Introduction
At present, >400,000 patients worldwide are newly
diagnosed with bladder cancer (BCa) each year (1). Among them, ~75% have non-muscle
invasive BCa (NMIBC) and 25% have muscle invasive bladder cancer
(MIBC), at the time of presentation (2). NMIBC is not immediately
life-threatening, but its propensity for recurrence leads to costly
clinical surveillance throughout life. The 5-year progression rate
of NMIBC is 10-30%; however, once progression occurs, the
cancer-specific survival rate falls to ~35% (3,4).
Thus, it is important to better understand the mechanisms
underlying BCa progression and to identify practical biomarkers for
early diagnosis, treatment and prognosis (5). In our previous study, next
generation sequencing and microRNA (miRNA/miR) microarray assays
were used to identify several differentially expressed miRNAs and
their target genes in BCa (6).
Among them, the expression levels of genes encoding collagen type
VI-α1 and 2 (COL6A1 and 2) were downregulated, while the expression
levels of miR-6124 and miR-4651 were upregulated in
BCa compared with in normal controls (6). Additionally, the expression of
cell-free miR-6124 in BCa urine was higher than that in
non-cancer patients with hematuria, indicating that urinary
miR-6124 may be a promising non-invasive diagnostic
biomarker for BCa (7).
The interaction between tumor cells and the
microenvironment can affect disease initiation and progression
(8). The extracellular matrix
(ECM) is an essential part of the tumor microenvironment (TME),
which interacts with cancer cells at each step of the metastatic
process (9). Collagen is the
major component of the ECM, and dysregulation of collagen is
associated with increased malignancy (10,11). COL6 is a major component of the
bladder ECM (12). In lung,
pancreatic, breast and ovarian cancer, COL6A1 and
COL6A2 act as oncogenes (13). In particular, COL6A1
expression is associated with a poor prognosis in some types of
cancer, including kidney, prostate, lung and cervical cancer
(14-17), and COL6A2 protein expression is
suppressed in UPII-SV40Tag mice (a model of invasive BCa) (18). A weighted gene co-expression
network analysis and protein-protein interaction network analysis
demonstrated that increased expression levels of six potential
biomarkers (COL3A1, fibronectin 1, COL5A1, fibrillin 1, COL6A1 and
thrombospondin 2) were significantly associated with a worse
overall survival in patients with BCa (19). In addition, a bioinformatic
analysis using three GSE datasets identified six collagen members
(COL1A1, COL1A2, COL5A2, COL6A1, COL6A2 and COL6A3) that were
upregulated in BCa, which all resided in the ECM-receptor
interaction signaling pathway (20). However, although COL6A1 and COL6A2
serve diverse roles in different types of cancer, the exact
functions of COL6A1 and COL6A2 in the pathogenesis and progression
of BCa remain unclear.
The present study examined the expression levels of
COL6A1 and COL6A2 in BCa using reverse
transcription-quantitative (RT-q) PCR. Additionally, the molecular
signaling cascades by which COL6A1 and COL6A2 may suppress
proliferation, migration and invasion of human BCa EJ cells
(MGH-U1) were investigated. To do this, the current study analyzed
different signaling pathways, cell cycle modulation and
transcription factors that regulate matrix metalloproteinase
(MMP)-2 and MMP-9.
Materials and methods
Patients and tissue samples
Table I lists the
baseline characteristics of the study subjects. A total of 237
bladder tissue samples were obtained from the National Biobank of
Korea at Chungbuk National University Hospital (Cheongju, South
Korea). Among these, 140 samples were from patients with primary
BCa (age range, 24-89 years) and were verified histologically as
transitional cell carcinoma. The remaining 97 samples (used as the
control set) comprised normal bladder mucosa samples (n=37), which
were taken from patients with continuous hematuria who were
suspicious of BCa (the final diagnoses were benign diseases, mostly
stress urinary incontinence) or normal tissues from the area
surrounding BCa (n=60); among them, 36 were obtained from the
aforementioned patients with BCa and 24 were from patients with BCa
not included in the present study (3 cm from tumor; age range,
19-89 years). To reduce the chances of confounding factors
affecting the analyses, patients diagnosed with concomitant
carcinoma in situ or carcinoma in situ lesions alone
were excluded. Voided urine cytology was tested before surgical
treatment to assist BCa diagnosis and/or prognosis. Fresh-frozen
specimens were obtained during surgical resection of transitional
cell carcinoma at Chungbuk National University Hospital (Cheongju,
South Korea) between April 2000 and October 2010. All tumors were
macro-dissected, typically within 15 min of surgical resection.
Each specimen was confirmed by pathological analysis of a part of
fresh-frozen specimens obtained from radical cystectomy and
transurethral resection of the bladder tumor. Tumors were staged
(2002 TNM Classification) and graded (2004 World Health
Organization Classification), according to standard criteria
(21). In cases of NMIBC (n=97),
transurethral resection (TUR) of the tumor was performed. If
incomplete, or when a high-grade or T1 tumor was detected, a second
TUR was performed 2-4 weeks after the initial resection. Patients
with intermediate- or high-risk NMIBC received one cycle of
intravesical Bacillus Calmette-Guérin immunotherapy. In cases of
MIBC (n=43), radical cystectomy and complete pelvic lymph node
dissection were performed. Patients with pT3 or pT4, or
node-positive disease (based on analysis of radical cystectomy
specimens), received at least four cycles of cisplatin-based
chemotherapy. Neither clinically metastatic disease, nor
non-cystectomy cases were excluded from the study. Each patient was
followed and managed according to standard recommendations
(22,23). Recurrence was defined as
recurrence of primary NMIBC of the same pathological stage.
Progression of NMIBC or MIBC was defined as progression of TNM
stage after disease relapse. The mean follow-up period for patients
with NMIBC was 72.95 months (range, 3.2-172.2 months). The mean
follow-up period for patients with MIBC was 36.18 months (range,
3.0-141.4 months). Collection and analysis of all samples were
approved by the Institutional Review Board of Chungbuk National
University (approval no. GR2010-12-010), and written informed
consent was obtained from each subject. The study methodologies
conformed to the standards set by the Declaration of Helsinki.
 | Table IClinicopathological features of
tissues from 140 patients with primary bladder cancer (97 with
NMIBC and 43 with MIBC) and 97 controls (surrounding normal tissues
and normal bladder mucosae). |
Table I
Clinicopathological features of
tissues from 140 patients with primary bladder cancer (97 with
NMIBC and 43 with MIBC) and 97 controls (surrounding normal tissues
and normal bladder mucosae).
