Introduction
Chemo- and immunotherapy resistance is, besides
incomplete cytoreductive surgery and tumor metastasis, a major
cause for recurrence and cancer therapy failure. Therapy resistance
can be either intrinsic where tumors are not sensitive to therapy
due to existing resistance-causing factors or acquired where
initially sensitive tumors become resistant during therapy. Major
causes for drug resistance have been extensively studied in the
past: elevated expression of drug efflux transporters, increased or
decreased DNA-damage repair, altered drug metabolism and
detoxification, mutated drug targets, altered survival/death
signaling, hypoxia, and presence of cancer stem cells (1,2).
Poly (ADP-ribose) polymerase (PARP)-inhibitors
(PARPi) such as olaparib and niraparib are successfully used as a
treatment of platinum-sensitive Breast Cancer Susceptibility Gene
(BRCA)-deficient tumors of the breast and the ovaries. They
are also effective in tumors with a 'BRCAness'-phenotype,
that is tumors with no mutations in BRCA genes but with
loss-of-function mutations in genes encoding other key players in
the homologous recombination repair (HRR) pathway. BRCA1 and
BRCA2 are crucial for the HRR process. PARPi are
small-molecule inhibitors of PARP enzymes 1, 2, and 3, which are
involved in detection and repair of single-strand breaks (SSB).
PARPi block PARP catalytic activity or 'trap' PARP molecules onto
the DNA, resulting in SSB persistence, replication fork stalling,
and accumulation of DNA double-strand breaks (DSB), eventually
leading to a 'synthetic lethality' phenotype in an HRR-deficient
(HRD) background (3-9). However, emerging evidence indicates
that PARPi are even effective in cells with functional HRR
(10).
Although BRCA-mutations are a favorable
predictor for PARPi sensitivity, a significant number of
BRCA-mutated cancer nevertheless fail to respond to PARPi
(11-14), either due to intrinsic resistance
or resistance acquisition during the treatment and possibly
enforced by the 'mutator phenotype' of HRD cells. Mechanisms of
PARPi resistance include the restoration of HRR through reversion
mutations in BRCA and RAD51, through BRCA1
promotor alterations, or through reconstitution of end-resection;
the occurrence of alterations of PARP1 that abolish PARPi-trapping;
the stabilization of the replication fork via depletion of
chromatin remodelers; and the increased PARPi efflux via
overexpression of multidrug resistance (MDR) pumps (15-17).
Reversion mutations (insertions or deletions) 'stamp
out' pathogenic BRCA or RAD51 mutations and thus
re-establish the reading frame, remove the deleterious mutation, or
cause a synonymous mutation, restoring functional BRCA and
RAD51 (18-20). Restoration of homologous
recombination (HR) with coinciding BRCA protein re-expression due
to de-methylation of BRCA1 was demonstrated in
patient-derived xenograft (PDX) models of PARPi-resistant breast
cancer (21) as well as in
patients with ovarian cancer (22). HR restoration can also occur by
loss of the end-resection repressor 53BP1 (23) or depletion of the shielding
complex component REV7 (24).
The occurrence of mutations in the PARP1 gene
is the most obvious cause for abolished PARPi-trapping: These
mutations may result in a PARP protein unable to become trapped in
response to PARPi. This has been found in an ovarian cancer patient
with olaparib de novo resistance: she had a PARP1 mutation
affecting a region critical for the cross-talk between the
DNA-binding and catalytic domains (25). Phosphorylation of PARP1 was also
found to reduce binding by PARPi, increase PARP1 enzymatic
activity, and thus confers resistance to PARPi (26).
The dysregulation or collapse of replication forks,
where for instance a variety of fork- and chromatin-remodeling
proteins promote MRE11-dependent nascent DNA strand degradation at
stalled replication forks, is one important feature of PARPi
cytotoxicity. Depletion of these remodelers prevented strand
degradation by MRE11, leading to the stabilization of the
replication fork, and resistance to olaparib in BRCA1/2
deficient cells (27,28).
MDR1 overexpression is a common feature for MDR and
the negative impact of MDR1 overexpression on olaparibsensitivity
was shown in a mouse model (29)
and also in breast and ovarian cancer cases where PARPi-resistance
could be attributed to fusions and rearrangements of genes located
near ABCB1 (30).
The issue whether PARPi exposure can generate an
acquired resistance phenotype in cancer cells has only been poorly
addressed, with controversial results been reported. Two studies
reported development of acquired PARPi resistance in ovarian cancer
cells by olaparib (31) and in
breast cancer cells by niraparib (32) whereas another study reported that
niraparib failed to induce mutations responsible for treatment
resistance (33).
Thus, it remains unclear whether PARPi-imposed
resistance acquisition is a common or a rather rare phenomenon,
whether it is cell line-dependent, whether it depends on the
mutational profile of genes implicated in DNA damage pathways (e.g.
BRCA, TP53) or on the genetic background in general,
whether it occurs preferably in cells that are intrinsically
PARPi-sensitive, and whether it is a permanent or rather a
transient (resistance phenotype is lost after removal of the
selection pressure) phenomenon. In addition, it is unclear whether
cells with an acquired PARPi resistance are cross-resistant to
PARPi-unrelated compounds such as platinum salts, anthracyclines
and even compounds not directly interacting with the DNA like
Paclitaxel.
To address these topics, the intrinsic sensitivity
of a panel of fourteen ovarian cancer cell lines to olaparib and
niraparib as well as to PARPi-unrelated compounds was first
determined, and then a subpanel of seven cell lines (six with and
one without TP53-mutations, and two with and five without
BRCA-mutations) was selected to investigate whether exposure
to escalating olaparib concentrations generates subcells with
acquired resistance to PARPi and/or PARPi-unrelated compounds.
Materials and methods
Cell culture and drugs
The following fourteen parental cell lines were
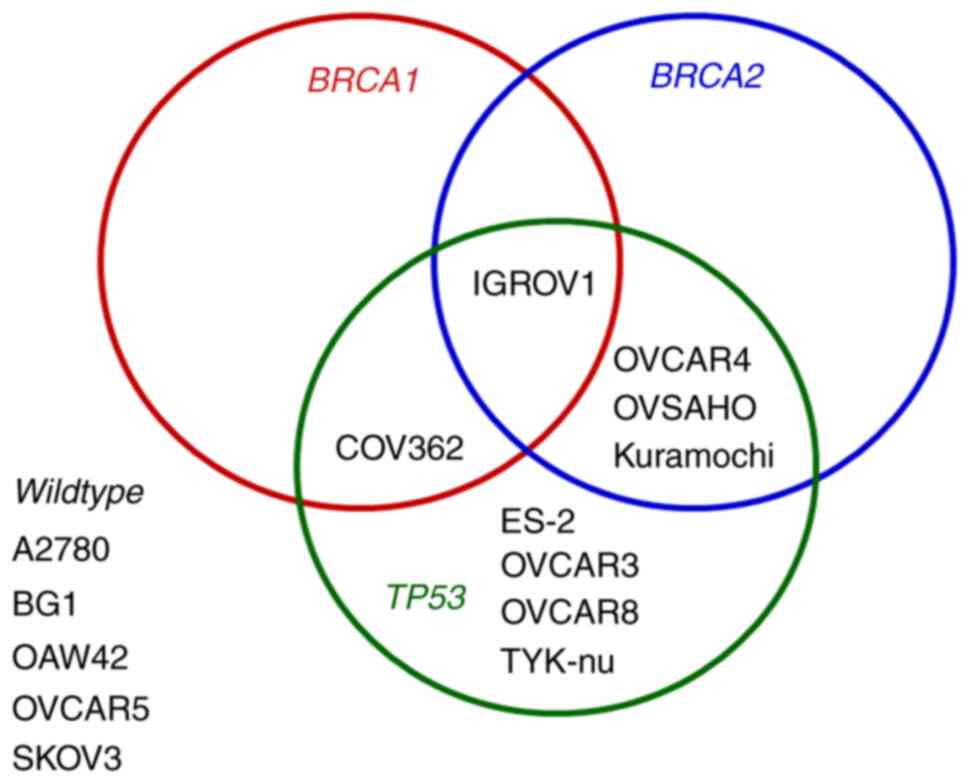
used: A2780, BG1, CaOV3, COV362, ES-2, IGROV1, Kuramochi, OVCAR3,
OVCAR4, OVCAR5, OVCAR8, OVSAHO, SKOV3, and TYK-nu. Nine of them are
TP53-mutated and five BRCA-mutated (Fig. 1). Cisplatin-resistant A2780/CP
were obtained from parental A2780 cells by stepwise exposure to
increasing cisplatin concentrations (35). The cisplatin-resistant TYK-nu(R)
cells were developed from parental TYK-nu cells by stepwise
exposure to cisplatin and obtained from JCRB Japan Cell Bank
(36). Paclitaxel-resistant
IGROV1-PXL were generated from parental IGROV1 cells through
stepwise exposure to escalating concentrations of paclitaxel in our
lab (37). All cell lines were
cultured in RPMI-1640 (cat. no. R8758) supplemented with 10% fetal
bovine serum (FBS; both from Sigma-Aldrich; Merck KGaA),
penicillin/streptomycin (100 U/ml/100 µg/ml; Sigma-Aldrich;
Merck KGaA) at 37°C in a 95% humidified atmosphere containing 5%
CO2. All cell lines are STR-profiled and routinely
tested for mycoplasma infection.
