The cancer testis antigen (CTA) is a large protein
family that is exclusively expressed in the testis, placenta and
certain types of malignant tumor, and is involved in the regulation
of critical processes during tumorigenesis and development
(1). Attributed to the
blood-testis barrier, CTAs are categorized as immunogenic
tumor-associated antigens and deemed optimal targets for the design
of therapeutic cancer vaccines (2). To date, >200 CTAs have been
identified and documented in the CT database (www.cta.lncc.br), and >100 gene families are highly
expressed in malignant tumors. Most genes in the same CTA subfamily
are located in adjacent positions on the chromosomes and the
encoded proteins generally share similar domains and structural
characteristics. In tumor tissues, members of the CTA subfamily are
frequently co-expressed and have similar cellular functions. For
instance, members of the melanoma antigen family (MAGE) have a
highly conserved domain, MAGE homology domain, (MHD), and have
essential roles in stress response and cancer progression (3,4). As
MAGEs are widely expressed in a wide range of malignancies,
vaccines targeting MAGEs have been developed in clinical trials to
treat various cancer types, including melanoma and lung cancer
(5-7). Therefore, a better understanding of
the structural and functional characteristics of the CTA family in
malignant tumors is helpful for developing reliable targets for
tumor immunotherapy.
Immunotherapeutic strategies targeting CTAs include
engineered T-cell receptor T-cell therapy, chimeric antigen
receptor T-cell therapy and vaccine-based therapy. Cancer vaccines
are and attractive complement or alternative to conventional cancer
treatments with great prophylactic and therapeutic potential
(8). Cancer vaccines stimulate
tumor-specific immune responses through delivering tumor antigens
into antigen-presenting cells (APCs) and induce vigorous antitumor
immunity to inhibit tumor growth, recurrence and metastasis
(9). Compared with other
immunotherapeutic strategies, cancer vaccines provide specific,
safe and tolerable control of cancer progression. Furthermore,
nanomaterials have been utilized to design vaccine platforms, which
improved the efficacy during antigen delivery, processing and
presentation to T cells (10). A
variety of cancer nanomaterial-based vaccines have been designed to
deliver peptide/adjuvant or nucleic acid of CTAs. In addition to
commonly used inorganic and organic materials (such as polymers and
liposomes), dipeptide-based nanotubes, nucleic acid nanostructures,
cell membranes and other biomimetic nanomaterials have also proven
effective as methods for delivering vaccine compositions to
targeted sites (11-14).
So far, cancer vaccines targeting the CTA family
exhibited promising efficacy in tumor control at preclinical and
clinical stages (15,16). However, the clinical translation of
cancer vaccines is hampered by relatively weak immunogenicity and a
suppressive tumor microenvironment (TME). Since members of the CTA
subfamily usually share homology in structure and expression
patterns, attention should be paid to improving the immunogenicity
of CTA vaccines. On the other hand, the application of
nanomaterials may also serve as an excellent approach to conquering
the suppressive TME and generate a profound antitumor response. In
fact, delivery of CTA antigens by nanomaterial-derived systems has
been demonstrated, with certain results of inhibiting tumor growth
and metastasis (13,17). The present review briefly
summarized the structural homology and distinct biological
functions of the six CTA subfamilies, providing a systematic
understanding of CTA antigens and a comprehensive approach for
designing cancer vaccines based on CTA (Table I; Fig.
1). The current status and challenges of cancer vaccines
targeting CTAs were also summarized, including the results and
adverse events of conventional vaccination (DNA vaccines, mRNA
vaccines and peptide vaccines). A proposal was made to develop a
nanomaterial-derived cancer vaccine, which holds great promise for
overcoming the suppressive immune microenvironment and achieving
co-delivery of multiple vaccine components.
Since first having been identified as a CTA in 1991,
the MAGE family is the largest CTA subfamily consisting of >40
members (18,19). Based on expression pattern and
chromosomal location, the human MAGE family is generally divided
into type I MAGEs and type II MAGEs (20-22).
Type I MAGEs, including MAGE-A, -B and -C subfamily members, are
considered CTAs due to their restricted expression pattern in adult
testicular germ cells and malignancies. Type II MAGEs, including
MAGE-D, -E, -F, -G, -H and -L subfamilies and Necdin, are observed
in various tissues, such as embryonic and various adult tissues,
such as the brain (20,21,23).
Given that type I MAGEs are the most studied CTAs in tumorigenesis
and anticancer treatments, the structural features and biofunctions
of type I MAGEs will be discussed in this subsection.