| Variable | Bladder cancer
| Control | P-value |
|---|
| NMIBC | MIBC |
|---|
| Mean age ± SD,
years | 63.45±13.79 | 67.60±9.84 | 61.98±14.32 | 0.083a |
| Sex, n (%) | | | | 0.975b |
| Male | 80 (82.5) | 36 (83.7) | 81 (83.5) | |
| Female | 17 (17.5) | 7 (16.3) | 16 (16.5) | |
| Operation, n
(%) | | | | <0.0001b |
| TUR-BT | 97 (100.0) | 17 (39.5) | | |
| Radical
cystectomy | 0 (0.0) | 26 (60.5) | | |
| Tumor size, n
(%) | | | | 0.003c |
| ≤1 cm | 16 (16.5) | 2 (4.7) | | |
| 2-3 cm | 37 (38.1) | 11 (25.6) | | |
| >3 cm | 37 (38.1) | 28 (65.1) | | |
| NA | 7 (7.3) | 2 (4.7) | | |
| Multiplicity, n
(%) | | | | 0.108c |
| Single | 52 (53.6) | 30 (69.8) | | |
| 2-7 | 28 (28.9) | 7 (16.3) | | |
| >7 | 11 (11.3) | 4 (9.3) | | |
| NA | 6 (6.2) | 2 (4.6) | | |
| Grade (2004 WHO
grading system), n (%) | | | | <0.0001b |
| Low | 72 (74.2) | 8 (18.6) | | |
| High | 25 (25.8) | 35 (81.4) | | |
| Stage, n (%) | | | | <0.001c |
| TaN0M0 | 26 (26.8) | | | |
| T1N0M0 | 71 (73.2) | | | |
| T2N0M0 | | 13 (30.2) | | |
| T3N0M0 | | 6 (14.0) | | |
| T≥4 or N≥1 or
M1 | | 24 (55.8) | | |
| Chemotherapy, n
(%) | | | | <0.0001b |
| No | 97 (100.0) | 23 (53.5) | | |
| Yes | 0 (0.0) | 20 (46.5) | | |
| BCG therapy, n
(%) | | | | <0.0001b |
| No | 56 (57.7) | 38 (88.4) | | |
| Yes | 40 (41.2) | 5 (11.6) | | |
| Recurrence, n
(%) | | | | |
| No | 59 (60.8) | - | | |
| Yes | 38 (39.2) | - | | |
| Progression, n
(%) | | | | 0.125b |
| No | 79 (81.4) | 30 (69.8) | | |
| Yes | 18 (18.6) | 13 (30.2) | | |
| Survival, n
(%) | | | | 0.009c |
| Alive | 64 (66.0) | 21 (48.8) | | |
|
Non-cancer-specific death | 18 (18.6) | 3 (7.0) | | |
| Cancer-specific
death | 15 (15.5) | 19 (44.2) | | |
| Mean follow-up time
(range), months | 72.95
(3.20-172.20) | 36.18
(3.00-141.40) | | |
BCa cell line
Human BCa EJ cells (MGH-U1) were kindly provided by
Dr Wun-Jae Kim (Department of Urology, Chungbuk National
University, Cheongju, South Korea) and were authenticated using
short tandem repeat analysis. After authentication, this cell line
matched 100% with T24 cells. Cells were grown at 37°C in a 5%
CO2 humidified incubator in DMEM (cat. no. 10-017-CV;
Corning, Inc) supplemented with 10% fetal bovine serum (FBS; cat.
no. 35-015-CV; Corning, Inc), 100 U/ml penicillin and 100
µg/ml streptomycin.
RNA extraction and RT-qPCR
Total RNA was extracted from tissues using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and stored at -80°C. Subsequently, cDNA was synthesized from
1 µg total RNA using the First Strand cDNA Synthesis kit
(Clontech Laboratories, Inc.) following the manufacturer's
protocol. Relative gene expression was analyzed using qPCR and the
2−ΔΔCq method (24).
qPCR was performed using a Rotor Gene 6000 instrument (Qiagen GmbH)
to amplify mRNA from tissue. Microtubes (Qiagen GmbH) containing
SsoFast EvaGreen Supermix (Bio-Rad Laboratories, Inc.) were used
for the qPCR reactions. The primers used for amplifying candidate
genes were as follows: COL6A1 sense, 5′-CGT GGA CCT GTT CTT
TGT G-3′ and antisense, 5′-CGT CAC TGT AGT GCA GCG-3′;
COL6A2 sense, 5′-CAG GAG GTC ATC TC GCC G-3′ and antisense,
5′-GTT CTG CAG CTG GCT GAT G-3′; miR-6124 sense, 5′-GGA AAA
GGA AGG GGG AGG A-3′; and miR-4651 sense, 5′-CGG GGU GGG UGA
GGU CGG GC-3′. A universal primer with a poly-A tail was used as
the miRNA antisense primer. Control GAPDH (used for target
mRNA normalization) primers were as follows: Sense, 5′-CAT GTT CGT
CAT GGG TGT GA-3′ and antisense, 5′-ATG GCA TGG ACT GTG GTC AT-3′.
Since there are no consensus housekeeping miRNAs to date, and the
utility of most commonly used miRNA internal controls, such as U6,
may lead to varied results (7,25,26), miRNA expression was normalized to
the total RNA concentration measured using Quant-iT™ RiboGreen™ RNA
Assay kit (Invitrogen; Thermo Fisher Scientific, Inc.).
Materials
Antibodies specific for p21WAF1 (cat. no.
sc-756), p27KIP1 (cat. no. sc-528), p53 (cat. no.
sc126), cyclin-dependent kinase (CDK) 2 (cat. no. sc-6248), CDK4
(cat. no. sc-23896), Cyclin D (cat. no. sc-8396), Cyclin E (s cat.
no. c-377100) and β-actin (cat. no. sc-47778) were purchased from
Santa Cruz Biotechnology, Inc. Antibodies specific for
extracellular signal-regulated kinase (ERK; cat. no. 9102), p38
MAPK (cat. no. 9212), Jun N-terminal kinase (JNK; cat. no. 9258),
AKT (cat. no. 9272), phospho-ERK (cat. no. 9101), phospho-p38 MAPK
(cat. no. 9211), phospho-JNK (cat. no. 9251) and phospho-AKT (cat.
no. 9271) were purchased from Cell Signaling Technology, Inc. All
primary antibodies were diluted in TBS at 1:1,000. Goat anti-rabbit
IgG-HRP (cat. no. sc-2004), goat anti-mouse IgG-HRP (cat. no.
sc-2005) and donkey anti-goat IgG-HRP (cat. no. sc-2020) were
purchased from Santa Cruz Biotechnology, Inc. All secondary
antibodies were diluted at 1:3,000. The nuclear extract kit and the
electrophoretic mobility shift assay (EMSA) kit (cat. nos. E3050
and E3300, respectively) were obtained from Promega Corporation.
The cDNA for COL6A1-pcNSD2, pcNSD2, COL6A2-pOTB7 and pOTB7 was
obtained from the Korea Human Gene Bank.
Transfection
Cells were transfected with cDNA using
Lipofectamine® 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Cells (1×105 cells/well) were seeded into
6-well plates and incubated for 24 h at 37°C. A total of 2
µg of each cDNA of COL6A1 and COL6A2, as well
as empty vectors (EVs; pcNSD2 and pOTB7) were mixed
with 1X OPTI-MEM (Thermo Fisher Scientific, Inc.) containing 15
µl Lipofectamine 2000 transfection reagent and incubated for
5 min at room temperature. The mixture was added to cells, which
were incubated for 48 h at 37°C before subsequent experiments. The
EV was used to evaluate if the elements on the EV alone (without
inserted gene cassette) would affect the transfection results.
MTT assay
Cell viability was measured using the assay.
Briefly, cells (5×103 cells/well) were plated in 96-well
plates and incubated for 24 h at 37°C. The medium was removed and
fresh medium containing 100 µl of 5 mg/ml MTT (cat. no.
M2128; Sigma-Aldrich; Merck KGaA) was added. After 2 h, the medium
was removed and 200 µl DMSO was added. Absorbance at 570 nm
was measured using a microplate reader.