Chemotherapeutic drugs were obtained from various
suppliers: Gloucester Pharmaceuticals Inc. (romidepsin); Labatec
(carboplatin); MedChemExpress (OTS167); Sigma-Aldrich; Merck KGaA
[cisplatin, doxorubicin, epirubicin, paclitaxel and suberoylanilide
hydroxamine (SAHA)]. They were dissolved in DMSO (niraparib,
olaparib and SAHA), methanol (paclitaxel), or water (carboplatin,
cisplatin, doxorubicin, epirubicin, OTS167 and romidepsin) and
stored as aliquots at −20°C.
Drug sensitivity and cell proliferation
rate
Drug sensitivity was determined by the MTT-assay and
the colony formation assay (CFA). For the MTT-assay, cells
(5,000-7,000 in 200 ml medium: density depends on the cell line)
were seeded into 96-well plates and treated with each drug for 72
h: carboplatin (range: 3-500 µM), cisplatin (0.5-50
µM), doxorubicin (3-3,000 nM), epirubicin (0.1-10
µM), niraparib (0.5-100 µM), olaparib (3-1,000
µM), OTS167 (10-1,000 nM), paclitaxel (0.5-300 nM),
romidepsin (0.1-100 nM), SAHA (0.5-300 µM).
Then MTT-dye (cat. no. M2128; Sigma-Aldrich; Merck
KGaA; final concentration: 0.5 mg/ml) was added for 3 h, followed
by removal of the medium and dissolution of the purple formazan
crystals with DMSO. The optical density (OD; absorbance at 540 nm)
was measured using the SynergyH1 Hybrid Reader (BioTek Instruments,
Inc.). Data (mean ± SD of at least four independent experiments
performed in quadruplets) are presented as the relative
proliferation as a function of time after seeding.
IC50-values were calculated by linear extrapolation. The
ratio of the IC50-values of the matched subcells and the
parental cells was calculated. Subcells were considered resistant
if the ratio was ≥2.0 or hypersensitive if ≤0.5.
For the CFA, 1,000 cells in 2-ml culture medium were
seeded into 12-well plates and exposed to olaparib on the next day
for 8-10 days: 0.1, 0.2, 0.5, 1 µM olaparib for A2780 cells;
4, 8, 16, 32 µM for IGROV1; 0.3, 1, 3, 10 µM for
OVCAR3; 1, 2, 5, 10 µM for OVCAR8. Then the medium was
removed, the colonies were fixed at room temperature for 30 min and
stained with 0.05% crystal violet (Sigma-Aldrich; Merck KGaA) in 4%
formalin (Formafix AG), the plates were then rinsed 3-4 times with
water and dried and images of the plates were captured (Fusion FX7
Edge Imaging System; Witek AG).
Cell proliferation rate was calculated from cell
counts at the seeding day and the harvesting day by the following
formula for exponential growth: Td = T1 × log(2)/log(N1/N0): where N0 is the number of
cells seeded at time T0, N1 the number of cells harvested after
time T1 and Td the doubling-time (proliferation rate).
Western blotting
Western blot analysis was used to assess the
expression of MDR1. Cell lysates were obtained from subconfluent
cultures at the time of harvest. Cells were lysed with RIPA buffer
(cat no. 9806; Cell Signaling Technology Europe). Protein
concentration was determined by the BCA Protein Assay (cat. no.
23227; Pierce; Thermo Fisher Scientific, Inc.). A total of 20
µg of protein were loaded and separated using sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (10% SDS-PAGE), followed
by blotting onto a polyvinylidene difluoride (PVDF) membrane (cat.
no. 162-0177; BioRad Laboratories, Inc.) according to standard
protocols. Membranes were blocked at room temperature for 60 min in
TBST/milk [Tris-buffered saline with 0.1% Tween® 20
(Sigma-Aldrich; Merck KGaA)] and containing 3% (w/v) fat-free milk
powder purchased from a local grocer). Cas9, EGFP, GAPDH, MDR1,
PARP1 and tubulin were detected with specific primary antibodies:
mouse anti-Cas9 (cat. no. 14697; Cell Signaling Technology, Inc.),
mouse anti-EGFP (cat. no. sc-9996; Santa Cruz Biotechnology, Inc.),
mouse anti-GAPDH (cat. no. sc-47724; Santa Cruz Biotechnology,
Inc.), mouse anti-MDR1 (cat. no. sc-13131; Santa Cruz
Biotechnology, Inc.), rabbit anti-PARP1 (cat. no. 9542; Cell
Signaling Technology, Inc.) and rabbit anti-tubulin antibody (cat.
no. 2148; Cell Signaling Technology, Inc.). All primary antibodies
were diluted 1:1,000 in TBST/milk, except MDR1 which was diluted
1:500). Following the primary incubation (at 4°C for overnight),
the membrane was incubated at room temperature for 3 h with the
matched secondary antibodyeither horseradish peroxidase
(HRP)-conjugated anti-mouse (cat. no. 7076) or HRPO-conjugated
anti-rabbit (cat. no. 7074; both from Cell Signaling Technology,
Inc.) antibody (both diluted 1:2,000 in TBST/milk) Complexes were
visualized by enhanced chemiluminescence (Dura West; Pierce; Thermo
Fisher Scientific, Inc.) and autoradiography (Fusion FX7 Edge
Imaging System, Witek AG).
Generation of cell lines with acquired
resistance
The following protocol was used to generate cells
with acquired resistance to olaparib. It is based on the selection
principle of clonal growth by repeated exposure of cells to
stepwise escalating drug concentrations, assuming that cells
acquire new features in an irreversible fashion by chronic drug
exposure, as previously described (38).
The seven cell lines subjected to this protocol were
selected according the following criteria. They exhibited different
status for TP53, BRCA1 and BRCA2 (Fig. 1) and displayed different intrinsic
olaparib sensitivity, ranging from relatively sensitive to
relatively resistant (Fig. 2A):
A2780, ES-2, IGROV1, OVCAR3, OVCAR4, OVCAR8, and TYK-nu). Briefly,
50,000 cells were seeded into six-well plates and exposed to
olaparib for 48 h, followed by replacement of the
olaparib-containing medium by olaparib-free medium in order to
allow viable cells to recover and expand to confluency. After
recovery, cells were re-seeded and exposed to a higher
concentration of olaparib for 48 h, again followed by replacement
of the olaparib-containing medium and recovery and expansion of
viable cells. These cycles of exposure with escalating olaparib
concentrations were repeated until no viable cells were left
anymore in the last step of the protocol. This protocol and in
particular the 48 h-exposure was selected because it was able to
produce histone deacetylase inhibitor resistance acquisition in our
previous studies (38-40). However, it is considered
worthwhile trying different experimental protocols and conditions
to induce PARPi resistance in future studies. The following matched
subcells were obtained: A2780-OLA, ES-2-OLA, IGROV1-OLA,
OVCAR3-OLA, OVCAR4-OLA, OVCAR8-OLA, TYK-nu-OLA. Protocol details
are summarized in Table I (the
start and end concentration of olaparib, the number of cycles and
the total olaparib escalation for each cell line). For each cell
line the protocol was performed twice.
 | Table IGeneration of subcells by escalating
olaparib concentrations. |
Table I
Generation of subcells by escalating
olaparib concentrations.
| Subcell line | Concentration start
(µM) | Concentration end
(µM) | Number of
cycles | Escalation
(fold) |
|---|
| A2780-OLA | 500 | 1080 | 2 | 2.16 |
| ES-2-OLA | 30 | 1800 | 5 | 60 |
| IGROV1-OLA | 200 | 1400 | 6 | 7 |
| OVCAR3-OLA | 250 | 800 | 8 | 3.3 |
| OVCAR4-OLA | 160 | 1200 | 4 | 7.5 |
| OVCAR8-OLA | 200 | 1200 | 4 | 6 |
| TYK-nu-OLA | 125 | 600 | 3 | 4.8 |
In order to monitor potential olaparib resistance
acquisition, matched subcells and parental cells were subjected to
MTT assays after each cycle. Resistance was defined if the ratio of
the IC50-values of the subcells and the parental cells
is ≥2.0.