Type I MAGEs are located on the X chromosome,
including MAGE-As (A1-A12) at q28, MAGE-Bs (B1-18) at Xp21 and Xp22
and MAGE-Cs (C1-3) at Xq27.2 (24,25).
Most type I MAGEs are broadly expressed in the testis and placenta
under normal physiological conditions, indicating their potential
roles in germ cell development (26,27).
It has been reported that MAGE-As are involved in embryonic and
spermatogenesis development, as well as participation in neuron
development (26,28,29,30).
Aberrant activation of MAGEs has been found in
various human cancers with different frequencies. It is noteworthy
that numerous MAGEs share co-expression patterns in tumors,
including MAGE-A1, -A9 and -A11 in laryngeal squamous cell
carcinoma with lymph node metastasis (71.0, 64.5 and 77.4%)
(31), MAGE-A9 and -A11 in breast
cancer (45 and 66.7%) (32) and
MAGE-A1, -A3 and -A11 in glioma (64.1, 51.3 and 57.7%) (33). As far as the regulatory mechanism
of expression is concerned, most MAGEs are activated by epigenetic
reprogramming in malignancies. DNA hypomethylation and histone
modification are thought to be responsible for the extensive
expression of MAGEs in tumors (3,34).
Treatment with the histone deacetylase inhibitor trichostatin A and
the DNA methylase inhibitor 5-aza-2'-deoxycytidine (5-aza-CdR)
synergistically activates the expression of MAGE-A1, -A2, -A3 and
-A12 in various cancer cells (35). Furthermore, bioinformatics has
confirmed that MAGE-A11 and MAGE-A6 were co-expressed in human
prostate cancer and formed a protein complex, which enhanced
MAGE-A11 stability by inhibiting the ubiquitination of MAGE-A11
(36).
Owing to the blood-testis barrier, an immune
response to MAGEs has been observed in numerous cancer types, which
has been summarized in several excellent reviews (12,37,38).
Heterogeneous humoral response against MAGE-A4 and -A10 was
detected in patients with melanoma, particularly in stage II
patients (39). Antibodies against
MAGE-A3 were detected in patients with multiple myeloma (MM) and
limited levels of autoantibodies against MAGE-B4 and -C2 were
detected in patients with non-small cell lung cancer (NSCLC), both
at a frequency of 3% (40,41). In patients with hepatocellular
carcinoma (HCC), a specific cellular response against MAGE-A1 and
-A3 was observed in 23.4% (11/47) and 32.76% (19/58) of patients,
respectively (42). More
importantly, researchers have demonstrated a significant
correlation between MAGE-A3-specific CD8+ T cells and tumor
regression in patients with melanoma (43). In breast cancer, MAGE-A10 was
considered the most prevalent CTA, which provoked a CD8+ T-cell
response (44).
In addition to the co-expression pattern in tumors,
most MAGEs also share significant homology in structure and are
involved in regulating tumorigenesis and cancer development
(45-47). The conserved signature domain
shared by MAGEs, which is called the MHD, consists of a stretch of
200 amino acids (48). All human
MHDs have 46% protein sequence identity and most of them possess a
conserved dileucine motif, particularly in MAGE-As with high
conservation at 70% (21,49). The major function of MAGEs is
interacting with E3 RING ubiquitin ligases to form MAGE-RING
ligases (MRLs) and regulate a myriad of processes. Shown by
targeted and global proteomics, different MAGEs recognize and bind
one specific RING ligase, which impacts ligase activity,
specification of novel substrates for ubiquitination and
subcellular relocation (22).
Specifically, MAGE-A2, -A3, -A6, and -C2 directly bind TRIM28 E3
ubiquitin ligase to reduce p53 and ZNFZ382 protein levels (49,50).
In addition to the major role of MRLs, MAGE-As also participate in
regulating Cullin-RING ligases (CRLs). MAGE-A11 interacts with S
phase kinase-associated protein (Skp2) to modulate substrate
specificity of Skp2 and its interaction with cyclin A, regulating
cell cycle progression (51).
Furthermore, MAGE-B2 serves as a methylation-driven gene
facilitating proliferation, migration and invasion of laryngeal
cancer cells (52,53).
Furthermore, MAGE-As also impact metabolism via
activation of signaling pathways. MAGE-As were proved to sustain
cancer cell growth when glycolysis was inhibited (29). Protein kinase AMP activated (AMPK)
signaling and autophagy are considered general adaptations of
cancer cells in response to metabolic stress during tumor
progression and metastasis (54).