Cell cycle analysis
Cells were harvested and fixed for 3 h at 4°C in 70%
ethanol. After washing the cells with ice-cold PBS, harvested cells
were incubated using the Muse™ Cell Cycle kit for 30 min at room
temperature to assess the relative number of cells at each phase of
the cell cycle according to the manufacturer's protocol. Samples
were analyzed using the Muse® Cell Cycle Analyzer (Merck
KGaA). Data was generated with the Muse Cell Cycle Software 1.5.0.0
(Merck KGaA).
Immunoblot analysis
Whole cell extracts from transfected cells were
prepared using lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM
EDTA, 1 mM DTT, 2.5 mM EGTA, 10 mM β-glycerophosphate, 0.1 mM
Na3VO4, 1 mM NaF, 0.1 mM PMSF, 0.1 mM 10%
glycerol, 0.1% Tween-20, 10 µg/ml leupeptin and 2
µg/ml aprotinin). Protein concentration was determined using
the BCA protein assay reagent kit (Thermo Fisher Scientific, Inc.).
Proteins (30-40 µg/lane) were separated by 4-20% SDS-PAGE
under denaturing conditions and then transferred onto
nitrocellulose membranes (Hybond; Cytiva). Non-specific binding to
the membrane was blocked by incubation in 5% skim milk for 2 h at
room temperature, after which the membranes were incubated
overnight with primary antibodies at 4°C. HRP-conjugated secondary
antibodies were applied to the membranes for 2 h at room
temperature. Immunocomplexes were analyzed using Western Lightning
Plus-ECL (PerkinElmer, Inc.). The values of the blots were measured
using the ImageJ software v1.50i (National Institutes of
Health).
Wound healing migration assay
Transfected cells (3×105) were seeded
into 6-well plates in 2 ml DMEM containing 10% FBS and then grown
to 90% confluency. To inhibit cell proliferation, cells were
pre-incubated for 2 h with 5 µg/ml mitomycin C
(Sigma-Aldrich; Merck KGaA). An assigned area of the cell surface
was scratched with a 2-mm pipette tip. After three washes with 1X
PBS, the plates were incubated with culture medium containing 10%
FBS (required for cells) for 24 h at 37°C. The number of cells
migrating into the wound surface was evaluated as the width of the
remaining wounded area relative to the initial wounded area.
Migration of cells into the scratched area was observed and
compared with that of control plates. Morphological changes were
photographed under an inverted light microscope at ×40
magnification.
Invasion assay
The cell invasion assay was performed using SPL
Insert Hanging 24 wells inserts (SPL Life Sciences). First, the
insert membrane was coated with 0.1% gelatin solution (cat. no.
G1393; Sigma-Aldrich; Merck KGaA) for 1 h at room temperature.
Next, cells (3.5×104) were resuspended in serum-free
DMEM and added to the upper chamber. DMEM containing 10% FBS was
added to the lower chamber and incubated for 24 h at 37°C. Cells
that had invaded the membrane were fixed with 4% formaldehyde for
30 min at room temperature and stained for 1 h at room temperature
with 0.1% crystal violet solution (2.5 mM crystal violet, 1% MeOH
and 4% paraformaldehyde). Non-invading cells on the upper surface
were removed using a cotton swab. Invasive cells were photographed
using an inverted light microscope at ×40 magnification.
Gelatin zymography
Culture medium from transfected cells was subjected
to electrophoresis on 8% polyacrylamide gel containing 1 mg/ml
gelatin. The gel was rinsed twice for 30 min at room temperature
with 2.5% Triton X-100 and then incubated in reaction buffer (150
mM NaCl, 50 mM Tris-HCl pH 7.5 and 10 mM CaCl2) at 37°C
overnight. The gel was stained with 0.2% Coomassie Blue, de-stained
with 10% acetic acid and 10% methanol in distilled water, and
photographed using a charge-coupled device camera (cat. no.
KCS3-63S; Korea Lab Tech) on a light box. Gelatinase activity was
visualized as a white zone on a dark-blue field.
Nuclear extracts and EMSA assays
Nuclear extracts were prepared using a Nuclear
Extraction kit (Promega Corporation) according to the
manufacturer's protocol. Briefly, cells were harvested by
centrifugation at 3,000 × g for 3 min at 25°C and resuspended in
buffer (50 mM KCl, 0.5% NP-40, 25 mM HEPES pH 7.8, 10 µg/ml
leupeptin, 20 µg/ml aprotinin, 125 µM DTT and 1 mM
PMSF in distilled water). After incubation for 30 min at 4°C, the
nuclear pellet was harvested by centrifugation at 10,000 × g for 1
min at 4°C and extracted using high salt buffer (500 mM KCl, 25 mM
HEPES pH 7.8, 10% glycerol, 10 µg/ml leupeptin, 20
µg/ml aprotinin, 125 µM DTT and 1 mM PMSF in
distilled water) for 30 min at 4°C. After centrifugation, the
supernatant containing the nuclear extract was obtained. The total
amount of nuclear protein was quantified using a BCA protein assay
reagent kit (Thermo Fisher Scientific, Inc.). The nuclear extract
(5 µg) was incubated at 25°C for 20 min with 5 fmol
(2×104 cpm) of end-labeled (32P ATP)
oligonucleotide in binding buffer (1 mM MgCl2, 0.5 mM
EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl pH 7.5, 5% glycerol
and 2 µg poly dI/dC·poly dI/dC) in a 10-µl reaction
volume. The oligonucleotide sequences were as follows: Activator
protein 1 (AP-1), 5′-CGC TTG ATG AGT CAG CCG GAA-3′ and 3′-GCG AAC
TAC TCA GTC GGC CTT-5′; NF-κB, 5′-AGT TGA GGG GAC TTT CCC AGG C-3′
and 3′-TCA ACT CCC CTG AAA GGG TCC G-5′; and specificity protein 1
(SP-1), 5′-ATT CGA TCG GGG CGG GGC GAG C-3′ and 3′-TAA GCT AGC CCC
GCC CCG CTC G-5′. Subsequently, the reaction products were mixed
with 1 µl loading buffer (250 mM Tris-HCl pH 7.5, 0.2%
bromophenol blue and 40% glycerol) and electrophoresed on a 4%
polyacrylamide gel at 4°C. The gel was dried and exposed to X-ray
film overnight. The density of the blots was measured using ImageJ
software v1.50i (National Institutes of Health). The Hela nuclear
extract was used as positive control. Unlabeled AP-1, NF-κB and
SP-1 oligonucleotides were used as competitors.
Construction of the protein-protein
interaction (PPI) network
To identify interactions between COL6A1, COL6A2 and
genes regulating cancer growth and progression in BCa, a PPI
network was constructed and analyzed using the Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING) (https://string-db.org/; v11.0b). The PPI networks
analyzed by STRING were based on the functional associations and
STRING implements well-known classification systems, such as Gene
Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)
(27). A PPI score of 0.4 (medium
confidence) was used for the analysis. Some important features
(derived from GO terms and KEGG pathways) associated with BCa were
selected and presented. False discovery rate (FDR)<0.05 and
P<0.05 were considered to be statistically significant.
Statistical analysis
The continuous variables were expressed as the mean
± SD. The normality of the data was estimated by One-Sample
Kolmogorov-Smirnov test and the statistical differences between
three groups (NMIBC, MIBC and controls) were tested by
Kruskal-Wallis test followed by Bonferroni's post-hoc test. The
groups were compared in terms of non-continuous variables using the
χ2 test. Gene expression values were natural
log-transformed and median-centered across samples. The
Kruskal-Wallis test was used to examine COL6A1 and
miR-6124 expression in NMIBC, MIBC and control tissues. For
the comparison of COL6A2 and miR-4651 expression,
one-way ANOVA was used since the datasets followed the normal
distribution. Both tests were followed by the Bonferroni
correction. The Mann-Whitney U test was used to examine COL6A1
expression in patients with NMIBC with stage Ta and T1.