Generation of CRISPR-Cas9-mediated
PARP1-knockout ovarian cancer cell lines
For molecular cloning, single guide RNAs (sgRNA)
targeting protein-coding genomic DNA sequences of PARP1
gene, exon 1 sg1_PARP1_5′-CGA GTC GAG TAC GCC AAG AG-3′, exon 2
sg2_PARP1 5′-TGG GTT CTC TGA GCT TCG GT-3′, and exon 1
sg3_PARP1_5′-GCA TCC CCA AGG ACT CGC TC-3′, were designed using
Benchling (Biology Software, 2021, retrieved from https://benchling.com). Single strand oligonucleotides
were purchased from Sigma-Aldrich; Merck KGaA and cloned into
LRG2.1 (cat. no. 108098; Addgene, Inc.) plasmid using the
BsmBI endonuclease restriction site. Annealed
oligonucleotides were ligated into the desired plasmid using the
T4-DNA ligase (Promega Corporation) for subsequent expression of
the sgRNA together with EGFP fluorescent protein. Ligations were
transformed into Stbl3 E. coli following ampicillin
selection using ZR Plasmid Miniprep-Classic plasmid purification
(Zymo Research Corp.) and Sanger DNA sequencing (Microsynth AG) was
used to confirm insertion of respective sgRNA using the human U6
primer (5′-GAG GGC CTA TTT CCC ATG ATT-3′).
Constitutively Cas9+ expressing
ovarian cancer cell lines were generated by lentiviral transduction
and subsequent puromycin selection. In brief, 293T cells (kindly
provided by Dr Neutzner, Department of Biomedicine, University
Hospital Basel) were seeded in a T75 flask at 50% confluency one
day before transfection for preparation of lentiviral particles. A
total of 4 µg of LRG2.1 (cat. no. 108098) or
pLenti-Cas9-P2A-Puro (ca. no. 110837), 2 µg of pMD2.G
(cat. no. 12259) and 2 µg of pCMVR8.74 (cat. no. 22036; all
from Addgene, Inc.) were co-transfected using 24 µl of
jetPEI reagent in 1 ml of 150 mM NaCl solution
(Polyplus-transfection; Chemie Brunschwig AG). Growth medium was
changed 24 h after transfection. Supernatant containing-lentivirus
particles was collected 48 h later and filtered with a
0.45-µm PVDF filter (Sartorius AG), aliquoted in cryotubes
and stored at −80°C until further use. OVCAR3, OVCAR5, and OVCAR8
cells were transduced with 1 ml of supernatant containing
pLenti-Cas9-P2A-Puro lentivirus particles and further selected with
1-3 µg/ml puromycin for one week. Selected
Cas9+ cell lines were kept in media containing 1
µg/ml puromycin. Cells lines stably expressing Cas9 protein
were then lentiviral-transduced either with sgRNAs targeting the
AAVS1 loci (mock) (41);
or PARP1 followed by sorting enrichment of EGFP+
cells using the BD FACSAria Cell Sorter (BD Biosciences).
To analyze and confirm CRISPR-mediated mutagenesis,
genomic DNA of Cas9+ non-gRNA transduced
(control) or transduced cells with sgRNAs targeting AAVS1
(mock) sg1_ AAVS1_5′-ACT GTT GAC GGC GGC GAT GT-3′, sg2_AAVS1_
5′-GCT GAT ACC GTC GGC GTT GG-3′; or PARP1 sg1_ PARP1_5′-CGA
GTC GAG TAC GCC AAG AG-3, sg2_PARP1 5′-TGG GTT CTC TGA GCT TCG
GT-3′, sg3_PARP1_5′-GCA TCC CCA AGG ACT CGC TC-3′ was extracted
using the DNeasy Blood & tissue Kit (cat. no. 69504 Qiagen) 3
and 6 days after transduction. The genomic locus targeted by
PARP1 was amplified using the forward 5′-GGG GGA GGG GTT GGG
GGT AAA A-3′ and reverse 5′-GCC TTC AAG CCC ACC ACC TCA C-3′
primers. PCRs were performed using 2xGoTAq green Master Mix
(Promega Corporation), 200 nM of each primer and 100 ng of genomic
DNA. PCR conditions were as follows: Initial DNA denaturation at
94°C for 5 min followed by 32 cycles of 95°C for 20 sec, 62°C for
15 sec and 72°C for 1 min and 30 sec with a final extension at 72°C
for 5 min. Amplicons were visualized on 1% agarose gel and purified
by Wizard SV gel and PCR Clean/up System (Promega Corporation).
Amplicons were analyzed using the Tracking of Indels by
Decomposition (TIDE) assay (42).
Statistical analysis
For all comparisons, the mean ± SD values were
calculated and statistical analysis was performed using the paired,
two-tailed Student's t-test (Microsoft Excel, version 2016).
P<0.05 was considered to indicate a statistically significant
difference. For correlation analyses, the Spearman's rank
correlation was calculated (Microsoft Excel).
Results
PARPi-sensitive ovarian cancer cells are
not only sensitive to platinum but also to other chemotherapeutic
compounds
At first the sensitivity of fourteen cell lines
(TP53-mutated, n=9; BRCA-mutated, n=5) (Fig. 1), to olaparib and niraparib was
determined. The results demonstrated that these cell lines display
a wide spectrum of olaparib and niraparib sensitivity, with ES-2
and BG1 as the most sensitive and COV362 and OVSAHO as the least
sensitive cells to olaparib (Fig.
2A), and with BG1 and TYK-nu the most sensitive and OVCAR8 and
COV362 the least sensitive cells to niraparib (Fig. 2B). Spearman's rank correlation
analysis (Fig. 2C) showed that
olaparib-sensitive cells were commonly also niraparibsensitive
(rs=0.582). They also indicated that among the fourteen cell lines,
those with BRCA-mutations (COV362, Kuramochi, OVCAR4,
IGROV1) tended to be less sensitive to both PARPi than those
without BRCA-mutations (Fig.
2A-C). No correlation was found between the proliferation rate
of ovarian cancer cells and their sensitivity to olaparib or
niraparib (Fig. 2D).
Then it was determined whether PARPi-sensitive cells
were, in addition to expectedly being sensitive to platinum
compounds, also sensitive to other classes of chemotherapeutic
compounds, such as representatives of the classes of
anthracyclines, taxanes, histone deacetylase (HDAC)-inhibitors
(HDACi), and maternal embryonic leucine zipper kinase
(MELK)-inhibitor. The latter three are not known to interact with
the DNA. MELK expression has recently been shown to correlate with
poor outcome in ovarian cancer and its inhibition by OTS167
abrogates proliferation and viability of ovarian cancer cells
(43). HDACi act epigenetically,
that is without directly interacting with the DNA, and induce
acetylation of histones and non-histone proteins and thus control
gene transcription, protein function, proliferation and apoptosis
(44). They have been shown to
inhibit the growth and spread of ovarian tumors and synergize with
platinum-based chemotherapy drugs (45), although their clinical usefulness
remains unclear (46). PARPi
sensitivity was not only identified in cells sensitive to
carboplatin and cisplatin (rs platinum compounds=0.542) but in
cells sensitive to doxorubicin and epirubicin (rs
anthracyclines=0.456), to paclitaxel (rs=0.667), and to romidsepsin
and SAHA (rs HDACi= 0.607). An association was also found between
PARPi and compounds that interact with the DNA (rs=0.587)
and compounds that do not interact with the DNA (rs=0.633). No
correlation was found for OTS167 (rs=0.248) (Fig. 3). Details are summarized in
Table II.
| Table IISpearman's rank correlation between
sensitivity to PARPi and other compounds. |
Table II
Spearman's rank correlation between
sensitivity to PARPi and other compounds.
