Introduction
Osteosarcoma (OS) is the most frequent primary
malignant bone tumor type, which predominantly affects children and
adolescents, and accounts for approximately 15% of all bone
malignancies (1,2). The current standard clinical
treatment for OS includes preoperative chemotherapy followed by
surgical removal of the primary tumor, combined with postoperative
chemotherapy. Since chemotherapy was introduced in the 1970s, the
5-year survival rate of OS has markedly improved from <20 to 70%
(3). Doxorubicin, cisplatin and
methotrexate are the most commonly used first-line chemotherapy
drugs in the treatment of OS (4,5).
Despite great advances in chemotherapy for OS, the survival rate
has reached a plateau and has remained unsatisfactory in the past
three decades, largely due to chemotherapy resistance (6,7).
Among patients with OS, >40% are not sensitive to chemotherapy
drugs, and the 5-year survival rate is only 16-20% (8). The emergence of chemoresistance
often leads to treatment failure and poor prognosis, and has,
therefore, become a major obstacle to improve OS therapeutic
effect. Thus, it is imperative to elucidate the underlying
molecular mechanisms involved in OS chemoresistance.
C-X-C motif chemokine receptor 4 (CXCR4) is a G
protein-coupled receptor (GPCR) to which C-X-C motif chemokine
ligand 12 (CXCL12) binds with high affinity (9). Accumulating evidence has indicated
that CXCR4 plays a crucial role in OS progression and metastasis
(10-12). In addition, CXCR4 has been shown
to be associated with poor survival of patients with OS, and is
considered an important clinical prognosis indicator (13). Increased attention has been paid
to CXCR4-mediated chemotherapy resistance in various types of tumor
(14-17). However, the association between
CXCR4 and OS chemoresistance remains unknown.
Autophagy is a catabolic process via which cells
eliminate and recycle their own damaged proteins and organelles to
provide energy. It has been demonstrated that autophagy plays a
dual role in the regulation of OS resistance (18). While moderate autophagy can lead
to drug resistance due to its cytoprotective effect, excessive
autophagy reverses drug resistance by inducing cell death (18). Previous findings revealed that
CXCR4 promoted OS growth and metastasis by activating the AKT
signaling pathway (10). Since
the PI3K/AKT/mTOR axis is one of the key regulators of autophagy,
it was hypothesized that CXCR4 is involved in OS resistance to
doxorubicin by regulating autophagy.
The aim of the present study was to investigate
whether CXCR4 blockade could enhance the sensitivity of OS to
doxorubicin by inducing autophagic cell death and whether CXCR4 is
a potential therapeutic target to reverse OS doxorubicin
resistance.
Materials and methods
Cell lines and culture
The murine LM8 and Dunn OS cell lines were kindly
donated by Dr Eugenie Kleinerman (MD Anderson Cancer Center,
University of Texas, Houston, TX, USA). The cell lines were
cultured in high-glucose DMEM (Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco), 100 U/ml penicillin and 100
µg/ml streptomycin. The cultures were incubated at 37°C in a
humidified atmosphere containing 5% CO2.
Reagents and antibodies
Doxorubicin (cat. no. S1208), rapamycin (cat. no.
S1039), bafilomycin A1 (cat. no. S1413) and AMD3100 (cat. no.
S8030) were purchased from Selleck Chemicals. Antibodies against
CXCR4 (cat. no. ab124824), PI3K (cat. no. ab40776), mTOR (cat. no.
ab134903) and phosphorylated (p)-mTOR (cat. no. ab109268) were
purchased from Abcam. Antibodies against beclin 1 (cat. no. 3738S),
light chain 3B (LC3B; cat. no. 12741S), cleaved-caspase 3 (cat. no.
9664S), p-PI3K (cat. no. 4228S), AKT (cat. no. 4685S), p-AKT (cat.
no. 4060S) and GAPDH (cat. no. 5174S) were purchased from Cell
Signaling Technology, Inc. Anti-P-glycoprotein antibody (P-gp; cat.
no. 49042) was purchased from Signalway Antibody LLC, while
anti-caspase 3 antibody (cat. no. 19677-1-AP) was purchased from
ProteinTech Group, Inc.
Cell viability assay
LM8 and Dunn cells (2×104 cells/ml) were
seeded in 96-well plates overnight at 37°C and then treated with
various concentrations of doxorubicin (0, 0.2, 0.4, 0.8, 1 and 10
µg/ml). A concentration of 0.2 µg/ml was equivalent
to 344.84 nM. After 48 h of incubation at 37°C, 10 µl Cell
Counting Kit-8 (CCK-8) solution (Dojindo Molecular Technologies,
Inc.) was added to each well for 1 h at 37°C. The optical density
(OD) was then measured using a microplate reader (EL800; BioTek
Instruments, Inc.) at 450 nm. The cell viability was calculated
using the equation: Cell viability (%)=(OD450nm of
treatment/OD450nm of control) ×100%. The IC50 was
calculated by GraphPad Prism 8.0 (GraphPad Software, Inc.).
Plasmid and small interfering RNA (siRNA)
transfection
Transfection was performed using
Lipofectamine® 2000 transfection reagent (Thermo Fisher
Scientific) according to the manufacturer's protocol. LM8 and Dunn
cells were transfected with 100 nM mouse siCXCR4 (5′-GCA UAG UCG
GCA AUG GAU UTT-3′) (Shanghai GenePharma Co., Ltd.) and 1
µg/ml plasmids encoding CXCR4 (Shanghai GenePharma Co.,
Ltd.). A scrambled siRNA used as the negative controls (5′-UUC UCC
GAA CGU GUC ACG UTT-3′). Both LM8 and Dunn cells were transfected
with mRFP-GFP-LC3 adenovirus (Asia-Vector Biotechnology) or
adenovirus vector containing RFP and GFP (Asia-Vector
Biotechnology) as control. Transfection efficiency was assessed by
western blotting.
Flow cytometry
LM8 and Dunn cells (5×104 cells/ml) were
cultured in 6-well plates for 24 h and treated with 0.2
µg/ml doxorubicin, or with siCXCR4/CXCR4 overexpression, or
with 0.2 µg/ml doxorubicin combined with siCXCR4/CXCR4
overexpression. After 48 h of incubation, cell apoptosis was
evaluated using an Annexin V-FITC apoptosis detection kit (BD
Biosciences), and was detected by flow cytometry (FC500; Beckman
Coulter). The cell apoptosis rate was calculated and analyzed with
FlowJo software (version: V10.5.2, Becton, Dickinson and
Company).
Western blotting
Total protein was extracted from cells using RIPA
lysis buffer (cat. no. P0013B, Beyotime Institute of Biotechnology)
containing phosphatase inhibitors, and was quantified with a BCA
protein assay kit (Beyotime Institute of Biotechnology). Equivalent
quantities of protein (40 µg/lane) were separated by 10-12%
SDS-PAGE at 80 V for 1.5 h and transferred to polyvinylidene
difluoride membranes. After blocking with TBS-0.05% Tween 20 (TBST)
containing 5% skimmed milk for 1 h at room temperature, the
membranes were incubated overnight at 4°C with primary antibodies
against CXCR4, beclin 1, LC3B, P-gp, cleaved caspase 3, caspase 3,
p-PI3K, PI3K, p-AKT, AKT, p-mTOR and mTOR (all diluted 1:1,000).
The membranes were rinsed with TBST three times and subsequently
incubated with Anti-rabbit IgG, HRP-linked Antibody (cat. no. 7074,
Cell Signaling Technology, 1:1,000 dilution) for 1 h at room
temperature. The protein bands were then visualized using an ECL
kit (cat. no. P0018S, Beyotime Institute of Biotechnology).
Semi-quantitative analysis of proteins was performed with Image J
software (National Institutes of Health, version 1.8.0).
Confocal microscopy analysis
LM8 and Dunn cells were cultured at 37°C in 6-well
plates and transfected with mouse CXCR4 siRNA or CXCR4-encoding
plasmids using Lipofectamine 2000 (Thermo Fisher Scientific)
according to the manufacturer's transfections for 48 h. Next, cells
were simultaneously treated with 0.2 µg/ml doxorubicin and
mRFP-GFP-LC3 adenovirus for 48 h. Subsequently, the cells were
fixed with 4% formaldehyde for 30 min and incubated with DAPI for 5
min. Images were obtained using a confocal laser scanning
microscope (Olympus Corporation) (magnification, ×400).