Correlations between COL6A1 and COL6A2 expression,
and miR-6124 and miR-4651 expression were examined by
calculating non-parametric Spearman's correlation coefficients. For
the in vitro tests, triplicate independent reads were
measured for all analyses and data were presented as the mean ± SD.
All data were evaluated in a blinded manner using factorial ANOVA
and Fisher's least significant difference test. Statistical
analysis was performed using SPSS v20.0 (IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
COL6A1 and COL6A2 mRNA expression in BCa
tissues
COL6A1 and COL6A2 mRNA expression in
both NMIBC and MIBC tissues was significantly lower than that in
controls (normal tissue surrounding BCa) (P<0.0001);
additionally, there was a significant difference between MIBC and
NMIBC tissues (P<0.05; Fig.
1A). By contrast, miR-6124 and miR-4651
expression, which interact with COL6A1 and COL6A2,
respectively, in both NMIBC and MIBC was significantly higher than
in controls (P<0.05; Fig. 1B).
Analysis of non-parametric Spearman's correlation coefficients
revealed a negative correlation between miR-6124 and
COL6A1 expression, and between miR-4651 and
COL6A2 expression (Fig.
1C). In particular, patients with NMIBC with stage T1 disease
exhibited lower COL6A1 expression than those with stage Ta
disease (P<0.05; Fig. S1). No
additional associations were identified between COL6A1 and
COL6A2 mRNA expression and clinicopathological
characteristics (data not shown).
 | Figure 1COL6A1 and COL6A2
expression and their interaction with miRNAs in BCa tissues. (A)
COL6A1 and COL6A2 mRNA expression in NMIBC, MIBC and
controls. (B) miR-6124 and miR-4651 expression, which
interact with COL6A1 and COL6A2, respectively, in
NMIBC, MIBC and controls. (C) Spearman's correlation presented
negative correlation between miR-6124 and COL6A1
expression and between miR-4651 and COL6A2
expression. Box plots represent the minimum score, first (lower)
quartile, median, third (upper) quartile and maximum score.
Controls represent normal tissue surrounding BCa. P-values were
calculated using the Kruskal-Wallis (COL6A1 and
miR-6124 expression) and one-way ANOVA (COL6A2 and
miR-4651 expression) with the Bonferroni correction.
****P<0.0001, **P<0.01 and
*P<0.05. BCa, bladder cancer; COL6A1/2, collagen type
VI-α1/2; NMIBC, non-muscle invasive BCa; MIBC, muscle invasive BCa;
miR, microRNA; Ln, natural logarithm. |
COL6A1 and COL6A2 inhibit the
proliferation of human BCa EJ cells (MGH-U1)
The expression levels of COL6A1 and
COL6A2 in transfected EJ cells (MGH-U1) were significantly
higher than those in the control and EV groups (P<0.05; Fig. 2A). In addition, COL6A1- and
COL6A2-treated cells exhibited lower cell viability than
untreated cells (P<0.05; Fig.
2B). These results demonstrated that COL6A1 and
COL6A2 inhibited the proliferation of human BCa EJ cells
(MGH-U1).
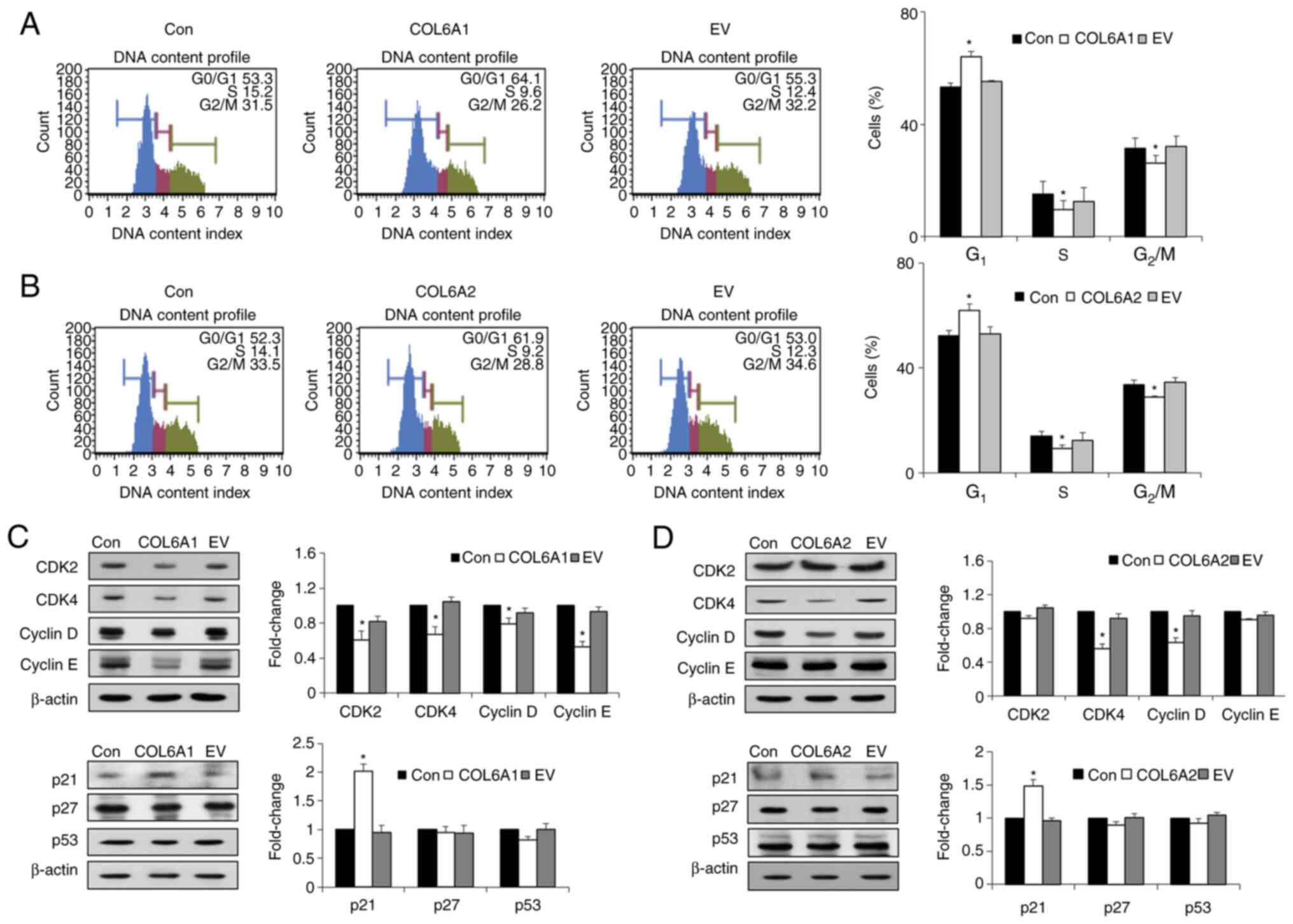
COL6A1 and COL6A2 trigger G1
cell cycle arrest in human BCa EJ cells (MGH-U1)
Cell cycle analysis revealed that COL6A1 and
COL6A2 induced significant accumulation of cells at the
G1 phase (Fig. 3A and
B). In addition, treatment with COL6A1 and COL6A2
significantly decreased the percentage of cells in both S and
G2/M phases (P<0.05; Fig. 3A and B). To better understand the
cell cycle-associated effects of COL6A1 and COL6A2, the expression
levels of major regulatory proteins that control progression of the
G1 phase were examined. Treatment with COL6A1
significantly decreased the expression levels of CDK2, CDK4, cyclin
E and cyclin D (P<0.05; Fig.