| Spearman's rank
correlation (rs) | Olaparib | Niraparib | Olaparib +
niraparib |
|---|
| Olaparib | | 0.582 | |
| Niraparib | 0.582 | | |
| Carboplatin | 0.358 | 0.460 | |
| Cisplatin | 0.602 | 0.510 | |
| Platinum | | | 0.542 |
| Doxorubicin | 0.376 | 0.386 | |
| Epirubicin | 0.210 | 0.513 | |
| Anthracyclines | | | 0.456 |
| Paclitaxel | 0.622 | 0.597 | |
| Taxanes | | | 0.667 |
| Romidepsin | 0.418 | 0.434 | |
| SAHA | 0.654 | 0.432 | |
| HDACi | | | 0.607 |
| OTS167 | 0.182 | -0.024 | |
| MELKi | | | 0.248 |
| DNAa | 0.458 | 0.589 | 0.587 |
| non-DNAb | 0.631 | 0.611 | 0.633 |
| ALLc | 0.490 | 0.610 | 0.656 |
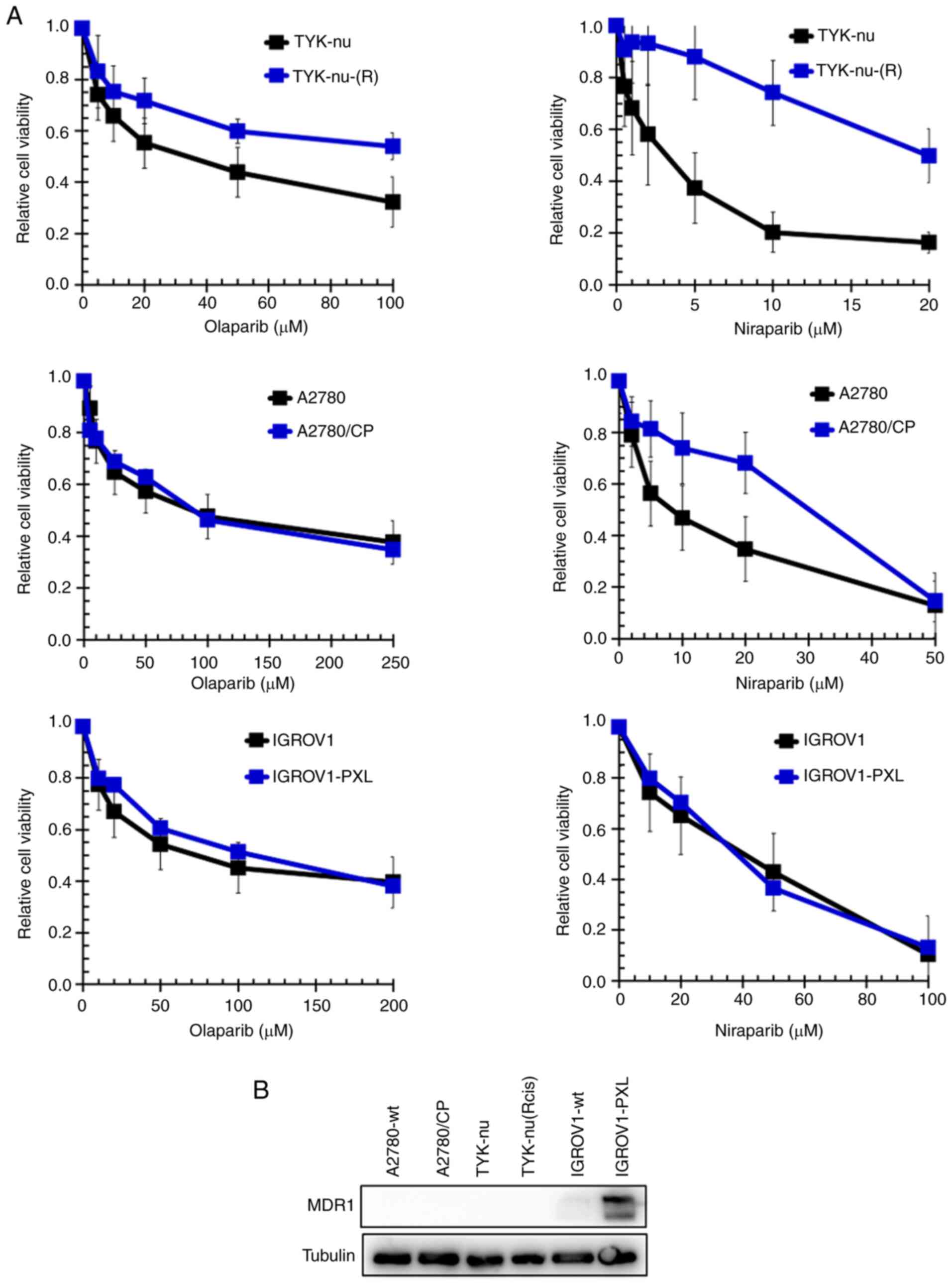
Next, the sensitivity to olaparib and niraparib of
cells with acquired cisplatin-resistance [TYK-nu(R) and A2780/CP
cells] or acquired paclitaxel-resistance (IGROV1-PXL cells) was
determined. TYK-nu(R) are 4-fold and A2780/CP are >9-fold
resistant to cisplatin, and IGROV1-PXL are 9.3-fold resistant to
paclitaxel (37). The results
(Fig. 4A) demonstrated that
TYK-nu(R) cells were cross-resistant to both PARPi (4-fold to
olaparib and 6-fold to niraparib), while A2780/CP cells were
cross-resistant to niraparib only (3.1-fold). By contrast,
IGROV1-PXL cells retained sensitivity to both olaparib and
niraparib. IGROV1-PXL cells express MDR1, but cisplatin-resistant
A2780/CP and TYK-nu(R) cells do not (Fig. 4B). Details are shown in Table III.
| Table IIIOlaparib/niraparib sensitivity of
cells with acquired cisplatin/paclitaxel resistance. |
Table III
Olaparib/niraparib sensitivity of
cells with acquired cisplatin/paclitaxel resistance.
| Cell line | Olaparib
(µM) | Niraparib
(µM) |
|---|
| TYK-nu | 40.6±24.9 | 3.3±2.0 |
| TYK-nu-(R) | 163.3±40.4 | 19.5±4.5 |
| Ratio | 4.0 | 6.0 |
| P-value | 0.019 | 0.016 |
| A2780 | 81.3±31.0 | 8.7±5.1 |
| A2780/CP | 85.1±13.3 | 29.7±5.6 |
| Ratio | 1.05 | 3.1 |
| P-value | 0.814 | 0.001 |
| IGROV1 | 80.4±26.1 | 43.0±17.8 |
| IGROV1-PXL | 98.8±38.5 | 41.8±28.6 |
| Ratio | 1.23 | 0.97 |
| P-value | 0.433 | 0.940 |
Olaparib does not induce acquired
resistance to PARPi or acquired cross-chemoresistance
Expanding on the previous studies which reported
opposing results on PARPi-imposed resistance acquisition (31-33), seven cell lines differing in their
status for TP53, BRCA1 and BRCA2, and also in
their intrinsic sensitivity to olaparib (from relatively sensitive
to relatively resistant) were selected (Figs. 1 and 2A). These cell lines were subjected to
the protocol of 'repeated exposure with olaparib concentration
escalation' as described in 'Materials and methods', yielding the
following subcell lines: A2780-OLA, ES-2-OLA, IGROV1-OLA,
OVCAR3-OLA, OVCAR4-OLA, OVCAR8-OLA and TYK-nu-OLA. The
proliferation rate and the sensitivity to olaparib, niraparib,
carboplatin, cisplatin, doxorubicin and paclitaxel were also
determined, demonstrating that the parental cells and their matched
subcells have comparable proliferation rates (Table IV).
| Table IVProliferation rates
(doubling-times). |
Table IV
Proliferation rates
(doubling-times).