Transmission electron microscopy
LM8 and Dunn cells (1×106) were fixed
with 2.5% glutaraldehyde at 4°C overnight and then fixed in 1%
buffered osmium tetroxide at room temperature for 1.5 h. Next, the
cells were dehydrated with increasing concentrations of ethanol
(25, 50, 70, 90 and 100%) for 5 min at each concentration, embedded
in epoxy resin for 48 h at 60°C and stained with uranyl acetate.
Representative areas were selected for ultrathin sectioning, as
detected by transmission electron microscopy (H-9500, Hitachi,
Ltd.) (magnification, ×12,000 and ×25,000).
Mouse tibia orthotopic tumor model
A total of 32 4-week-old female C3H mice (16-18 g)
were purchased from the Shanghai SLAC Laboratory Animal Co. Ltd.,
housed under standard conditions with a 12-h light-dark cycle, and
fed with pelleted mouse food and water which were provided ad
libitum. All the animal procedures were performed in accordance
with a protocol approved by the Animal Care and Use Committee of
Shanghai Tenth People's Hospital (approval no. SHDSYY-2020-3650).
LM8 cells (5×105) in 10 µl PBS were injected into
the tibia medullary cavity to establish an orthotopic OS model.
Then, 2 weeks after injection of tumor cells, the mice were
randomly allocated to four groups: Control (n=8), 5 mg/kg AMD3100
(n=8), 1 mg/kg doxorubicin (n=8) and 5 mg/kg AMD3100 plus 1 mg/kg
doxorubicin (n=8). Each mouse in the treatment groups received 100
µl AMD3100 or doxorubicin by tail vein injection every 2
days. The control mice were injected instead with 100 µl
PBS. The tumor volume and body weight of each mouse were measured
at each injection time point using the formula: Tumor
volume=(length × width2)/2. Humane endpoints were
reached when the xenograft tumor reached >10% of the animal's
body weight or the tumor diameter was >20 mm. After eight
consecutive injections, the mice that reached study endpoints were
anesthetized with 40 mg/kg pentobarbital injected
intraperitoneally, while pentobarbital at a dose of 100 mg/kg was
administered for euthanasia. Death was verified by the cessation of
a heartbeat and dilated pupils. Tumors were dissected, weighed and
stored in liquid nitrogen or fixed in formalin for
immunohistochemical analysis. The maximum tumor diameter and volume
observed in this study were 19.2 mm and 3,819.11 mm3,
respectively.
Immunohistochemical staining
Tumor samples were fixed in 4% paraformaldehyde
overnight at room temperature, embedded in paraffin, sliced into
4-µm sections, and then deparaffinized in xylene, rehydrated
with graded alcohol and incubated in 3% H2O2
to block endogenous peroxidase activity for 10 min. The slides were
then boiled for 30 min in 10 mM sodium citrate for antigen
retrieval, blocked in 5% BSA (Beyotime Institute of Biotechnology)
for 30 min, and incubated with antibodies against CXCR4 (1:400),
beclin 1 (1:400), LC3B (1:200) and P-gp (1:200) at 4°C overnight.
Next, the slides were washed three times with PBS and incubated
with horseradish peroxidase-conjugated goat anti-rabbit antibody
(1:400; cat. no. ab6721; Abcam) for 30 min at room temperature.
Immunoreactivity was visualized using a 3,3′-diaminobenzidine kit
(Beyotime Institute of Biotechnology). The areal density of each
image was quantified by Image-Pro Plus 6.0 software (MEDIA
CYBERNETICS, USA) for statistical analysis.
Statistical analysis
All data are presented as the mean ± SD from ≥3
independent experiments. Statistical analysis of differences
between two groups was performed using unpaired, two-tailed
Student's t-test with GraphPad Prism 8.0 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
CXCR4 silencing sensitizes OS cells to
doxorubicin by regulating apoptosis and P-gp
To determine whether CXCR4 affects OS doxorubicin
resistance, CXCR4 expression was first downregulated in
CXCR4-positive LM8 cells, and CXCR4 expression was upregulated in
CXCR4-negative Dunn cells by siRNA and lentiviral transfection,
respectively. The differential expression of CXCR4 in the two cells
has been described previously (10). It was firstly confirmed that CXCR4
expression was significantly decreased by siCXCR4 in LM8 cells and
increased by lentiviral transfection in Dunn cells. LM8 and Dunn
cells were then treated with various concentrations of doxorubicin
(0, 0.2, 0.4, 0.8, 1 and 10 µg/ml) for 48 h, and cell
viability was then measured by CCK-8 assay. The results showed that
the IC50 value of CXCR4-knockdown LM8 cells (0.3879
µg/ml) was obviously reduced compared with that of LM8 cells
(0.5891 µg/ml). Conversely, the IC50 value of
CXCR4-overexpressed Dunn cells (0.6661 µg/ml) was much
higher than that of Dunn cells (0.4581 µg/ml) (Fig. 1A).
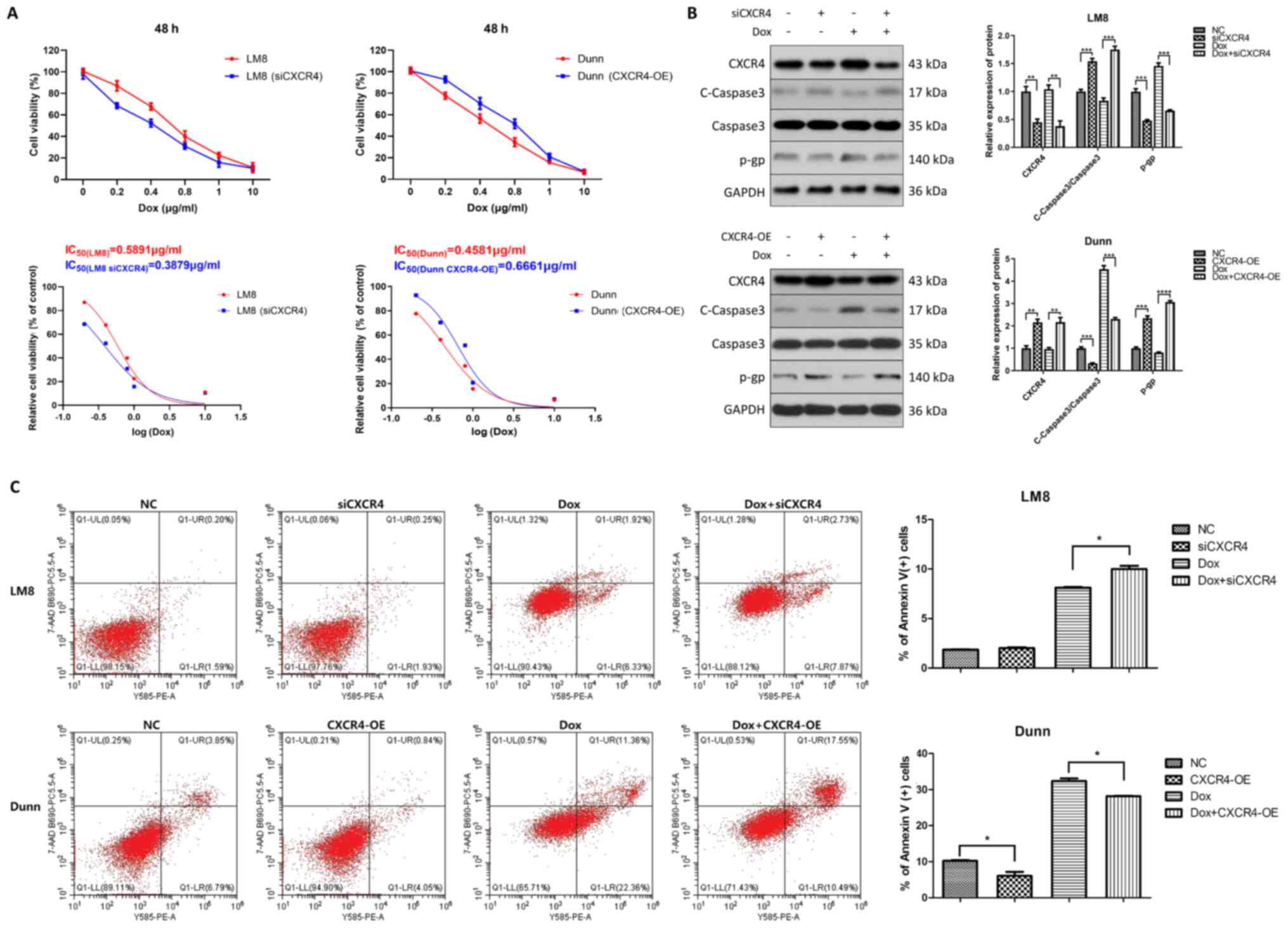 | Figure 1CXCR4 is implicated in the regulation
of osteosarcoma doxorubicin resistance. (A) CXCR4 was downregulated
in CXCR4-positive LM8 cells by small interfering RNA and
upregulated in CXCR4-negative Dunn cells by lentiviral
transfection. LM8 and Dunn cells were treated with various
concentrations of doxorubicin (0, 0.2, 0.4, 0.8, 1 and 10
µg/ml) for 48 h, and cell viability was measured by Cell
Counting Kit-8 assay. Dose-response curves were generated with
GraphPad Prism 8.0 software, and half maximal inhibitory
concentrations of doxorubicin were obtained for each group. (B) LM8
and Dunn cells were cultured in the presence of 0.2 µg/ml
doxorubicin (with or without CXCR4 knockdown/overexpression) for 48
h, and the expression levels of the apoptosis-related protein
cleaved caspase 3, caspase 3 and the drug resistant-related protein
P-glycoprotein were then determined by western blotting. The
protein bands were quantified and subjected to statistical
analysis. (C) LM8 and Dunn cells were cultured with 0.2
µg/ml doxorubicin (with or without CXCR4
knockdown/overexpression) for 48 h, and the apoptosis ratios for
each group (percentage of Annexin V+ cells) were
determined by flow cytometry. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. CXCR4, C-X-C motif chemokine receptor
4. |
The expression of the apoptosis-related protein
caspase 3 and the multidrug resistance-related P-gp was determined.