3C), while COL6A2 significantly decreased the expression
levels of CDK4 and cyclin D compared with the control group
(P<0.05; Fig. 3D).
Furthermore, the cyclin kinase inhibitor (CKI) p21WAF1
was upregulated by both COL6A1- and COL6A2-treated
human BCa EJ cells (MGH-U1) compared with untreated controls
(P<0.05; Fig. 3C and D). There
were no differences between treated and control cells with respect
to the expression levels of other CKIs, such as p27KIP1
and p53 (Fig. 3C and D). Thus,
the current results indicated that COL6A1 and COL6A2 induced the
arrest of BCa cells at the G1 phase via the
p21WAF1-CDK2/cyclin E, p21WAF1-CDK4/cyclin D
axes and the p21WAF1-CDK4/cyclin D axis,
respectively.
COL6A1 and COL6A2 induce phosphorylation
of p38 MAPK and inhibit phosphorylation of AKT in human BCa EJ
cells (MGH-U1)
BCa progression involves phosphorylation of early
signaling pathway molecules, such as MAPKs, ERK, c-JNK, p38 MAPK
and AKT (28). To investigate
whether COL6A1 and COL6A2 affect the levels of the phosphorylated
forms of MAPKs and AKT, immunoblot analysis was performed. The
results revealed that both COL6A1 and COL6A2 significantly induced
phosphorylation of p38 MAPK in human BCa EJ cells (MGH-U1)
(P<0.05), but did not alter phosphorylation of ERK and JNK
(Fig. 4). By contrast,
phosphorylation of AKT was significantly inhibited in both
COL6A1- and COL6A2-treated human BCa EJ cells
(MGH-U1) (P<0.05; Fig. 4).
These results suggested that COL6A1 and COL6A2 may inhibit BCa cell
proliferation by upregulating p38 MAPK phosphorylation and
downregulating AKT phosphorylation.
COL6A1 and COL6A2 inhibit migration and
invasion of human BCa EJ cells (MGH-U1)
Wound healing migration was assessed by examining
movement of cells into a wounded area. Compared with the control
group, treatment with COL6A1 and COL6A2 significantly
suppressed migration of human BCa EJ cells (MGH-U1) into the
wounded area (P<0.05; Fig.
5A). The invasive potential is shown in Fig. 5B. The number of COL6A1- and
COL6A2-treated cells that invaded through the Transwell
basement membrane was lower than that of the control group
(P<0.05; Fig. 5B).
COL6A1 and COL6A2 suppress the activity
of MMP-2 and MMP-9 by repressing the transcription factors NF-κB,
AP-1 and SP-1 in human BCa EJ cells (MGH-U1)
The enzymatic activity of MMP-2 and MMP-9, both of
which are essential for degradation of the ECM and subsequent
migration and invasion of tumor cells, was analyzed in human BCa EJ
cells (MGH-U1) by gelatin zymography. The proteolytic activities of
both MMP-2 and MMP-9 in COL6A1- and COL6A2-treated
cells were significantly lower than those in untreated cells
(control group) (P<0.05; Fig. 6A
and B). Next, the transcriptional activity of MMP-2 and MMP-9
was examined by measuring the binding activities of the
transcriptional responsive elements NF-κB, AP-1 and
SP-1 via EMSA. As shown in Fig.
6C, the binding activity of NF-κB was significantly inhibited
in the presence of COL6A1, while COL6A2 significantly inhibited the
binding activity of AP-1 and SP-1 (P<0.05; Fig. 6D).
Association between COL6A1, COL6A2 and
genes regulating cancer cell proliferation and migration in
BCa
Interactions between COL6A1, COL6A2, p38 MAPK, AKT,
MMP-2, MMP-9 and IL-1β were depicted using the STRING web tool
(Fig. S2). COL6A1 regulated both
MMP-2 and MMP-9, which interacted with IL-1β, p38 MAPK and AKT,
whereas COL6A2 only regulated MMP-2. Subsequently, BCa-associated
features, including extracellular matrix organization (FDR,
0.0002), positive regulation of cell migration (FDR, 0.00047),
negative regulation of apoptotic signaling (FDR, 0.0011), pathways
involved in bladder cancer (FDR, 6.93×10−6) and pathways
involved in cancer (FDR, 0.00012) were extracted from GO terms and
KEGG pathways, each of them depicted in different colors in
Fig. S2.
Discussion
Cancer is not only a disease caused by tumor cells,
but also a disease of imbalance. Thus, the role of the TME is
critical. Collagen is one of the main ECM proteins, and its
degradation and/or re-deposition affects the TME by regulating ECM
remodeling to promote tumor progression (29). The traditional view of collagen is
that it acts as a passive barrier to tumor cells (29). However, a number of studies have
indicated that several types of cancer exhibit high expression
levels of COL6 (19,20,29,30); it should be noted that the
traditional role of COL6 is to block tumor cells in the TME
(31). The present study revealed
that COL6A1 and COL6A2 expression was downregulated
in BCa tissues. During tumorigenesis, COL6 acts directly on tumor
cells through several pathways, including upregulation of
transcription factors, growth factors, protein kinases and
angiogenic factors, and through activation of the AKT/glycogen
synthase kinase-3β/β-catenin-T-cell factor/lymphoid enhancer factor
signaling pathway (29).
Indirectly, COL6 promotes tumor growth and progression by cleaving
the ETP peptide, which induces epithelial-mesenchymal transition
and fibrosis via the TGF-β-dependent signaling pathway (29). By contrast, ETP in the TME
initiates tumor inflammation by recruiting macrophages and
increasing TNF-α and interleukin (IL)-6 expression, or by promoting
tumor angiogenesis by recruiting endothelial cells and upregulating
the expression levels of CD31, VEGF receptor 2 and
hypoxia-inducible factor 1α (29). COL6 comprises three major
polypeptide chains (α1, 2 and 3), which are encoded by distinct
genes (COL6A1, COL6A2 and COL6A3,
respectively). Recent database-dependent studies have identified
COL6A1, COL6A2 and COL6A3 as prognostic biomarkers for BCa
(19,20,30). However, the mechanism underlying
the involvement of COL6 in BCa tumorigenesis or progression is
unclear.