| Cell line | Time (h) | Ratio
(OLA/par) | P-value |
|---|
| ES-2 | 35.4±7.9 | | |
| ES-2-OLA | 30.4±4.8 | 0.86 | 0.003 |
| IGROV1 | 23.7±3.6 | | |
| IGROV1-OLA | 27.7±8.7 | 1.17 | 0.015 |
| OVCAR3 | 43.6±9.8 | | |
| OVCAR3-OLA | 48.1±8.3 | 1.10 | 0.480 |
| OVCAR4 | 32.9±10.4 | | |
| OVCAR4-OLA | 27.3±5.1 | 0.83 | 0.020 |
| OVCAR8 | 25.3±2.5 | | |
| OVCAR8-OLA | 27.6±3.4 | 1.09 | 0.139 |
| A2780 | 21.5±2.6 | | |
| A2780-OLA | 23.8±2.3 | 1.11 | 0.060 |
| TYK-nu | 36.6±7.4 | | |
| TYK-nu-OLA | 34.8±3.7 | 0.95 | 0.747 |
The MTT-assay results demonstrated that in none of
these seven cell lines, stepwise 48 h-exposure of cells to
escalating olaparib produced acquired PARPi resistance or an
acquired cross-resistance to platinum compounds, doxorubicin, and
paclitaxel (Fig. 5A), meaning
that resistance factors (ratio from the IC50-values of
the resistant subcells vs. the parental cells) >2.0 were not
found. Detailed data are summarized in Table V. The failure to produce acquired
PARPi-resistance was confirmed by CFAs for A2780, IGROV1, OVCAR3
and OVCAR8 (cell lines that form distinct colonies rather than
proliferating as a confluent monolayer), where the clonogenic
potential of the-OLA cells was not different from that of their
parental cells in response to olaparib (Fig. 5B). The absence of an acquired
resistance phenotype in all cells in this setup also indicated that
it was not relevant whether or not TP53 and/or BRCA
were mutated. OVCAR8 cells were also continuously (instead of 48 h
as for olaparib) exposed to three cycles of escalating Niraparib
and resistance acquisition to Niraparib, Olaparib, Carboplatin and
Doxorubicin was not observed (data not shown).
| Table VDrug sensitivity
(IC50-values) of subcells exposed to escalating
olaparib. |
Table V
Drug sensitivity
(IC50-values) of subcells exposed to escalating
olaparib.
| Olaparib
(µM) | Niraparib
(µM) | Carboplatin
(µM) | Cisplatin
(µM) | Paclitaxel
(nM) | Doxorubicin
(nM) |
|---|
| A2780 | 150.8±29.1 | 20.4±3.3 | 130.8±18.5 | 10.8±1.0 | 6.1±3.9 | 15.1±6.8 |
| A2780-OLA | 86.3±47.2 | 14.6±7.6 | 103.5±8.5 | 6.9±1.1 | 3.3±1.5 | 6.9±1.7 |
| Ratio | 0.57 | 0.72 | 0.79 | 0.64 | 0.54 | 0.46 |
| P-value | 0.068 | 0.236 | 0.052 | 0.002 | 0.255 | 0.092 |
| ES-2 | 71.4±56.6 | 13.5±12.0 | 90.6±49.5 | 6.6±1.2 | 31.6±33.5 | 102.6±70.8 |
| ES-2-OLA | 185.7±75.9 | 16.0±8.6 | 73.4±27.3 | 6.2±2.6 | 20.0±14.7 | 99.8±69.5 |
| Ratio | 2.60 | 1.18 | 0.81 | 0.94 | 0.63 | 0.97 |
| P-value | 0.00003 | 0.493 | 0.442 | 0.752 | 0.895 | 0.307 |
| IGROV1 | 166.5±68.2 | 39.0±15.5 | 72.7±47.5 | 4.88±2.62 | 2.4±0.1 | 99.3±49.5 |
| IGROV1-OLA | 135.6±51.0 | 38.0±8.3 | 83.3±28.4 | 8.17±2.50 | 3.3±1.6 | 90.0±21.4 |
| Ratio | 0.81 | 0.98 | 1.15 | 1.67 | 1.38 | 0.91 |
| P-value | 0.952 | 0.846 | 0.528 | 0.01 | 0.546 | 0.748 |
| OVCAR3 | 84.0±24.0 | 34.9±3.6 | 41.3±10.6 | 2.42±0.77 | 1.8±0.3 | 120.0±31.7 |
| OVCAR3-OLA | 70.6±54.9 | 51.9±14.1 | 36.6±7.3 | 2.34±0.82 | 1.6±0.4 | 110.0±75.7 |
| Ratio | 0.84 | 1.48 | 0.89 | 0.97 | 0.89 | 0.92 |
| P-value | 0.261 | 0.0004 | 0.450 | 0.877 | 0.653 | 0.771 |
| OVCAR4 | 114.0±46.2 | 32.4±14.0 | 287.5±53.0 | 22.3±3.2 | 9.1±11.1 | 89.3±16.8 |
| OVCAR4-OLA | 82.6±33.1 | 33.2±11.9 | 235.0±91.9 | 18.3±3.2 | 9.5±9.2 | 102.7±36.7 |
| Ratio | 0.72 | 1.02 | 0.82 | 0.82 | 1.04 | 1.15 |
| P-value | 0.255 | 0.925 | 0.572 | 0.336 | 0.97 | 0.61 |
| OVCAR8 | 124.0±35.3 | 39.8±18.7 | 115.3±25.9 | 13.4±1.1 | 3.5±1.7 | 150.7±9.3 |
| OVCAR8-OLA | 140.0±51.0 | 34.8±13.4 | 93.3±25.2 | 10.9±3.6 | 3.4±1.5 | 115.3±66.0 |
| Ratio | 1.13 | 0.87 | 0.81 | 0.81 | 0.99 | 0.88 |
| P-value | 0.547 | 0.607 | 0.351 | 0.347 | 0.981 | 0.728 |
| TYK-nu | 87.3±40.5 | 8.0±3.7 | 33.8±25.7 | 1.19±0.52 | 17.1±11.2 | 110.5±41.7 |
| TYK-nu-OLA | 96.3±55.3 | 9.5±5.8 | 43.0±42.4 | 1.89±1.64 | 26.9±25.6 | 127.5±60.1 |
| Ratio | 1.10 | 1.18 | 1.27 | 1.59 | 1.58 | 1.15 |
| P-value | 0.883 | 0.734 | 0.822 | 0.652 | 0.686 | 0.777 |
Genomic deletion of PARP1 does not affect
sensitivity to PARPi and other chemotherapeutic compounds
To determine whether the abundance of PARP1 protein
expression affects drug sensitivity, PARP1-knockouts of
three ovarian cancer cell lines with differential
olaparib-sensitivity (OCVAR3 > OVCAR8 > OVCAR5) were produced
using the CRISPR-Cas9 technology. To this aim, three
different sgRNAs targeting exons 1 and 2 were designed in order to
perform CRISPR-Cas9-mediated mutagenesis of PARP1 in
these three cell lines (Fig. 6A).
Downstream western blot analysis demonstrated reduction of PARP1
protein expression in all three PARP1-knockout cell lines
(PARP1) in comparison with the non-transduced (control) and
the AAVS1-transduced (mock) cells (Fig. 6B). The gene-editing of the
PARP1 loci was further confirmed by the TIDE assay (42) (Fig.
6C).
Sensitivity of the OVCAR3, OVCAR5 and OVCAR8
PARP1-knockout cells and their respective mock cells to
olaparib and niraparib, carboplatin and cisplatin, doxorubicin and
paclitaxel was determined by MTT-assays. The results revealed that
loss of PARP1 in these cell lines does not affect sensitivity to
these drugs (Table VI).
| Table VIDrug sensitivity
(IC50-values) of parental and PARP1-knockout cells. |
Table VI
Drug sensitivity
(IC50-values) of parental and PARP1-knockout cells.