As revealed by western blot analysis, both CXCR4 knockdown and
doxorubicin treatment increased cleaved caspase 3 and reduced P-gp
levels in LM8 cells. Furthermore, the highest expression of cleaved
caspase 3 and lowest expression of P-gp were found in the
doxorubicin combined with CXCR4 knockdown group in LM8 cells. By
contrast, CXCR4 overexpression reduced doxorubicin-induced cleaved
caspase 3 activation and increased P-gp levels in Dunn cells
(Fig. 1B).
To further explore CXCR4-mediated OS doxorubicin
resistance, the effect of CXCR4 regulation on apoptosis induced by
doxorubicin was investigated in LM8 and Dunn cells by flow
cytometry. CXCR4-postive LM8 cells were cultured for 48 h in the
presence of 0.2 µg/ml doxorubicin, CXCR4 silencing or 0.2
µg/ml doxorubicin combined with CXCR4 silencing.
CXCR4-negative Dunn cells were cultured for 48 h in the presence of
0.2 µg/ml doxorubicin, CXCR4 overexpression or 0.2
µg/ml doxorubicin combined with CXCR4 overexpression. The
percentage of apoptotic LM8 cells in the doxorubicin group was
8.12±0.12 vs. 10.2±0.35% in the doxorubicin combined with siCXCR4
group, which indicated that CXCR4 knockdown increased
doxorubicin-induced apoptosis in LM8 cells. By contrast, the
percentage of apoptotic Dunn cells in the doxorubicin group was
32.52±1.14 vs. 28.19±0.20% in the doxorubicin combined with
CXCR4-overexpression group, which indicated that CXCR4
overexpression partially reversed doxorubicin-induced apoptosis in
Dunn cells (Fig. 1C). These
findings suggested that CXCR4 silencing enhanced the sensitivity of
LM8 cells to doxorubicin by inducing apoptosis and reducing P-gp
levels. By contrast, CXCR4 overexpression reduced the sensitivity
of Dunn cells to doxorubicin by inhibiting apoptosis and inducing
P-gp expression.
CXCR4 silencing induces autophagy,
whereas CXCR4 overexpression inhibits autophagy in OS cells
To investigate the role of autophagy in
CXCR4-mediated OS doxorubicin resistance, western blotting was
performed to detect the expression levels of the autophagy-related
proteins beclin 1 and LC3B after CXCR4 regulation in LM8 and Dunn
cells. It is well known that the conversion of LC3B-I to LC3B-II is
necessary for autophagosome formation. Therefore, LC3B-II detection
has been widely used in autophagy-related research (18). In LM8 cells, both CXCR4 silencing
and doxorubicin treatment increased beclin 1 and LC3B-II
expression, and the highest expression of beclin 1 and LC3B-II was
observed in the doxorubicin combined with CXCR4 silencing group. By
contrast, CXCR4 overexpression reduced beclin 1 and LC3B-II
expression in Dunn cells compared with that in the control group,
and partially reversed the beclin 1 and LC3B-II expression induced
by doxorubicin (Fig. 2A).
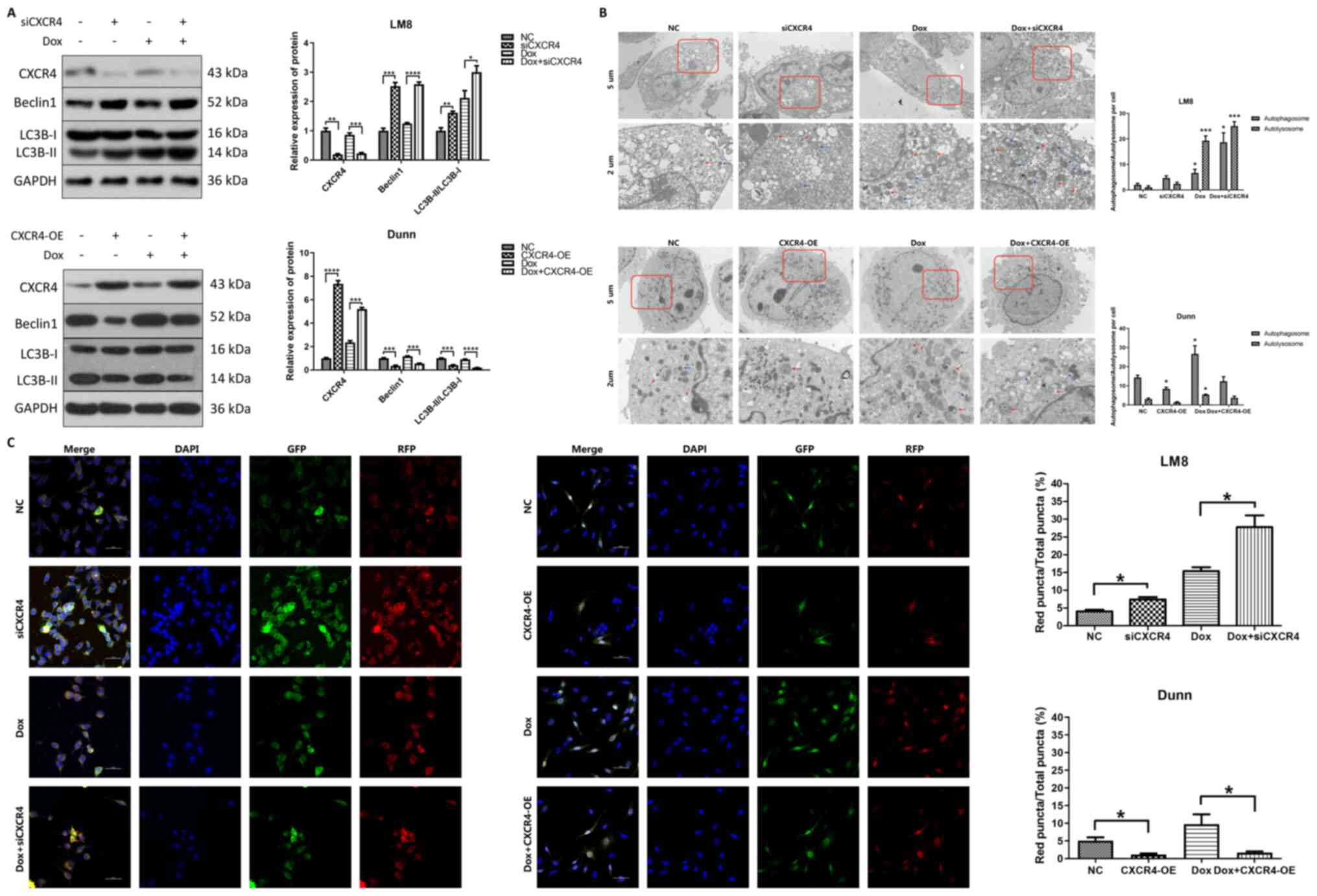 | Figure 2CXCR4 silencing induces autophagy in
LM8 cells, while CXCR4 overexpression suppresses autophagy in Dunn
cells. (A) The expression levels of CXCR4, and of the
autophagy-related proteins beclin 1 and LC3B were determined by
western blotting, and the protein bands were quantified and
subjected to statistical analysis. The protein bands of GAPDH for
CXCR4 silencing and overexpression here are the same as Fig. 1B, as all protein bands in the
Fig. 1B and Fig. 2A are from the same blot. (B)
Autophagosomes and autolysosomes were detected by transmission
electron microscopy in LM8 and Dunn cells after CXCR4 silencing and
overexpression respectively. Red arrows indicated autophagosomes,
while blue arrows indicated autolysosomes. The number of
autophagosomes and autolysosomes was calculated and subjected to
statistical analysis. (C) LM8 and Dunn cells were transfected with
mRFP-GFP-LC3 adenovirus before treatment. The colocalization of RFP
and GFP puncta was examined by confocal microscopy. Yellow puncta
represented autophagosomes, while red represented autolysosomes.