One of the main hallmarks of cancer is a
dysregulated cell cycle, which leads to abnormal cell proliferation
and division (32,33). p21WAF1 protein is an
established CDK inhibitor that plays an important role in cell
cycle arrest at the G1 phase by inhibiting DNA
replication via its interaction with proliferating cell nuclear
antigen (34). The CDK4-cyclin D
complex is responsible for early G1 phase progression,
while the CDK2-cyclin E complex is thought to act at late
G1 phase; the activity of both complexes is inhibited by
p21WAF1 (35). The
present study demonstrated that COL6A1 and COL6A2 exert a strong
inhibitory effect on the proliferation of BCa cells by inducing
cell cycle arrest. COL6A1 and COL6A2 increased p21WAF1
expression, followed by suppression of CDK4-cyclin D and
CDK2-cyclin E complexes and, finally, arrest of BCa cells at the
G1 phase. The phosphorylation states of MAPKs, such as
ERK1/2, JNK, p38 MAPK, and AKT, are crucial to the mechanisms that
regulate cell cycle progression and cell proliferation (36,37). p38 MAPK serves a dual role in
regulating cell survival or cell death depending on the stimulus
(38). AKT plays an
anti-apoptotic role, and suppression of AKT may induce
phosphorylation of p21, which in turn inhibits cell growth and
proliferation, and activates pro-apoptotic pathways (39). The present study revealed that
both COL6A1 and COL6A2 induce phosphorylation of p38 MAPK in BCa
cells; however, they impeded activation of AKT. Phosphorylation of
ERK and JNK was unchanged. Thus, COL6A1 and COL6A2 may regulate BCa
cell growth and proliferation by activating p38 MAPK
phosphorylation and by preventing AKT phosphorylation, leading to
p21 upregulation and decreased expression of CDK-cyclin complexes,
resulting in cell cycle arrest at the G1 phase. In
addition, migration and invasion of BCa cells treated with COL6A1
and COL6A2 was inhibited, suggesting that COL6A1 and COL6A2 prevent
BCa metastasis. MMPs, such as MMP-2 and MMP-9, accelerate
proteolytic degradation of the ECM, thereby enabling migration and
invasion of cancer cells, which in turn promotes tumor metastasis
(8,9). Thus, MMP-2 and MMP-9 expression, and
their transcription factors NF-κB, AP-1 and SP-1 were evaluated in
COL6A1- and COL6A2-treated BCa cells. COL6A1
inhibited both MMP-2 and MMP-9 by regulating NF-κB; COL6A2
inhibited MMP-2 and MMP-9 by decreasing the expression of AP-1 and
SP-1. This indicates that COL6A1 and COL6A2 may inhibit BCa
metastasis.
Several studies have revealed an association
between classical signaling pathways involved in major tumor
events, such as cancer cell proliferation, invasion and migration
(40-42). Active p38 MAPK signaling regulates
BCa cell migration or invasion by regulating MMP-2/-9 activity
(40). MMP-2 triggers
VEGF-induced angiogenesis in lung tumors by activating the PI3K/AKT
signaling pathway (41), while
PI3K/AKT downregulates p38 MAPK (42). Accordingly, a search using STRING
in the present study identified interactions between COL6A1,
COL6A2, p38 MAPK, AKT, MMP-2, MMP-9 and IL-1β, which is a potent
pro-inflammatory cytokine that interacts with MMPs to regulate
collagen synthesis (43). It was
demonstrated that COL6A1 regulated both MMP-2 and MMP-9, which
interacted with IL-1β, p38 MAPK and AKT, whereas COL6A2 only
regulated MMP-2.
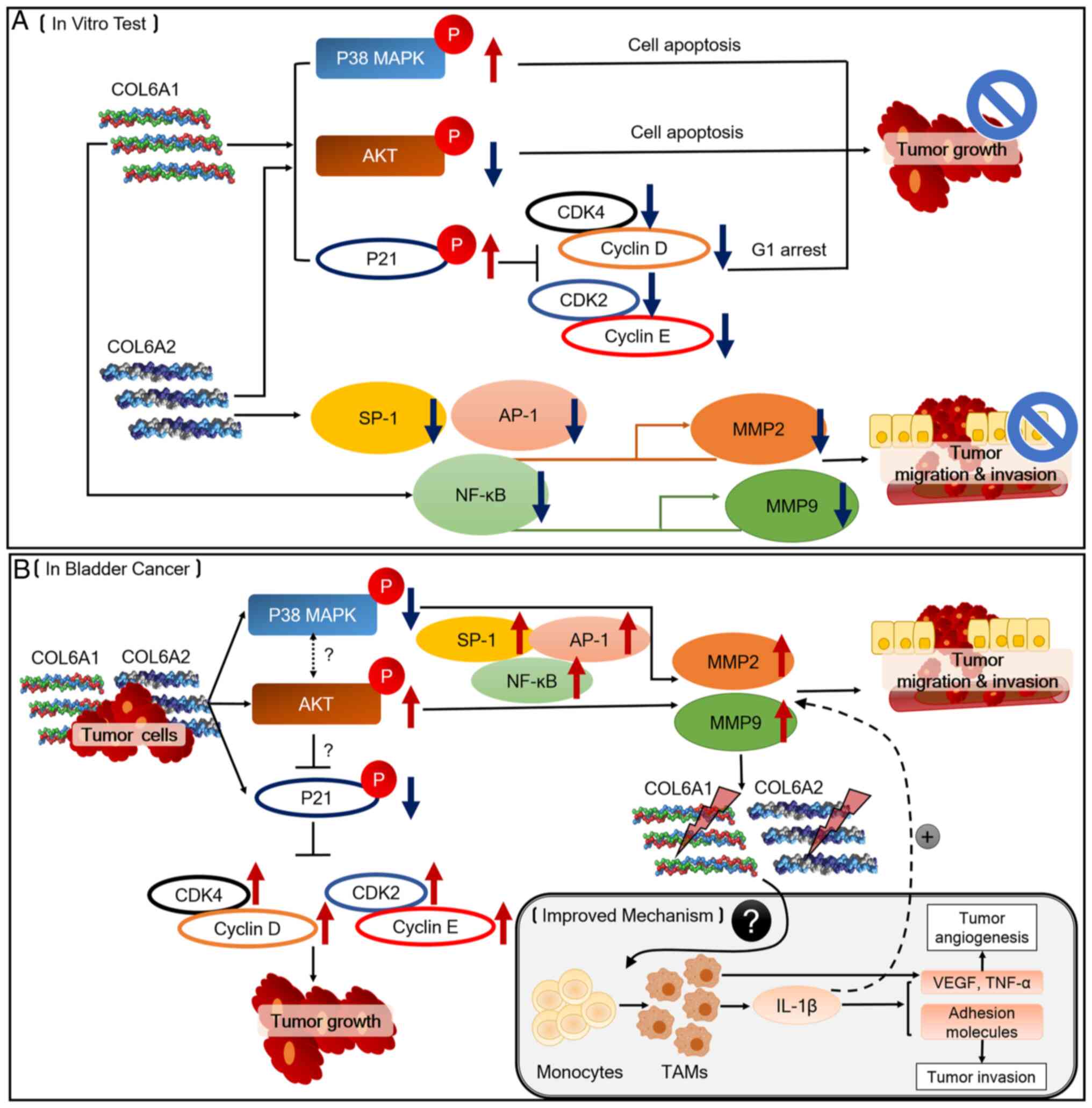
The antitumor effects of COL6A1 and COL6A2 on BCa
cells are summarized in Fig. 7.
Fig. 7A outlines the potential
roles of COL6A1 and COL6A2 as BCa tumor suppressors, and Fig. 7B outlines the proposed mechanisms
by which COL6A1 and COL6A2 may be involved in BCa pathogenesis and
progression, as well as the mechanisms associated with the TME.
During BCa occurrence and development, the ECM undergoes remodeling
via collagen degradation to further coordinate the behavior of
cancer cells. Degraded COL6A1 and COL6A2 may then inhibit p38 MAPK
and p21, whereas they may activate AKT signaling. According to
previous studies (37-39), de-phosphorylation of p38 MAPK
induces AKT phosphorylation, which inhibits p21 phosphorylation.