| Olaparib
(µM) | Niraparib
(µM) | Carboplatin
(µM) | Cisplatin
(µM) | Paclitaxel
(nM) | Doxorubicin
(nM) |
|---|
| OVCAR3-mock | 110.0±85.44 | 29.7±7.6 | 27.4±12.9 | 2.4±2.2 | 3.5±1.4 | 32.3±25.8 |
| OVCAR3-ko
(PARP1) | 112.7±96.0 | 36.0±8.2 | 25.6±11.4 | 2.4±2.2 | 3.5±0.4 | 20.0±433.0 |
| Ratio | 1.02 | 1.18 | 0.94 | 0.99 | 1.01 | 0.63 |
| P-value | 0.973 | 0.493 | 0.845 | 0.986 | 0.972 | 0.518 |
| OVCAR5-mock | 307.3±169.1 | 34.0±3.0 | 154.0±18.3 | 6.6±1.2 | 2.4±0.1 | 99.3±49.5 |
| OVCAR5-ko
(PARP1) | 276.7±140.1 | 36.3±6.0 | 111.7±20.6 | 6.5±2.1 | 3.3±1.6 | 90.0±21.4 |
| Ratio | 0.90 | 1.07 | 0.73 | 0.98 | 1.38 | 0.91 |
| P-value | 0.821 | 0.592 | 0.057 | 0.947 | 0.546 | 0.748 |
| OVCAR8-mock | 495.0±261.6 | 51.5±17.0 | 252.5±103.7 | 13.2±4.3 | 16.7±15.8 | 1060±1166 |
| OVCAR8-ko
(PARP1) | 392.5±83.4 | 45.0±10.2 | 198.0±58.8 | 12.9±2.2 | 10.6±8.2 | 513±344 |
| Ratio | 0.79 | 0.87 | 0.78 | 0.98 | 0.63 | 0.48 |
| P-value | 0.501 | 0.544 | 0.405 | 0.922 | 0.591 | 0.507 |
Discussion
In the present study, the potential of olaparib to
induce acquired resistance to PARPi and to PARPi-unrelated
compounds was investigated. The results demonstrated i) that
olaparib exposure did neither induce acquired resistance to PARPi
nor induce cross-resistance to PARPi-unrelated compounds such as
platinum salts, paclitaxel and doxorubicin; ii) that intrinsic
PARPi resistance not only associates with resistance to platinum
salts but also with resistance to other chemotherapeutic compounds
like doxorubicin and epirubicin, paclitaxel and romidepsin and
SAHA; and that iii) cells with acquired cisplatin-resistance are
PARPi cross-resistant.
The key finding of the present study is the failure
to generate subcells with a detectable acquired resistance
phenotype both to PARPi and to PARPi-unrelated compounds after
escalating olaparib (Table V).
This is consistent with a previous study reporting that long-term
treatment with niraparib did not cause genetic alterations and did
not increase the mutation load in wildtype and
BRCA1-defective breast cancer cells to allow the genetic
evolution of resistance (33).
The present findings are, however, opposed to two
other studies reporting the occurrence of acquired PARPi resistance
after long-term exposure of ovarian cancer cells to olaparib
(31) and in high-grade serous
ovarian carcinoma, patient-derived xenograft modes following
treatment with niraparib (32).
In the former study, olaparib-imposed acquired resistance to
olaparib and niraparib was associated with the activation of the
Wnt-signaling pathway. This was found in PEO1 cells, which are
mutated in both TP53 and BRCA2 (and hence were cells
representative for ovarian cancer) as well as in OVCA433 cells,
which were TP53-mutated but BRCA2-wildt-ype. In the
latter study, acquired PARPi resistance was associated with RAD51C
promoter methylation loss, indicating that PARPi treatment can
cause demethylation of RAD51C and that a single alteration was
sufficient to confer PARPi resistance (32). Whether olaparib-induced acquired
PARPi resistance also associated with cross-resistance to
PARPi-unrelated compounds was not reported in these studies
(31,32).
The study by Yamamoto et al (31), also suggested that PARPi
resistance acquisition can occur regardless of the
BRCA2-status. It was also considered that if the status of
BRCA1, BRCA2, and TP53 in our subcell panel is
relevant for resistance acquisition, but failed to produce acquired
resistance in all cell lines investigated, that is regardless of
whether they are mutated in TP53 (ES-2, OVCAR3, OVCAR8,
TYK-nu) or TP53 and BRCA2 (IGROV1, OVCAR4) or whether
they are wild-type for both (A2780). Intriguingly, not even the
triple TP53/BRCA1/BRCA2-mutant IGROV1 cells, which carry
also mutations in numerous DNA repair-associated genes including
ARID1A, ATR, BLM, MRE11, MLH1, MSH2, MSH3, MSH6, RAD50 and
RAD52, developed acquired resistance after olaparib escalation.
It thus appears that whether or not olaparib-imposed resistance
acquisition occurs does not necessarily depend on the TP53
and BRCA mutation status and that, if it occurs, it may
rather be a matter of cell line dependence and/or of the mutational
background of potential oncogenic genes.
There are other important findings. One is that
intrinsically PARPi-sensitive ovarian cancer cells were not only
cross-sensitive to platinum salts as expected (7,47,48) but notably tended to be also
cross-sensitive to anthracyclines and even to DNA-unrelated
compounds like HDACi, and paclitaxel (Table II). These findings suggested an
association between PARPi sensitivity and a general
chemo-sensitivity of cancer cells, that is beyond platinum
sensitivity.
Anthracyclines intercalate into DNA and directly
interfere with topoisomerase II, resulting in DSB and eventually in
cell death (49).
BRCA-deficient cells have been revealed to be sensitive to
doxorubicin (50) and doxorubicin
may reduce PARP activity and PARP1 expression (51). It appears that doxorubicin can
mimic the effect of PARPi by reducing the abundance and function of
PARP. Similarly, HDACi may acetylate PARP and increase PARP binding
to chromatin, resembling PARP-trapping to DNA and hence mimicking
the effect of PARPi, which eventually leads to decreased repair of
cytotoxic DSBs (52) or HDACi
induce hyperacetylation of the nuclear HSP90 and cause depletion of
HR-related proteins, thus conferring BRCAness and defective
DNA damage and HR response in wild-type BRCA1 breast cancer
cells (53). Paclitaxel interacts
with microtubules and inhibits cytokinesis and is not known to be
implicated in DNA-repair (54).
PARP has also various functions in mitosis, and PARP inhibition may
give rise to various mitotic defects (55), possibly imitating the effect of
the microtubule poison.
Similarly, cells with acquired platinum-resistance
were cross-resistant to PARPi, whereas cells with acquired
paclitaxel resistance remain PARPi-sensitive (Table III). The absence of PARPi
cross-resistance in cells with acquired paclitaxel-resistance is
obvious as PARPi and paclitaxel act by different mechanisms.
Moreover, the observed PARPi cross-resistance in cells with
acquired cisplatin-resistance is predictable since resistance to
platinum-based chemotherapy is a strong predictor for PARPi
resistance (12). However, it is
unclear why A2780/CP cells were cross-resistant to niraparib but
not olaparib, whereas TYK-nu(R) cells are cross-resistant to both.
Although olaparib was reported to be a MDR1 substrate (56), an involvement of MDR1 does not
explain the present observations. Cisplatin-resistant A2780/CP and
TYK-nu(R) cells and their respective parental cells do not express
MDR1, indicating that PARPi cross-resistance in A2780/CP and
TYK-nu(R) cells is MDR1-independent. Similarly, MDR1-expressing
paclitaxel-resistant IGROV1- PXL cells retain PARPi sensitivity,
also indicating that MDR1 is not involved.
Another interesting observation was that at least in
our cell line panel cells with BRCA-mutations tended to be
PARPi-resistant rather than PARPi-sensitive as compared with cell
lines with no BRCA mutations, which is opposed to the
synthetic lethality concept (3,4).
Taken together, our in vitro results argue
against olaparib as a likely inducer of acquired PARPi resistance
and cross-resistance to other chemotherapeutic compounds. Not
ignoring that the in vivo and in vitro situation may
be different, but nevertheless assuming certain transferability
into a clinical context, the present results would mean that an
olaparib-based therapy would not produce PARPi- or
chemotherapy-resistant cells and that 'any other' chemotherapy or
even a therapy with a different type of PARPi could follow a
PARPi-based therapy. They also suggested to extend the current view
of PARPi efficacy into a broader context, that is beyond
BRCAness, meaning that PARPi can be an option to treat
cancers regardless of a BRCAness phenotype. However, this is
rather speculative and should be evaluated in clinical trials.
Moreover, although not observed, an acquired hypersensitivity
phenotype would be even more intriguing in this respect and perhaps
be an add-on to the idea that so-called 'exceptional responders'
may be an alternative strategy to better identify novel molecular
determinants of (hyper)sensitivity to these agents PARPi (57).
The failure to induce acquired PARPi resistance
seems predictable, because PARPi are, in contrast to platinum
compounds (58), unlikely
mutagenic. On the other hand, it cannot be ruled out that other
genetic changes or cellular events may occur (33) which in our experimental setup
remain phenotypically undetectable, maybe because the present
experimental protocol was not sufficiently stringent to produce or
to select for subcells with acquired resistance. Similarly, it
appears too simple to just consider BRCA- and
TP53-mutations or the HRD-status (10) to delineate the sensitivity of
different drugs to PARPi and to other chemotherapeutics. Rather,
the entire mutational profile of the potential oncogenes for each
individual cell line should be taken into consideration.