The percentage of red fluorescence was calculated and analyzed with
ImageJ software (National Institutes of Health, version 1.8.0).
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. CXCR4, C-X-C
motif chemokine receptor 4; LC3B, light chain 3B; RFP, red
fluorescent protein; GFP, green fluorescent protein. |
Transmission electron microscopy, which is
considered the golden standard for autophagy detection, was used to
observe the ultrastructure of autophagosomes and autolysosomes in
OS cells. In LM8 cells, compared with that of the control group,
the number of autophagosomes and autolysosomes was obviously
increased in both the CXCR4 silencing group and the
doxorubicin-treated group, and were further increased in the
doxorubicin combined with CXCR4 silencing group. In Dunn cells, the
number of autophagosomes and autolysosomes was markedly decreased
in the doxorubicin combined with CXCR4 overexpression group
compared with that in the doxorubicin group (Fig. 2B).
Considering that autophagy is a dynamic process,
mRFP-GFP-LC3 was utilized to observe autophagic flux via confocal
microscopy. Specifically, autophagosomes were labeled as yellow
puncta, while autolysosomes were labeled as red puncta. In LM8
cells, the percentage of red fluorescence in the CXCR4 silencing
group (7.40±1.11%) was higher than that of the control group
(4.07±0.72%). The percentage of red fluorescence in the doxorubicin
combined with CXCR4 silencing group (27.77±5.75%) was further
increased compared with that of the doxorubicin group
(15.40±1.92%). In Dunn cells, the percentage of red fluorescence
was reduced in the CXCR4 overexpression group (1.17±0.51%) compared
with that of the control group (5.10±1.60%). The percentage of red
fluorescence was significantly decreased in the doxorubicin
combined with CXCR4 overexpression group (1.67±0.64%) compared with
that of the doxorubicin group (9.73±4.81%) (Fig. 2C). These results indicated that
CXCR4 silencing induced autophagic flux activation in LM8 cells,
whereas CXCR4 overexpression suppressed autophagic flux activation
in Dunn cells.
CXCR4-mediated autophagic cell death
reverses OS doxorubicin resistance
Due to the dual role of autophagy in the regulation
of OS chemoresistance, cytoprotective autophagy leads to drug
resistance, while autophagic cell death reverses drug resistance
(18). To further determine
whether CXCR4 silencing increases doxorubicin sensitivity by either
inhibiting cytoprotective autophagy or inducing autophagic cell
death, the autophagy inhibitor bafilomycin A1 and the autophagy
activator rapamycin were used to observe the effect of autophagy on
chemoresistance prior to CXCR4 regulation. Western blot analysis
revealed that pretreatment with bafilomycin A1 reduced beclin 1,
LC3B-II and cleaved caspase 3 levels, and increased P-gp levels in
LM8 cells. Conversely, pretreatment with rapamycin increased beclin
1, LC3B-II and cleaved caspase 3 levels, and reduced P-gp levels in
Dunn cells (Fig. 3A).
Additionally, CCK-8 assay and flow cytometry indicated that
pretreatment with bafilomycin A1 promoted cell proliferation in
vitro and reversed the apoptosis induced by CXCR4 silencing
with or without doxorubicin in LM8 cells. By contrast, rapamycin
inhibited cell proliferation in vitro and enhanced
doxorubicin-induced apoptosis in Dunn cells (Fig. 3B and C). These results
demonstrated that CXCR4 silencing enhances OS doxorubicin
sensitivity by inducing autophagic cell death.
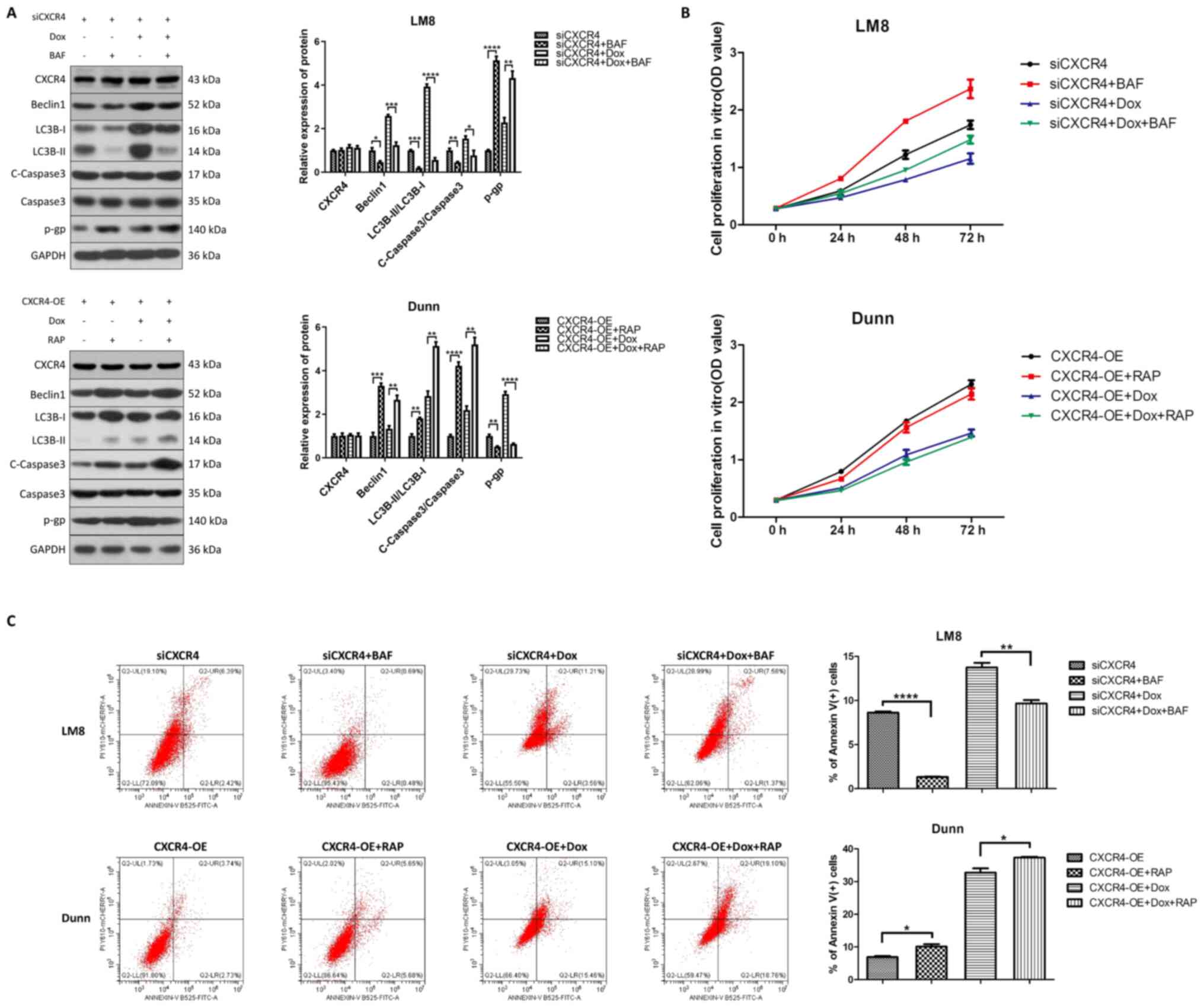 | Figure 3CXCR4 silencing reverses osteosarcoma
doxorubicin resistance by inducing autophagic cell death. (A) LM8
(CXCR4 knockdown) and Dunn (CXCR4 overexpression) cells were
pretreated with bafilomycin A1 (200 nM) and rapamycin (200 nM),
respectively, for 6 h, and then treated with or without 0.2
µg/ml doxorubicin for 48 h. The expression levels of beclin
1, light chain 3B, cleaved caspase 3, caspase 3 and P-glycoprotein
were determined by western blotting, and the protein bands were
semi-quantified and subjected to statistical analysis. (B) Cell
proliferation in each group was detected by Cell Counting Kit-8
assay. (C) The apoptosis ratios for each group (percentage of
Annexin V+ cells) were determined by flow cytometry.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. CXCR4, C-X-C
motif chemokine receptor 4. |
OS doxorubicin resistance regulated by
the CXCR4/autophagy axis is dependent on the PI3K/AKT/mTOR
signaling pathway
To further determine whether the CXCR4/autophagy
axis modulates OS doxorubicin resistance via the PI3K/AKT/mTOR
signaling pathway, one of the most important regulators of
autophagy, western blotting was performed to detect the
phosphorylation levels of PI3K, AKT and mTOR in LM8 and Dunn cells
after doxorubicin treatment and CXCR4 regulation. In LM8 cells,
CXCR4 silencing reduced the phosphorylation of PI3K, AKT and mTOR.