These events trigger tumor growth, migration and invasion by
activating MMPs induced by SP-1, AP-1 and NF-κB. Furthermore,
increased expression levels of MMPs may decrease the expression
levels of COL6A1 and COL6A2. MMP-derived collagen fragments may
recruit monocytes and trigger them to differentiate into
tumor-associated macrophages (TAMs) that secret factors such as
IL-1β, which are responsible for tumor progression (31). Further studies should focus on the
roles of COL6A1 or COL6A2 on TAMs during BCa progression.
However, there is a limitation in the present
study. Only one BCa cell line was used, the human BCa EJ cells
(MGH-U1). Human BCa EJ cells (MGH-U1) are considered to reflect
high grade BCa, and they are widely used in invasive BCa studies
(44-48). In the present study, both
COL6A1 and COL6A2 mRNA expression was significantly
lower in NMIBC and MIBC compared with controls, and these genes
were more highly expressed in MIBC than in NMIBC. Accordingly, it
may be inferred that the human BCa EJ cells (MGH-U1) are a superior
option that may be able to cover results of COL6A1 and 2 from both
NMIBC and MIBC. Nevertheless, the number of cell lines used in the
present study is inadequate. However, to the best of our knowledge,
this is the first research paper to excavate the association
between COL6 and BCa, which may provide a reference for studying
the functions of COL6 in BCa. As the scaffold of the TME, collagens
serve an important role in carcinogenesis and cancer progression.
Thus, it would be interesting to explore the functions of collagens
in BCa. Following the present study, future studies should use more
reasonable cohorts (including more cases of bladder carcinoma
tissues and cell lines) to verify the detailed mechanisms.
In conclusion, the current study demonstrated a
novel mechanism by which COL6A1 and COL6A2 may affect BCa
pathogenesis and progression. Along with the present findings,
further study of the roles of the COL6A1 and COL6A2
mRNA-interacting miRNAs miR-6124 and miR-4651 may
help determine whether these miRNAs definitely act as oncomiRNAs in
BCa.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
XMP, IYK, SJY and WJK made contributions to
conception and design of the study. XMP and BH performed all
experiments. XMP, BH, YJB, JYL, YHC, SKM and EJC contributed to
data acquisition and analysis. PJ, SPS, HWK, WTK, YSH and YSL
contributed to data interpretation. XMP and BH drafted the
manuscript. YHC, IYK, SKM, EJC, SJY and WJK revised the manuscript
critically for important intellectual content, and gave final
approval of the version to be published. XMP, BH, SKM and WJK
assessed and confirmed the authenticity of all the raw data. WJK
agreed to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved, and provided
funding. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Institutional Review
Board at Chungbuk National University (approval no. GR2010-12-010).
The collection and analysis of all samples were approved by the
Institutional Review Board at Chungbuk National University, and
written informed consent was obtained from each subject. All
samples derived from the National Biobank of Korea at Chungbuk
National University Hospital (Cheongju, South Korea) were obtained
with informed consent under institutional review board-approved
protocols.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Ms. Eun-Ju Shim
from the National Biobank of Korea at Chungbuk National University
Hospital (Cheongju, South Korea) for the sample preparations and
excellent technical assistance.
Funding
The present study was supported by the Basic Science Research
Program through the National Research Foundation of Korea funded by
the Ministry of Education (grant nos. 202 0R1F1A1068488 and
2020R1I1A3062508) and by the Osong Medical Innovation Foundation
funded by Chungcheongbuk (grant no. 2020017001).
Abbreviations:
|
AP-1
|
activator protein 1
|
|
BCa
|
bladder cancer
|
|
CDK
|
cyclin-dependent kinase
|
|
CKI
|
cyclin kinase inhibitor
|
|
COL6A1/2
|
collagen type VI-α1/2
|
|
ECM
|
extracellular matrix
|
|
EMSA
|
electrophoretic mobility shift
assay
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FBS
|
fetal bovine serum
|
|
IL
|
interleukin
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MIBC
|
muscle invasive BCa
|
|
MMP
|
matrix metalloproteinase
|
|
NMIBC
|
non-MIBC
|
|
SP-1
|
specificity protein 1
|
|
TAM
|
tumor-associated macrophage
|
|
TME
|
tumor microenvironment
|
|
TUR
|
transurethral resection
|
References
|
1
|
Chavan S, Bray F, Lortet-Tieulent J,
Goodman M and Jemal A: International variations in bladder cancer
incidence and mortality. Eur Urol. 66:59–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sylvester RJ, van der Meijden AP,
Oosterlinck W, Witjes JA, Bouffioux C, Denis L, Newling DW and
Kurth K: Predicting recurrence and progression in individual
patients with stage Ta T1 bladder cancer using EORTC risk tables: A
combined analysis of 2596 patients from seven EORTC trials. Eur
Urol. 49:466–477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shariat SF, Karakiewicz PI, Palapattu GS,
Lotan Y, Rogers CG, Amiel GE, Vazina A, Gupta A, Bastian PJ,
Sagalowsky AI, et al: Outcomes of radical cystectomy for
transitional cell carcinoma of the bladder: A contemporary series
from the Bladder Cancer Research Consortium. J Urol. 176(6 Pt 1):
2414–2422. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tilki D, Reich O, Karakiewicz PI, Novara
G, Kassouf W, Ergün S, Fradet Y, Ficarra V, Sonpavde G, Stief CG,
et al: Validation of the AJCC TNM substaging of pT2 bladder cancer:
Deep muscle invasion is associated with significantly worse
outcome. Eur Urol. 58:112–117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cancer Genome Atlas Research Network:
Comprehensive molecular characterization of urothelial bladder
carcinoma. Nature. 507:315–322. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JY, Yun SJ, Jeong P, Piao XM, Kim YH,
Kim J, Subramaniyam S, Byun YJ, Kang HW, Seo SP, et al:
Identification of differentially expressed miRNAs and
miRNA-targeted genes in bladder cancer. Oncotarget. 9:27656–27666.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Piao XM, Jeong P, Kim YH, Byun YJ, Xu Y,
Kang HW, Ha YS, Kim WT, Lee JY, Woo SH, et al: Urinary cell-free
microRNA biomarker could discriminate bladder cancer from benign
hematuria. Int J Cancer. 144:380–388. 2019. View Article : Google Scholar
|
|
8
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Venning FA, Wullkopf L and Erler JT:
Targeting ECM disrupts cancer progression. Front Oncol. 5:2242015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levental KR, Yu H, Kass L, Lakins JN,
Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et
al: Matrix crosslinking forces tumor progression by enhancing
integrin signaling. Cell. 139:891–906. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arnold SA, Rivera LB, Miller AF, Carbon
JG, Dineen SP, Xie Y, Castrillon DH, Sage EH, Puolakkainen P,
Bradshaw AD and Brekken RA: Lack of host SPARC enhances vascular
function and tumor spread in an orthotopic murine model of
pancreatic carcinoma. Dis Models Mech. 3:57–72. 2010. View Article : Google Scholar
|
|
12
|
Aitken KJ and Bägli DJ: The bladder
extracellular matrix. Part I: Architecture, development and
disease. Nat Rev Urol. 6:596–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cescon M, Gattazzo F, Chen P and Bonaldo
P: Collagen VI at a glance. J Cell Sci. 128:3525–3531. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wan F, Wang H, Shen Y, Zhang H, Shi G, Zhu
Y, Dai B and Ye D: Upregulation of COL6A1 is predictive of poor
prognosis in clear cell renal cell carcinoma patients. Oncotarget.
6:27378–27387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu YP, Wan FN, Shen YJ, Wang HK, Zhang GM
and Ye DW: Reactive stroma component COL6A1 is upregulated in
castration-resistant prostate cancer and promotes tumor growth.