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AF mainly conceived the study, performed the
experiments, wrote and edited the manuscript. NM, AT and MD
performed the experiments. RC performed the experiments and edited
the manuscript. FJ and VHS made substantial contributions to the
conception and design of the study and critically reviewed and
edited the manuscript. FJ and RC confirm the authenticity of all
the raw data. All authors reviewed and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was in part supported by the Swiss National
Science Foundation (grant no. CRSII5_171037).
References
|
1
|
Longley DB and Johnston PG: Molecular
mechanisms of drug resistance. J Pathol. 205:275–292. 2005.
View Article : Google Scholar
|
|
2
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar
|
|
3
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar
|
|
4
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar
|
|
5
|
McCabe N, Turner NC, Lord CJ, Kluzek K,
Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka
MZ, et al: Deficiency in the repair of DNA damage by homologous
recombination and sensitivity to poly(ADP-ribose) polymerase
inhibition. Cancer Res. 66:8109–8115. 2006. View Article : Google Scholar
|
|
6
|
Kim G, Ison G, McKee AE, Zhang H, Tang S,
Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al: FDA approval
summary: Olaparib monotherapy in patients with deleterious germline
BRCA-mutated advanced ovarian cancer treated with three or more
lines of chemotherapy. Clin Cancer Res. 21:4257–4261. 2015.
View Article : Google Scholar
|
|
7
|
Lord CJ and Ashworth A: BRCAness
revisited. Nat Rev Cancer. 16:110–120. 2016. View Article : Google Scholar
|
|
8
|
Ashworth A and Lord CJ: Synthetic lethal
therapies for cancer: What's next after PARP inhibitors? Nat Rev
Clin Oncol. 15:564–576. 2018. View Article : Google Scholar
|
|
9
|
Litton JK, Rugo HS, Ettl J, Hurvitz SA,
Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin
M, et al: Talazoparib in patients with advanced breast cancer and a
germline BRCA mutation. N Engl J Med. 379:753–763. 2018. View Article : Google Scholar
|
|
10
|
Smeby J, Kryeziu K, Berg KCG, Eilertsen
IA, Eide PW, Johannessen B, Guren MG, Nesbakken A, Bruun J, Lothe
RA and Sveen A: Molecular correlates of sensitivity to PARP
inhibition beyond homologous recombination deficiency in
pre-clinical models of colorectal cancer point to wild-type TP53
activity. EBioMedicine. 59:1029232020. View Article : Google Scholar
|
|
11
|
Audeh MW, Carmichael J, Penson RT,
Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN,
Oaknin A, Loman N, et al: Oral poly(ADP-ribose) polymerase
inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and
recurrent ovarian cancer: A proof-of-concept trial. Lancet.
376:245–251. 2010. View Article : Google Scholar
|
|
12
|
Fong PC, Yap TA, Boss DS, Carden CP,
Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S,
Messiou C, et al: Poly(ADP)-ribose polymerase inhibition: Frequent
durable responses in BRCA carrier ovarian cancer correlating with
platinum-free interval. J Clin Oncol. 28:2512–2519. 2010.
View Article : Google Scholar
|
|
13
|
Gelmon KA, Tischkowitz M, Mackay H,
Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M,
Gilks B, et al: Olaparib in patients with recurrent high-grade
serous or poorly differentiated ovarian carcinoma or
triple-negative breast cancer: A phase 2, multicentre, open-label,
non-randomised study. Lancet Oncol. 12:852–861. 2011. View Article : Google Scholar
|
|
14
|
Jiang X, Li X, Li W, Bai H and Zhang Z:
PARP inhibitors in ovarian cancer: Sensitivity prediction and
resistance mechanisms. J Cell Mol Med. 23:2303–2313. 2019.
View Article : Google Scholar
|
|
15
|
Noordermeer SM and van Attikum H: PARP
inhibitor resistance: A tug-of-war in BRCA-mutated cells. Trends
Cell Biol. 29:820–834. 2019. View Article : Google Scholar
|
|
16
|
Lee EK and Matulonis UA: PARP inhibitor
resistance mechanisms and implications for post-progression
combination therapies. Cancers (Basel). 12:20542020. View Article : Google Scholar
|
|
17
|
Dias MP, Moser SC, Ganesan S and Jonkers
J: Understanding and overcoming resistance to PARP inhibitors in
cancer therapy. Nat Rev Clin Oncol. 18:773–791. 2021. View Article : Google Scholar
|
|
18
|
Lin KK, Harrell MI, Oza AM, Oaknin A,
Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo LT, et al:
BRCA reversion mutations in circulating tumor DNA predict primary
and acquired resistance to the PARP inhibitor rucaparib in
high-grade ovarian carcinoma. Cancer Discov. 9:210–219. 2019.
View Article : Google Scholar
|
|
19
|
Mayor P, Gay LM, Gornstein E, Morley S,
Frampton GM, Heilmann A, Sun J, Chung J, Daniel S, Ramkissoon S, et
al: BRCA1/2 reversion mutations revealed in breast and gynecologic
cancers sequenced during routine clinical care using tissue or
liquid biopsy. J Clin Oncol. 35:55512017. View Article : Google Scholar
|
|
20
|
Kondrashova O, Nguyen M, Shield-Artin K,
Tinker AV, Teng NN, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin
M, et al: Secondary somatic mutations restoring RAD51C and RAD51D
associated with acquired resistance to the PARP inhibitor rucaparib
in high-grade ovarian carcinoma. Cancer Discov. 7:984–998. 2017.
View Article : Google Scholar
|
|
21
|
Brugge PT, Kristel P, Van Der Burg E, Boon
U, de Maaker M, Lips E, Mulder L, de Ruiter J, Moutinho C,
Gevensleben H, et al: Mechanisms of therapy resistance in
patient-derived xenograft models of BRCA1-deficient breast cancer.
J Natl Cancer Inst. 108: View Article : Google Scholar : 2016.
|
|
22
|
Kondrashova O, Topp M, Nesic K, Lieschke
E, Ho GY, Harrell MI, Zapparoli GV, Hadley A, Holian R, Boehm E, et
al: Methylation of all BRCA1 copies predicts response to the PARP
inhibitor rucaparib in ovarian carcinoma. Nat Commun. 9:39702018.
View Article : Google Scholar
|
|
23
|
Bunting SF, Callén E, Wong N, Chen HT,
Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao
L, et al: 53BP1 inhibits homologous recombination in
Brca1-deficient cells by blocking resection of DNA breaks. Cell.
141:243–254. 2010. View Article : Google Scholar
|
|
24
|
Xu G, Chapman JR, Brandsma I, Yuan J,
Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M,
et al: REV7 counteracts DNA double-strand break resection and
affects PARP inhibition. Nature. 521:541–544. 2015. View Article : Google Scholar
|
|
25
|
Pettitt SJ, Krastev DB, Brandsma I, Dréan
A, Song F, Aleksandrov R, Harrell M, Menon M, Brough, Campbell J,
et al: Genome-wide and high-density CRISPR-Cas9 screens identify
point mutations in PARP1 causing PARP inhibitor resistance. Nat
Commun. 9:18492018. View Article : Google Scholar
|
|
26
|
Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL,
Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR IV, et al: Blocking
c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of
PARP inhibitors. Nat Med. 22:194–201. 2016. View Article : Google Scholar
|
|
27
|
Chaudhuri AR, Callen E, Ding X, Gogola E,
Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et
al: Replication fork stability confers chemoresistance in
BRCA-deficient cells. Nature. 535:382–387. 2016. View Article : Google Scholar
|
|
28
|
Taglialatela A, Alvarez S, Leuzzi G,
Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan
R, et al: Restoration of replication fork stability in BRCA1- and
BRCA2-deficient cells by inactivation of SNF2-family fork
remodelers. Mol Cell. 68:414–430. 2017. View Article : Google Scholar
|
|
29
|
Rottenberg S, Jaspers JE, Kersbergen A,
van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M,
Zevenhoven J, Lau A, et al: High sensitivity of BRCA1-deficient
mammary tumors to the PARP inhibitor AZD2281 alone and in
combination with platinum drugs. Proc Natl Acad Sci USA.