In Dunn cells, CXCR4 overexpression induced the phosphorylation of
PI3K, AKT and mTOR (Fig. 4).
These findings indicated that CXCR4 silencing induced autophagic
cell death to reverse OS doxorubicin resistance by suppressing the
PI3K-AKT-mTOR signaling pathway.
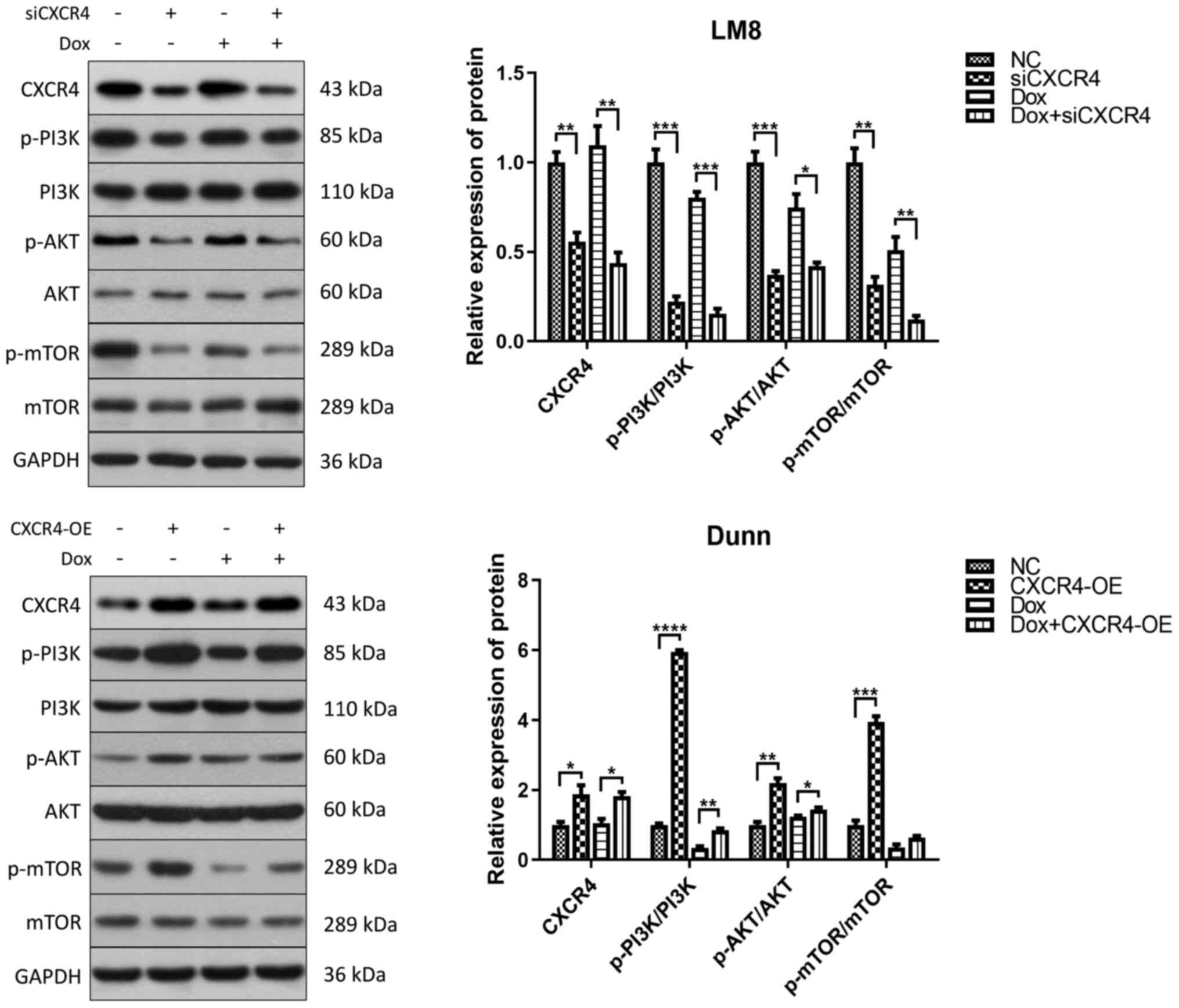 | Figure 4CXCR4 silencing induces autophagic
cell death by suppressing the PI3K-AKT-mTOR signaling pathway LM8
and Dunn cells were cultured in the presence of 0.2 µg/ml
doxorubicin (with or without CXCR4 knockdown/overexpression) for 48
h, and the expression levels of p-PI3K, PI3K, p-AKT, AKT, p-mTOR
and mTOR were determined by western blotting. The protein bands
were semi-quantified and subjected to statistical analysis.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. CXCR4, C-X-C
motif chemokine receptor 4; p-, phosphorylated. |
AMD3100 enhances the antitumor effect of
doxorubicin in an orthotopic OS mouse model
To investigate whether CXCR4 inhibition could
reinforce the cytotoxicity of doxorubicin in vivo, a C3H
mouse orthotopic model was established through intratibial
injection of LM8 cells. Mice were randomly allocated to four groups
and then treated by tail vein injection with PBS, 5 mg/kg AMD3100,
1 mg/kg doxorubicin and AMD3100 plus doxorubicin. After eight
consecutive injections, both treatment with AMD3100 and doxorubicin
resulted in significant tumor growth inhibition compared to that of
the PBS group. Notably, AMD3100 showed a markedly enhanced
antitumor effect compared with that of doxorubicin (Fig. 5A). In the dynamic observation of
mouse body weight, a different trend was found in the four groups:
The body weight in the PBS group increased linearly, while weight
gain was delayed in the AMD3100 and doxorubicin groups, and no
obvious weight change was observed in the AMD3100 combined with
doxorubicin group (Fig. 5B). The
results indicated that, compared with that of the PBS group, after
eight consecutive administrations of AMD3100, doxorubicin and
AMD3100 plus doxorubicin resulted in a notably decreased tumor
volume (50.3, 66.3 and 89.9%, respectively), and induced a weight
loss of 45.9, 67.9 and 82.4%, respectively. (Fig. 5C). Immunohistochemical staining
demonstrated that AMD3100 prominently increased beclin 1 and LC3B
expression, and decreased P-gp expression in AMD3100 plus
doxorubicin-treated tumor tissues (Fig. 5D). Decreased expression of p-PI3K,
p-Akt and p-mTOR was also observed in AMD3100 plus
doxorubicin-treated tumor tissues compared with that of other
groups (Fig. 5E). These findings
suggested that AMD3100 facilitated the antitumor effect of
doxorubicin on tumor growth in vivo.