Oncotarget. 6:14488–14496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou T, Tong C, Kazobinka G, Zhang W, Huang
X, Huang Y and Zhang Y: Expression of COL6A1 predicts prognosis in
cervical cancer patients. Am J Transl Res. 8:2838–2844.
2016.PubMed/NCBI
|
|
17
|
Chiu KH, Chang YH, Wu YS, Lee SH and Liao
PC: Quantitative secretome analysis reveals that COL6A1 is a
metastasis-associated protein using stacking gel-aided purification
combined with iTRAQ labeling. J Proteome Res. 10:1110–1125. 2011.
View Article : Google Scholar
|
|
18
|
Stone R II, Sabichi AL, Gill J, Lee IL,
Adegboyega P, Dai MS, Loganantharaj R, Trutschl M, Cvek U and
Clifford JL: Identification of genes correlated with early-stage
bladder cancer progression. Cancer Prev Res (Phila). 3:776–786.
2010. View Article : Google Scholar :
|
|
19
|
Shi S and Tian B: Identification of
biomarkers associated with progression and prognosis in bladder
cancer via co-expression analysis. Cancer Biomark. 24:183–193.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu H, Chen H, Wang J, Zhou L and Liu S:
Collagen stiffness promoted non-muscle-invasive bladder cancer
progression to muscle-invasive bladder cancer. OncoTargets Ther.
12:3441–3457. 2019. View Article : Google Scholar
|
|
21
|
Oosterlinck W, Lobel B, Jakse G, Malmström
PU, Stöckle M and Sternberg C; European Association of Urology
(EAU) Working Group on Oncological Urology: Guidelines on bladder
cancer. Eur Urol. 41:105–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Witjes JA, Compérat E, Cowan NC, De Santis
M, Gakis G, Lebret T, Ribal MJ, Van der Heijden AG and Sherif A;
European Association of Urology: EAU guidelines on muscle-invasive
and metastatic bladder cancer: Summary of the 2013 guidelines. Eur
Urol. 65:778–792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alfred Witjes J, Lebret T, Compérat EM,
Cowan NC, De Santis M, Bruins HM, Hernandez V, Espinós EL, Dunn J,
Rouanne M, et al: Updated 2016 EAU guidelines on muscle-invasive
and metastatic bladder cancer. Eur Urol. 71:462–475. 2017.
View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Yun SJ, Jeong P, Kang HW, Kim YH, Kim EA
and Yan C: Urinary MicroRNAs of prostate cancer: Virus-Encoded
hsv1-miRH18 and hsv2-miR-H9-5p could be valuable diagnostic
markers. Int Neurourol J. 19:74–84. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiang M, Zeng Y, Yang R, Xu H, Chen Z,
Zhong J, Xie H, Xu Y and Zeng X: U6 is not a suitable endogenous
control for the quantification of circulating microRNAs. Biochem
Biophys Res Commun. 454:210–214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47(D1): D607–D613. 2019.
View Article : Google Scholar
|
|
28
|
Knowles MA and Hurst CD: Molecular biology
of bladder cancer: New insights into pathogenesis and clinical
diversity. Nat Rev Cancer. 15:25–41. 2015. View Article : Google Scholar
|
|
29
|
Chen P, Cescon M and Bonaldo P: Collagen
VI in cancer and its biological mechanisms. Trends Mol Med.
19:410–417. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin Z, Yao J, Xie N, Cai L, Qi S, Zhang Z
and Li B: Melittin constrains the expression of identified key
genes associated with bladder cancer. J Immunol Res.
2018:50381722018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fang M, Yuan J, Peng C and Li Y: Collagen
as a double-edged sword in tumor progression. Tumor Biol.
35:2871–2882. 2014. View Article : Google Scholar
|
|
32
|
Otto T and Sicinski P: Cell cycle proteins
as promising targets in cancer therapy. Nat Rev Cancer. 17:93–115.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jeggo PA, Pearl LH and Carr AM: DNA
repair, genome stability and cancer: A historical perspective. Nat
Rev Cancer. 16:35–42. 2016. View Article : Google Scholar
|
|
34
|
Shamloo B and Usluer S: p21 in cancer
research. Cancers. 11:11782019. View Article : Google Scholar :
|
|
35
|
Donjerkovic D and Scott DW: Regulation of
the G1 phase of the mammalian cell cycle. Cell Res. 10:1–16. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kamiyama M, Naguro I and Ichijo H: In vivo
gene manipulation reveals the impact of stress-responsive MAPK
pathways on tumor progression. Cancer Sci. 106:785–796. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH
and Hung MC: Cytoplasmic localization of p21 Cip1/WAF1 by
Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat
Cell Biol. 3:245–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kumar B, Koul S, Petersen J, Khandrika L,
Hwa JS, Meacham RB, Wilson S and Koul HK: p38 mitogen-activated
protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer
by modulation of MMP-2 and MMP-9 activity. Cancer Res. 70:832–841.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chetty C, Lakka SS, Bhoopathi P and Rao
JS: MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated
PI3K/AKT signaling in A549 lung cancer cells. Int J Cancer.
127:1081–1095. 2010. View Article : Google Scholar :
|
|
42
|
Giannı̀ M, Kopf E, Bastien J,
Oulad-Abdelghani M, Garattini E, Chambon P and Rochette-Egly C:
Down-regulation of the phosphatidylinositol 3-kinase/Akt pathway is
involved in retinoic acid-induced phosphorylation, degradation, and
transcriptional activity of retinoic acid receptor gamma 2. J Biol
Chem. 277:24859–24862. 2002. View Article : Google Scholar
|
|
43
|
Liu W, Ding I, Chen K, Olschowka J, Xu J,
Hu D, Morrow GR and Okunieff P: Interleukin 1beta (IL1B) signaling
is a critical component of radiation-induced skin fibrosis. Radiat
Res. 165:181–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang F, Tang J, Li P, Si S, Yu H, Yang X,
Tao J, Lv Q, Gu M, Yang H and Wang Z: Chloroquine enhances the
radiosensitivity of bladder cancer cells by inhibiting autophagy
and activating apoptosis. Cell Physiol Biochem. 45:54–66. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Devanand P, Kim SI, Choi YW, Sheen SS, Yim
H, Ryu MS, Kim SJ, Kim WJ and Lim IK: Inhibition of bladder cancer
invasion by Sp1-mediated BTG2 expression via inhibition of DNA
methyltransferase 1. FEBS J. 281:5581–5601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu D, Tao J, Xu B, Qing W, Li P, Lu Q and
Zhang W: Phosphatidylinositol 3-kinase inhibitor LY294002
suppresses proliferation and sensitizes doxorubicin chemotherapy in
bladder cancer cells. Urol Int. 86:346–354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wahafu W, Gai J, Song L, Ping H, Wang M,
Yang F, Niu Y and Xing N: Increased H2S and its
synthases in urothelial cell carcinoma of the bladder, and enhanced
cisplatin-induced apoptosis following H2S inhibition in
EJ cells. Oncol Lett. 15:8484–8490. 2018.PubMed/NCBI
|
|
48
|
Silvers CR, Liu YR, Wu CH, Miyamoto H,
Messing EM and Lee YF: Identification of extracellular
vesicle-borne periostin as a feature of muscle-invasive bladder
cancer. Oncotarget. 7:23335–23345. 2016. View Article : Google Scholar : PubMed/NCBI
|