105:17079–17084. 2008. View Article : Google Scholar
|
|
30
|
Christie EL, Fereday S, Doig K, Pattnaik
S, Dawson SJ and Bowtell DDL: Reversion of BRCA1/2 germline
mutations detected in circulating tumor DNA from patients with
high-grade serous ovarian cancer. J Clin Oncol. 35:1274–1280. 2017.
View Article : Google Scholar
|
|
31
|
Yamamoto TM, McMellen A, Watson ZL,
Aguilera J, Ferguson R, Nurmemmedov E, Thakar T, Moldovan GL, Kim
H, Cittelly DM, et al: Activation of Wnt signaling promotes
olaparib resistant ovarian cancer. Mol Carcinog. 58:1770–1782.
2019. View Article : Google Scholar
|
|
32
|
Nesic K, Kondrashova O, Hurley RM, McGehee
CD, Vandenberg CJ, Ho GY, Lieschke E, Dall G, Bound N, Shield-Artin
K, et al: Acquired RAD51C promoter methylation loss causes PARP
inhibitor resistance in high grade serous ovarian carcinoma. Cancer
Res. 81:4709–4722. 2021. View Article : Google Scholar
|
|
33
|
Póti Á, Berta K, Xiao Y, Pipek O, Klus GT,
Ried T, Csabai I, Wilcoxen K, Mikule K, Szallasi Z and Szüts D:
Long-term treatment with the PARP inhibitor niraparib does not
increase the mutation load in cell line models and tumour
xenografts. Br J Cancer. 119:1392–1400. 2018. View Article : Google Scholar
|
|
34
|
Domcke S, Sinha R, Levine DA, Sander C and
Schultz N: Evaluating cell lines as tumour models by comparison of
genomic profiles. Nat Commun. 4:21262013. View Article : Google Scholar
|
|
35
|
Behrens BC, Hamilton TC, Masuda H,
Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young
RC and Ozols RF: Characterization of a cis-diamminedichloroplat
inum(II)-resistant human ovarian cancer cell line and its use in
evaluation of platinum analogues. Cancer Res. 47:414–148. 1987.
|
|
36
|
Yoshiya N, Adachi S, Misawa Y, Yuzawa H,
Honda T, Kanazawa K, Takeuchi S and Tanaka K and Tanaka K:
Isolation of cisplatin-resistant subline from human ovarian cancer
cell line and analysis of its cell-biological characteristics.
Nihon Sanka Fujinka Gakkai Zasshi. 41:7–14. 1989.In Japanese.
|
|
37
|
Montavon C, Stricker GR, Schoetzau A,
Heinzelmann-Schwarz V, Jacob F and Fedier A: Outcome in serous
ovarian cancer is not associated with LATS expression. J Cancer Res
Clin Oncol. 145:2737–2749. 2019. View Article : Google Scholar
|
|
38
|
Fedier A, Dedes KJ, Imesch P, Von Bueren
AO and Fink D: The histone deacetylase inhibitors suberoylanilide
hydroxamic (Vorinostat) and valproic acid induce irreversible and
MDR1-independent resistance in human colon cancer cells. Int J
Oncol. 31:633–741. 2007.
|
|
39
|
Dedes KJ, Dedes I, Imesch P, von Bueren
AO, Fink D and Fedier A: Acquired vorinostat resistance shows
partial cross-resistance to 'second-generation' HDAC inhibitors and
correlates with loss of histone acetylation and apoptosis but not
with altered HDAC and HAT activities. Anticancer Drugs. 20:321–333.
2009. View Article : Google Scholar
|
|
40
|
Imesch P, Dedes KJ, Furlato M, Fink D and
Fedier A: MLH1 protects from resistance acquisition by the histone
deacetylase inhibitor trichostatin A in colon tumor cells. Int J
Oncol. 35:631–640. 2009.
|
|
41
|
Shin S, Kim SH, Shin SW, Grav LM, Pedersen
LE, Lee JS and Lee GM: Comprehensive analysis of genomic safe
harbors as target sites for stable expression of the heterologous
gene in HEK293 cells. ACS Synth Biol. 9:1263–1269. 2020. View Article : Google Scholar
|
|
42
|
Brinkman EK, Chen T, Amendola M and van
Steensel B: Easy quantitative assessment of genome editing by
sequence trace decomposition. Nucleic Acids Res. 42:e1682014.
View Article : Google Scholar
|
|
43
|
Kohler RS, Kettelhack H,
Knipprath-Mészaros AM, Fedier A, Schoetzau A, Jacob F and
Heinzelmann-Schwarz V: MELK expression in ovarian cancer correlates
with poor outcome and its inhibition by OTSSP167 abrogates
proliferation and viability of ovarian cancer cells. Gynecol Oncol.
145:159–166. 2017. View Article : Google Scholar
|
|
44
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar
|
|
45
|
Lapinska K, Housman G, Byler S, Heerboth
S, Willbanks A, Oza A and Sarkar S: The effects of histone
deacetylase inhibitor and calpain inhibitor combination therapies
on ovarian cancer cells. Anticancer Res. 36:5731–7542. 2016.
View Article : Google Scholar
|
|
46
|
Matulonis U, Berlin S, Lee H, Whalen C,
Obermayer E, Penson R, Liu J, Campos S, Krasner C and Horowitz N:
Phase I study of combination of vorinostat, carboplatin, and
gemcitabine in women with recurrent, platinum-sensitive epithelial
ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother
Pharmacol. 76:417–423. 2015. View Article : Google Scholar
|
|
47
|
Armstrong DK, Bundy B, Wenzel L, Huang HQ,
Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA and
Gynecologic Oncology Group: Gynecologic oncology group:
Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl
J Med. 354:34–43. 2006. View Article : Google Scholar
|
|
48
|
Pennington KP, Walsh T, Harrell MI, Lee
MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord
AS, et al: Germline and somatic mutations in homologous
recombination genes predict platinum response and survival in
ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer
Res. 20:764–775. 2014. View Article : Google Scholar
|
|
49
|
Nitiss JL: Targeting DNA topoisomerase II
in cancer chemotherapy. Nat Rev Cancer. 9:338–350. 2009. View Article : Google Scholar
|
|
50
|
Adams SF, Marsh EB, Elmasri W, Halberstadt
S, Vandecker S, Sammel MD, Bradbury AR, Daly M, Karlan B and Rubin
SC: A high response rate to liposomal doxorubicin is seen among
women with BRCA mutations treated for recurrent epithelial ovarian
cancer. Gynecol Oncol. 123:486–491. 2011. View Article : Google Scholar
|
|
51
|
Zaremba T, Thomas H, Cole M, Plummer ER
and Curtin NJ: Doxorubicin-induced suppression of poly(ADP-ribose)
polymerase-1 (PARP-1) activity and expression and its implication
for PARP inhibitors in clinical trials. Cancer Chemother Pharmacol.
66:807–812. 2010. View Article : Google Scholar
|
|
52
|
Robert C and Rassool FV: HDAC inhibitors:
Roles of DNA damage and repair. Adv Cancer Res. 116:87–129. 2012.
View Article : Google Scholar
|
|
53
|
Ha K, Fiskus W, Choi DS, Bhaskara S,
Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM,
et al: Histone deacetylase inhibitor treatment induces 'BRCAness'
and synergistic lethality with PARP inhibitor and cisplatin against
human triple negative breast cancer cells. Oncotarget. 5:5637–5650.
2014. View Article : Google Scholar
|
|
54
|
Weaver BA: How taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar
|
|
55
|
Slade D: Mitotic functions of
poly(ADP-ribose) polymerases. Biochem Pharmacol. 167:33–43. 2019.
View Article : Google Scholar
|
|
56
|
McCormick A and Swaisland H: In vitro
assessment of the roles of drug transporters in the disposition and
drug-drug interaction potential of olaparib. Xenobiotica.
47:903–915. 2017. View Article : Google Scholar
|
|
57
|
Wheeler DA, Takebe N, Hinoue T, Hoadley
KA, Cardenas MF, Hamilton AM, Laird PW, Wang L, Johnson A, Dewal N,
et al: Molecular features of cancers exhibiting exceptional
responses to treatment. Cancer Cell. 39:38–53. 2021. View Article : Google Scholar
|
|
58
|
Sanderson BJ, Ferguson LR and Denny WA:
Mutagenic and carcinogenic properties of platinum-based anticancer
drugs. Mutat Res. 355:59–70. 1996. View Article : Google Scholar
|