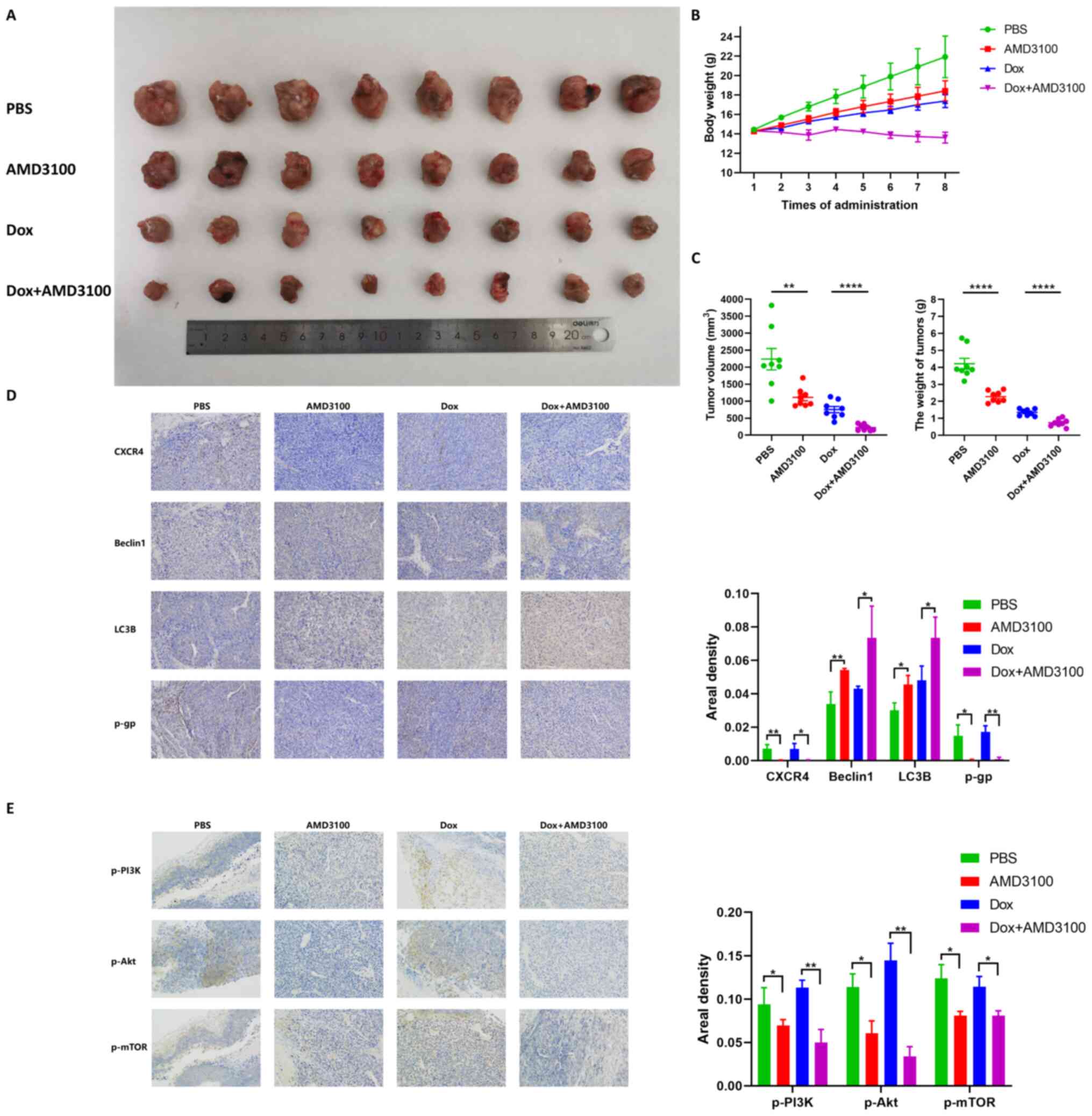 | Figure 5AMD3100 enhances the antitumor effect
of doxorubicin in an orthotopic OS mouse model. (A) Macroscopic
appearance of OS tumors in the tibia of C3H mice after 8
consecutive injections of PBS, 5 mg/kg AMD3100, 1 mg/kg doxorubicin
and AMD3100 plus doxorubicin. (B) The body weight of the mice in
each group was measured at every injection, and the growth curve
was obtained. (C) The volume and weight of the tumors in the four
treatment groups were determined. (D) The expression levels of
C-X-C motif chemokine receptor 4, beclin 1, light chain 3B and
P-glycoprotein in tumor tissues were detected by
immunohistochemical staining, and the areal density was quantified
and subjected to statistical analysis. (E) The expression of
p-PI3K, p-AKT and p-mTOR in tumor tissues was detected by
immunohistochemical staining, and the areal density was quantified
and subjected to statistical analysis. *P<0.05,
**P<0.01, ****P<0.0001. OS,
osteosarcoma; p-, phosphorylated. |
Discussion
Tumor recurrence, distant metastasis and
chemoresistance are three important factors contributing to
treatment failure and poor prognosis in OS (7,19).
It has already been shown that CXCR4 plays a crucial role in OS
survival and metastasis, and targeting CXCR4 is an effective
strategy for OS (10-12). However, whether CXCR4 is involved
in the regulation of OS chemoresistance and its specific mechanism
have not yet been elucidated. The present study first reported that
CXCR4 silencing could increase the sensitivity of LM8 cells to
doxorubicin by inducing autophagic cell death, while CXCR4
overexpression oppositely increased the chemoresistance of Dunn
cells to doxorubicin by inhibiting autophagic cell death. The
results further revealed that the negative correlation between
CXCR4 and autophagy was dependent on the PI3K/AKT/mTOR signaling
pathway. These findings indicate that CXCR4 abrogation could
overcome the chemoresistance of OS cells via autophagic cell
death.
To observe the effect of CXCR4 on OS doxorubicin
resistance, the IC50 of doxorubicin in OS LM8 and Dunn
cells after CXCR4 regulation was calculated. As shown in our
previous study, CXCR4 is highly expressed in LM8 cells and lowly
expressed in Dunn cells (10).
Therefore, CXCR4 silencing and overexpression were performed in LM8
and Dunn cells, respectively. In LM8 cells, the IC50
value of doxorubicin decreased when CXCR4 expression was inhibited.
Conversely, in Dunn cells, the IC50 value of doxorubicin
increased when CXCR4 expression was upregulated.
Observation of apoptosis and caspase family protein
activation induced by chemotherapy drugs is another method to
evaluate chemosensitivity. Since CXCR1 knockdown increased
cisplatin-induced apoptosis and caspase 3 activation in Saos2 and
Saos2-lung cells, Han et al (20) concluded that CXCR1 knockdown
enhanced the sensitivity of OS to cisplatin. Consistent with their
findings, the present study found that CXCR4 silencing facilitated
doxorubicin-induced apoptosis and caspase 3 activation in LM8
cells, while CXCR4 overexpression partially reversed
doxorubicin-induced apoptosis and caspase 3 activation in Dunn
cells.
P-gp, also known as MDR1, which is encoded by
ATP-binding cassette subfamily B member 1, contributes to
chemoresistance in various types of cancer (21). Wang et al (22) demonstrated that raddeanin A
restored doxorubicin chemosensitivity in OS drug-resistant U2OSR
and KHOSR cells by downregulating MDR1. To further identify whether
MDR1/P-gp is involved in CXCR4-mediated doxorubicin resistance,
western blotting was utilized to detect changes in P-gp expression
after CXCR4 regulation. A positive correlation between CXCR4 and
P-gp was found in LM8 and Dunn cell lines. Specifically, both CXCR4
silencing alone and CXCR4 silencing combined with doxorubicin
reduced P-gp expression compared with the findings in LM8 cells. On
the contrary, both CXCR4 overexpression alone and CXCR4
overexpression combined with doxorubicin increased P-gp expression
compared with the findings in Dunn cells. It can be concluded that
combination treatment of CXCR4 silencing and doxorubicin exerts an
enhanced cytotoxic effect on LM8 cells, while CXCR4 overexpression
partially reverses doxorubicin-induced cell death in Dunn
cells.
Autophagy is a catabolic process through which cells
eliminate and recycle their own damaged proteins and organelles to
provide energy. It can be activated under stressful conditions such
as hypoxia, starvation and cytotoxicity induced by chemotherapeutic
drugs to maintain cell survival (23). Autophagy has long been regarded as
a cytoprotective process contributing to OS chemoresistance, and a
number of studies have focused on the role of autophagy inhibition
in OS chemosensitization. Huang et al (24) found that the chemotherapeutic
drugs doxorubicin, cisplatin and methotrexate induced high mobility
group box 1 (HMGB1) expression in MG-63, Saos2 and U2OS cells,
while downregulation of HMGB1 sensitized OS cells to
chemotherapeutic drugs by suppressing the autophagy-related protein
beclin 1. Kim et al (25)
demonstrated that glial cell line-derived neurotrophic factor
receptor α1 promoted OS cisplatin resistance by inducing autophagy.
