Khosravi-Far R:
Apoptotic cell signaling in cancer progression and therapy. Integr
Biol (Camb). 3:279–296. 2011. View Article : Google Scholar
|
|
38
|
Liu F, Wang B, Wang J, Ling X, Li Q, Meng
W and Ma J: Oxymatrine inhibits proliferation and migration while
inducing apoptosis in human glioblastoma cells. Biomed Res Int.
2016:17841612016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Y, Zhao Z, Chen Y, Lv Z, Ding X, Wang
R, Xiao H, Hou C, Shen B, Feng J, et al: An
epithelial-to-mesenchymal transition-inducing potential of
granulocyte macrophage colony-stimulating factor in colon cancer.
Sci Rep. 7:82652017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim BR, Kang MH, Kim JL, Na YJ, Park SH,
Lee SI, Kang S, Joung SY, Lee SY, Lee DH, et al: RUNX3 inhibits the
metastasis and angiogenesis of colorectal cancer. Oncol Rep.
36:2601–2608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zucker S and Vacirca J: Role of matrix
metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis
Rev. 23:101–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhu J and Thompson CB: Metabolic
regulation of cell growth and proliferation. Nat Rev Mol Cell Biol.
20:436–450. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen Y, Liu P, Sun P, Jiang J, Zhu Y, Dong
T, Cui Y, Tian Y, An T, Zhang J, et al: Oncogenic MSH6-CXCR4-TGFB1
feedback loop: A novel therapeutic target of photothermal therapy
in glioblastoma multiforme. Theranostics. 9:1453–1473. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tanaka S, Kobayashi W, Haraguchi M,
Ishihata K, Nakamura N and Ozawa M: Snail1 expression in human
colon cancer DLD-1 cells confers invasive properties without
N-cadherin expression. Biochem Biophys Rep. 8:120–126. 2016.
|
|
45
|
Chen JB, Zhu YW, Guo X, Yu C, Liu PH, Li
C, Hu J, Li HH, Liu LF, Chen MF, et al: Microarray expression
profiles analysis revealed lncRNA OXCT1-AS1 promoted bladder cancer
cell aggressiveness via miR-455-5p/JAK1 signaling. J Cell Physiol.
234:13592–13601. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zheng R, Liang J, Lu J, Li S, Zhang G,
Wang X, Liu M, Wang W, Chu H, Tao G, et al: Genome-wide long
non-coding RNAs identified a panel of novel plasma biomarkers for
gastric cancer diagnosis. Gastric Cancer. 22:731–741. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yin L, Chen Y, Zhou Y, Deng G, Han Y, Guo
C, Li Y, Zeng S and Shen H: Increased long noncoding RNA LASP1-AS
is critical for hepatocellular carcinoma tumorigenesis via
upregulating LASP1. J Cell Physiol. 234:13493–13509. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li YJ, Wang Y and Wang YY: MicroRNA-99b
suppresses human cervical cancer cell activity by inhibiting the
PI3K/AKT/mTOR signaling pathway. J Cell Physiol. 234:9577–9591.
2019. View Article : Google Scholar
|
|
49
|
Yu X, Yuan Z, Yang Z, Chen D, Kim T, Cui
Y, Luo Q, Liu Z, Yang Z, Fan X, et al: The novel long noncoding RNA
u50535 promotes colorectal cancer growth and metastasis by
regulating CCL20. Cell Death Dis. 9:7512018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li Y, Zhang H, Zhao Y, Wang C, Cheng Z,
Tang L, Gao Y, Liu F, Li J, Li Y, et al: A mandatory role of
nuclear PAK4-LIFR axis in breast-to-bone metastasis of ERα-positive
breast cancer cells. Oncogene. 38:808–821. 2019. View Article : Google Scholar
|
|
51
|
Du ZH, Bi FF, Wang L and Yang Q:
Next-generation sequencing unravels extensive genetic alteration in
recurrent ovarian cancer and unique genetic changes in
drug-resistant recurrent ovarian cancer. Mol Genet Genomic Med.
6:638–647. 2018. View Article : Google Scholar :
|
|
52
|
Koveitypour Z, Panahi F, Vakilian M,
Peymani M, Seyed Forootan F, Nasr Esfahani MH and Ghaedi K:
Signaling pathways involved in colorectal cancer progression. Cell
Biosci. 9:972019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Suman S, Kurisetty V, Das TP, Vadodkar A,
Ramos G, Lakshmanaswamy R and Damodaran C: Activation of AKT
signaling promotes epithelial-mesenchymal transition and tumor
growth in colorectal cancer cells. Mol Carcinog. 53(Suppl 1):
E151–E160. 2014. View Article : Google Scholar
|
|
54
|
Yang X, Zheng YT and Rong W: Sevoflurane
induces apoptosis and inhibits the growth and motility of colon
cancer in vitro and in vivo via inactivating Ras/Raf/MEK/ERK
signaling. Life Sci. 239:1169162019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao GX, Ying YX, Weng SQ, Zhang S, Chen
Y, Shen XZ, Dong L and Chen S: CAPS1 promotes colorectal cancer
metastasis via snail mediated epithelial mesenchymal
transformation. Oncogene. 38:4574–4589. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhu D, Zhou J, Zhao J, Jiang G, Zhang X,
Zhang Y and Dong M: ZC3H13 suppresses colorectal cancer
proliferation and invasion via inactivating Ras-ERK signaling. J
Cell Physiol. 234:8899–8907. 2019. View Article : Google Scholar
|
|
57
|
Zheng M, Zhou Q, Liu X, Wang C and Liu G:
CTHRC1 overexpression promotes cervical carcinoma progression by
activating the Wnt/PCP signaling pathway. Oncol Rep. 41:1531–1538.
2019.PubMed/NCBI
|
|
58
|
Wang Y, Lee M, Yu G, Lee H, Han X and Kim
D: CTHRC1 activates pro-tumorigenic signaling pathways in
hepatocellular carcinoma. Oncotarget. 8:105238–105250. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu J, Li W, Liu S, Zheng X, Shi L, Zhang
W and Yang H: Knockdown of collagen triple helix repeat containing
1 (CTHRC1) inhibits epithelial-mesenchymal transition and cellular
migration in glioblastoma cells. Oncol Res. 25:225–232. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gu L, Liu L, Zhong L, Bai Y, Sui H, Wei X,
Zhang W, Huang P, Gao D, Kong Y and Lou G: Cthrc1 overexpression is
an independent prognostic marker in gastric cancer. Hum Pathol.
45:1031–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hou M, Cheng Z, Shen H, He S, Li Y, Pan Y,
Feng C, Chen X, Zhang Y, Lin M, et al: High expression of CTHRC1
promotes EMT of epithelial ovarian cancer (EOC) and is associated
with poor prognosis. Oncotarget. 6:35813–35829. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Abdifetah O and Na-Bangchang K:
Pharmacokinetic studies of nanoparticles as a delivery system for
conventional drugs and herb-derived compounds for cancer therapy: A
systematic review. Int J Nanomedicine. 14:5659–5677. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li C, Cai G, Song D, Gao R, Teng P, Zhou
L, Ji Q, Sui H, Cai J, Li Q and Wang Y: Development of
EGFR-targeted evodiamine nanoparticles for the treatment of
colorectal cancer. Biomater Sci. 7:3627–3639. 2019. View Article : Google Scholar : PubMed/NCBI
|