However, further research confirmed that the dual role of autophagy
in OS chemoresistance, since cytoprotective autophagy contributes
to chemoresistance while autophagic cell death reverses
chemoresistance (18).
Recently, attention has been paid to autophagic cell
death, which is defined as cell death mediated by autophagy rather
than by apoptosis or necrosis (26). An intricate crosstalk between
autophagy and apoptosis is probably involved in the mechanism of
cell death. Generally, the interaction between autophagy and
apoptosis is mostly negative in the sense that autophagy blocks
apoptosis induction while apoptosis-related caspase activation
suppresses the autophagic process. Notably, the induction of
autophagic cell death inversely facilitates apoptosis activation
(26). Previous findings revealed
that autophagy inhibition could induce apoptosis. Wang et al
(27) reported that the antitumor
drug combretastatin A-4 (CA-4) could induce cytoprotective
autophagy, and combined with the autophagy inhibitor chloroquine,
it exerted a synergistic cytotoxic effect on OS cells, since
chloroquine further enhanced CA-4-induced apoptosis by elevating
the levels of the apoptosis-related proteins poly (ADP-ribose)
polymerase (PARP) and caspase 3. Another study found that the
microtubule-disrupting agent CYT997 induced both apoptosis and
autophagy in OS. Furthermore, pretreatment with the autophagy
inhibitor 3-methyladenine (3-MA) enhanced the antitumor effect of
CYT997 by increasing cell apoptosis and the levels of the
apoptosis-related protein PARP (28). By contrast, honokiol, which is
extracted from Magnolia trees, was found to exhibit an antitumor
effect on HOS and U2OS cells by inducing both apoptosis and
autophagy. In addition, honokiol-induced cell death was more
obviously reversed by the autophagy inhibitor 3-MA compared with
that induced by the apoptosis inhibitor Z-VAD-FMK, which indicated
that honokiol-induced cell death was largely dependent on
autophagic cell death (29).
Autophagic cell death induced by tanshinone IIA and diallyl
disulfide exerted an inhibitory effect on 143B and MG-63 cells,
respectively (30,31). Consistent with these findings, the
present study observed that CXCR4 silencing combined with or
without doxorubicin treatment induced autophagy, as shown by
increased expression of beclin 1 and LC3B-II, a larger number of
autophagosomes and autolysosomes, and autophagic flux activation.
By contrast, CXCR4 overexpression blocked autophagy, as shown by
reduced expression of beclin 1 and LC3B-II, a lower number of
autophagosomes and autolysosomes, and autophagic flux inactivation.
To determine whether CXCR4-mediated autophagy has a pro-survival or
pro-death effect, the autophagy inhibitor bafilomycin A1 and the
autophagy activator rapamycin were employed to detect the effect of
CXCR4-mediated autophagy on cell death induced by doxorubicin. The
results revealed that bafilomycin A1 reversed apoptosis induced by
CXCR4 silencing with or without doxorubicin in LM8 cells, while
rapamycin enhanced doxorubicin-induced apoptosis in Dunn cells,
which indicated that the enhanced doxorubicin cytotoxicity caused
by CXCR4 silencing was, at least in part, dependent on autophagic
cell death.
The mechanisms by which GPCRs regulate autophagy
include second messengers such as cAMP and Ca2+, and
downstream signal molecules such as ERK1/2 and mTOR complex 1
modulation (32). The role of
CXCR4 in the regulation of autophagy is paradoxical, since the
positive CXCR4-autophagy loop is involved in drug resistance and
metastasis, and CXCR4 induced by reactive oxygen species stimulated
autophagy formation, which further contributed to drug resistance
in mantle cell lymphoma (14).
This result was consistent with those of another study, which found
that CXCR4 activation decreased the sensitivity of acute myeloid
leukemia cells to cytarabine by inducing autophagy (16). In addition, the autophagy
inhibitor polymeric chloroquine reduced CXCR4-mediated metastasis
in U2OS cells by promoting the internalization of surface CXCR4,
which blocked its binding with the extracellular CXCL12 (33). In addition, the negative
regulation of CXCR4 in autophagy has also been reported. Coly et
al (34) found that CXCR4
activation led to a decrease in the number of autophagosomes in 293
and U87 cells, which indicated that CXCR4 exerted its
anti-autophagy effect by activating calpains, which prevented the
formation of pre-autophagosomal vesicles. Similar to these results,
the present study also observed the anti-autophagy effect of CXCR4
on two OS cell lines. CXCR4 silencing accelerated autophagy
activation in LM8 cells by inhibiting the PI3K/AKT/mTOR signaling
pathway, which is a well-known negative regulator of autophagy.
Inversely, CXCR4 overexpression suppressed autophagy in Dunn cells
by activating the PI3K/AKT/mTOR signaling pathway.
Tumor recurrence, distant metastasis and drug
resistance are the three main reasons contributing to OS treatment
failure (7). AMD3100, a widely
used CXCR4-specific antagonist, only blocks CXCR4 and not any other
C-X-C or C-C chemokine receptors. Thus, it is commonly applied in
cancer research targeting CXCR4 (35). In our previous study, AMD3100 was
able to inhibit OS growth and metastasis in vivo (10). However, whether CXCR4 is also
involved in OS chemoresistance remains unknown. In the present
study, it was found that AMD3100 increased doxorubicin-induced
tumor suppression in an OS orthotopic mouse model.
The limitation of this study is the absence of data
on human OS cell lines. Given that LM8 and Dunn cells were used in
our previous study to demonstrate the role of CXCR4 in the growth
and metastasis of OS (10), and
our present study focuses on CXCR4-mediated OS chemoresistance,
thus we use the LM8 and Dunn cells to perform this study.
In conclusion, the present study shows that CXCR4
blockade enhances the sensitivity of OS to doxorubicin by inducing
autophagic cell death by inhibiting the PI3K/AKT/mTOR signaling
pathway (Fig. 6). Taken together,
these findings elucidate a novel molecular mechanism of CXCR4 in OS
doxorubicin resistance regulation. Targeting the CXCR4/autophagy
axis may be a promising therapeutic strategy to overcome OS
chemotherapy resistance.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YXL designed the study and wrote the manuscript. KYL
drew the graphical abstract image. JYL and ZFZ performed the in
vitro experiments. TYX and DY performed the animal experiments.
QMG, LF and GDL collected and analyzed the data. HYY and KYL
revised the manuscript critically. All the authors read and
approved the final manuscript. YXL, JYL, ZFZ, TYX, DY, QMG, LF,
GDL, HYY and KYL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The animal procedures were performed in accordance
with a protocol approved by the Animal Care and Use Committee of
Shanghai Tenth People's Hospital (approval no.
SHDSYY-2020-3650).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We thank Dr Eugenie Kleinerman (MD Anderson Cancer
Center, University of Texas, Houston, USA) for kindly donating the
murine LM8 and Dunn OS cell lines.
Funding
This study was supported by the Shanghai Tenth People's Hospital
Climbing Talent Program (2021SYPDRC062).
Abbreviations:
|
OS
|
osteosarcoma
|
|
CXCR4
|
C-X-C motif chemokine receptor 4
|
|
CXCL12
|
C-X-C motif chemokine ligand 12
|
|
P-gp
|
P-glycoprotein
|
|
MDR1
|
multidrug resistance 1
|
|
HMGB1
|
high mobility group box 1
|
|
CA-4
|
combretastatin A-4
|
|
3-MA
|
3-methyladenine
|
References
|
1
|
Morrow JJ, Bayles I, Funnell APW, Miller
TE, Saiakhova A, Lizardo MM, Bartels CF, Kapteijn MY, Hung S,
Mendoza A, et al: Positively selected enhancer elements endow
osteosarcoma cells with metastatic competence. Nat Med. 24:176–185.
2018. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Isakoff MS, Bielack SS, Meltzer P and
Gorlick R: Osteosarcoma: Current treatment and a collaborative
pathway to success. J Clin Oncol. 33:3029–3035. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Buondonno I, Gazzano E, Tavanti E, Chegaev
K, Kopecka J, Fanelli M, Rolando B, Fruttero R, Gasco A, Hattinger
C, et al: Endoplasmic reticulum-targeting doxorubicin: A new tool
effective against doxorubicin-resistant osteosarcoma. Cell Mol Life
Sci. 76:609–625. 2019. View Article : Google Scholar
|
|
5
|
Gazzano E, Buondonno I, Marengo A, Rolando
B, Chegaev K, Kopecka J, Saponara S, Sorge M, Hattinger CM, Gasco
A, et al: Hyaluronated liposomes containing H2S-releasing
doxorubicin are effective against
P-glycoprotein-positive/doxorubicin-resistant osteosarcoma cells
and xenografts. Cancer Lett. 456:29–39. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu J, Wang H, Hu Y, Zhang YS, Wen L, Yin
F, Wang Z, Zhang Y, Li S, Miao Y, et al: Inhibition of CaMKIIα
activity enhances antitumor effect of fullerene C60 nanocrystals by
suppression of autophagic degradation. Adv Sci (Weinh).
6:18012332019. View Article : Google Scholar
|
|
7
|
Chen R, Wang G, Zheng Y, Hua Y and Cai Z:
Drug resistance-related microRNAs in osteosarcoma: Translating
basic evidence into therapeutic strategies. J Cell Mol Med.
23:2280–2292. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roundhill EA, Jabri S and Burchill SA:
ABCG1 and Pgp identify drug resistant, self-renewing osteosarcoma
cells. Cancer Lett. 453:142–157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liao YX, Zhou CH, Zeng H, Zuo DQ, Wang ZY,
Yin F, Hua YQ and Cai ZD: The role of the CXCL12-CXCR4/CXCR7 axis
in the progression and metastasis of bone sarcomas (Review). Int J
Mol Med. 32:1239–1246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liao YX, Fu ZZ, Zhou CH, Shan LC, Wang ZY,
Yin F, Zheng LP, Hua YQ and Cai ZD: AMD3100 reduces CXCR4-mediated
survival and metastasis of osteosarcoma by inhibiting JNK and Akt,
but not p38 or Erk1/2, pathways in in vitro and mouse experiments.
Oncol Rep. 34:33–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu Y, Tang L, Zhao S, Sun B, Cheng L,
Tang Y, Luo Z, Lin Z, Zhu J, Zhu W, et al: CXCR4-mediated
osteosarcoma growth and pulmonary metastasis is suppressed by
MicroRNA-613. Cancer Sci. 109:2412–2422. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pollino S, Palmerini E, Dozza B,
Bientinesi E, Piccinni-Leopardi M, Lucarelli E, Righi A, Benassi MS
and Pazzaglia L: CXCR4 in human osteosarcoma malignant progression.
The response of osteosarcoma cell lines to the fully human CXCR4
antibody MDX1338. J Bone Oncol. 17:1002392019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li YJ, Dai YL, Zhang WB, Li SJ and Tu CQ:
Clinicopathological and prognostic significance of chemokine
receptor CXCR4 in patients with bone and soft tissue sarcoma: A
meta-analysis. Clin Exp Med. 17:59–69. 2017. View Article : Google Scholar
|
|
14
|
Chen Z, Teo AE and McCarty N: ROS-Induced
CXCR4 signaling regulates mantle cell lymphoma (MCL) cell survival
and drug resistance in the bone marrow microenvironment via
autophagy. Clin Cancer Res. 22:187–199. 2016. View Article : Google Scholar
|
|
15
|
Dragoj M, Milosevic Z, Bankovic J, Tanic
N, Pesic M and Stankovic T: Targeting CXCR4 and FAK reverses
doxorubicin resistance and suppresses invasion in non-small cell
lung carcinoma. Cell Oncol (Dordr). 40:47–62. 2017. View Article : Google Scholar
|
|
16
|
Hu X, Mei S, Meng W, Xue S, Jiang L, Yang
Y, Hui L, Chen Y and Guan MX: CXCR4-mediated signaling regulates
autophagy and influences acute myeloid leukemia cell survival and
drug resistance. Cancer Lett. 425:1–12. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Y, Liang HM, Lv YQ, Tang SM and Cheng
P: Blockade of SDF-1/CXCR4 reduces adhesion-mediated
chemoresistance of multiple myeloma cells via interacting with
interleukin-6. J Cell Physiol. 234:19702–19714. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liao YX, Yu HY, Lv JY, Cai YR, Liu F, He
ZM and He SS: Targeting autophagy is a promising therapeutic
strategy to overcome chemoresistance and reduce metastasis in
osteosarcoma. Int J Oncol. 55:1213–1222. 2019.PubMed/NCBI
|
|
19
|
Lenna S, Bellotti C, Duchi S, Martella E,
Columbaro M, Dozza B, Ballestri M, Guerrini A, Sotgiu G, Frisoni T,
et al: Mesenchymal stromal cells mediated delivery of photoactive
nanoparticles inhibits osteosarcoma growth in vitro and in a murine
in vivo ectopic model. J Exp Clin Cancer Res. 39:402020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han XG, Du L, Qiao H, Tu B, Wang YG, Qin
A, Dai KR, Fan QM and Tang TT: CXCR1 knockdown improves the
sensitivity of osteosarcoma to cisplatin. Cancer Lett. 369:405–415.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Robey RW, Pluchino KM, Hall MD, Fojo AT,
Bates SE and Gottesman MM: Revisiting the role of ABC transporters
in multidrug-resistant cancer. Nat Rev Cancer. 18:452–464. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Z, Wang C, Zuo D, Zhang T, Yin F,
Zhou Z, Wang H, Xu J, Mao M, Wang G, et al: Attenuation of STAT3
phosphorylation promotes apoptosis and chemosensitivity in Human
osteosarcoma induced by raddeanin A. Int J Biol Sci. 15:668–679.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Amaravadi RK, Kimmelman AC and Debnath J:
Targeting autophagy in cancer: Recent advances and future
directions. Cancer Discov. 9:1167–1181. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L, Tang D and Ni J: Targeting HMGB1-mediated
autophagy as a novel therapeutic strategy for osteosarcoma.
Autophagy. 8:275–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim M, Jung JY, Choi S, Lee H, Morales LD,
Koh JT, Kim SH, Choi YD, Choi C, Slaga TJ, et al: GFRA1 promotes
cisplatin-induced chemoresistance in osteosarcoma by inducing
autophagy. Autophagy. 13:149–168. 2017. View Article : Google Scholar :
|
|
26
|
Marino G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang H, Li W, Xu J, Zhang T, Zuo D, Zhou
Z, Lin B, Wang G, Wang Z, Sun W, et al: NDRG1 inhibition sensitizes
osteosarcoma cells to combretastatin A-4 through targeting
autophagy. Cell Death Dis. 8:e30482017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Z, Yin F, Xu J, Zhang T, Wang G, Mao
M, Wang Z, Sun W, Han J, Yang M, et al: CYT997(Lexibulin) induces
apoptosis and autophagy through the activation of mutually
reinforced ER stress and ROS in osteosarcoma. J Exp Clin Cancer
Res. 38:442019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang K, Chen Y, Zhang R, Wu Y, Ma Y, Fang
X and Shen S: Honokiol induces apoptosis and autophagy via the
ROS/ERK1/2 signaling pathway in human osteosarcoma cells in vitro
and in vivo. Cell Death Dis. 9:1572018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yen JH, Huang ST, Huang HS, Fong YC, Wu
YY, Chiang JH and Su YC: HGK-sestrin 2 signaling-mediated autophagy
contributes to antitumor efficacy of Tanshinone IIA in human
osteosarcoma cells. Cell Death Dis. 9:10032018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yue Z, Guan X, Chao R, Huang C, Li D, Yang
P, Liu S, Hasegawa T, Guo J and Li M: Diallyl disulfide induces
apoptosis and autophagy in Human osteosarcoma MG-63 cells through
the PI3K/Akt/mTOR pathway. Molecules. 24:26652019. View Article : Google Scholar :
|
|
32
|
Wauson EM, Dbouk HA, Ghosh AB and Cobb MH:
G protein-coupled receptors and the regulation of autophagy. Trends
Endocrinol Metab. 25:274–282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu F, Li J, Xie Y, Sleightholm RL and
Oupicky D: Polymeric chloroquine as an inhibitor of cancer cell
migration and experimental lung metastasis. J Control Release.
244:347–356. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coly PM, Perzo N, Le Joncour V, Lecointre
C, Schouft MT, Desrues L, Tonon MC, Wurtz O, Gandolfo P, Castel H
and Morin F: Chemotactic G protein-coupled receptors control cell
migration by repressing autophagosome biogenesis. Autophagy.
12:2344–2362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Tannous BA, Poznansky MC and Chen
H: CXCR4 antagonist AMD3100 (plerixafor): From an impurity to a
therapeutic agent. Pharmacol Res. 159:1050102020. View Article : Google Scholar : PubMed/NCBI
|