MAGE-A3/6 was reported to be involved in ubiquitination of AMPK α 1
catalytic subunit through direct interaction, and is also degraded
by CRL4-DDB1 and CUL4 associated factor 12 to regulate autophagy
and cellular adaptation to nutrition stress (55).
Mapped at chromosome band Xp11.2, SSX RNAs may be
detected in the testis and thyroid at a rather low level, but the
proteins are observed only in the testis, particularly in early
spermatogenic cells (57,60,61).
SSX proteins are distributed in the nucleus and the homology
between SSX and Kruppel-associated box (KRAB) domain indicates
their role as transcriptional repressors (59,62,63).
In addition, SSX proteins are expressed in undifferentiated
mesenchymal stem cells but are downregulated after the
differentiation of osteocytes and adipocytes, suggesting their
involvement in stem cell differentiation (64).
Members of the SSX family are widely co-expressed in
various tumors, including seminomas (SSX1/2/4, 58%) (65), melanoma (SSX2/3/4, 40%) (66) and sarcomas (11.8-94.1%) (67). It was revealed that SSX proteins
are normally expressed in spermatogonia cells, mainly due to
genome-wide demethylation (68).
Of note, similar demethylation patterns were observed in tumor cell
lines and tumor tissues (69).
Immune responses against SSXs have been widely
reported in several types of cancer. Recently, T-cell responses to
SSX2 were reported in 10.64% of patients with early- or
advanced-stage HCC (42).
Antibodies against SSX2 have been detected in patients with breast
cancer (2%) and MM who have received allogeneic stem cell
transplantation (41,70). Furthermore, the peptide epitope of
SSX2 was identified to have the potential to react with anti-SSX2
antibodies in the serum of patients with breast cancer and to
induce a specific T-cell response in vitro (71). In addition to SSX2, immune response
against SSX4 is present in gynecological cancer (72). In epithelial ovarian cancer,
antibodies against SSX2 and SSX4 have been detected in 2 out 120
patients and specific T-cell response to SSX4 was also identified
with SSX4-derived epitopes. These early findings demonstrated the
immunogenicity of SSXs and provide SSXs as the primary CTAs used to
design cancer vaccines (73).
Similar to MAGEs, high homology is also observed
between SSXs. Two main domains are characterized in SSXs: The
N-terminal portion with high homology to KRAB consisting of 75
highly charged amino acids (66)
and the SSX repressive domain (SSXRD) formed by 33 amino acids at
the C-terminus (62).
Functionally, SSXs have a significant oncogenic role
in various tumor types through their KRAB and SSXRD domains.
Translocation of the SSXRD domain to the C-terminal end of SYT
occurs in SS to form the SYT-SSX fusion oncogene, and the chimeric
products of SYT-SSXs exhibit aberrant activity to promote cellular
transformation during SS development (74,75).
Transactivation of SYT-SSX1/SSX2 proteins leads to transcriptional
activation in tumors, whereas unrearranged SSX1/SSX2 proteins have
an inhibitory effect due to the repressive KRAB domain at the
N-terminus (62). PcG proteins are
observed in various types of cancer and associated with
pericentromeric heterochromatin region and tumor development
(76). Through interaction with
various PcG factors via KRAB and SSXRD domains, SS18-SSX and SSX
negatively regulate genes involved in multicellular
differentiation, stem cell renewal, embryonic development in
drosophila and vertebrates, and tumor progression (66,77,78).
Researchers have found that SSX genes are co-localized with B
cell-specific Moloney murine leukemia virus insertion site 1
(Bmi1), which is a core factor of polycomb repressive complex 1
(PRC1). In addition, there is an intrinsic nucleolar localization
signal induced by cellular stress, which consequently leads to
dissociation of SSX from Bmi1, resulting in downregulation of SSX
protein activity (79).
GAGE antigens are typical CTAs frequently expressed
in various types of cancer, as well as germ cells in the testis and
ovary. The GAGE family comprises at least 16 highly conserved
genes.
GAGEs are mapped to chromosome X p11.23 and each of
them is located in one of an identical number of highly conserved
tandem repeats (80). GAGE
proteins are distributed in nuclei and cytoplasm of spermatogonia
and primary spermatocytes (81).
Beyond the testis, GAGEs expression was also found in primordial
germ cells of the gonad primordium, which is maintained until
adulthood (82). GAGE proteins
were indicated to be expressed in human ectodermal and mesodermal
derivatives, implying that they are related to maintaining
ground-state pluripotency (83).
GAGEs are aberrantly activated in 76% glioblastoma
and negatively associated with the 2-year overall survival (OS)
rate (84). GAGE genes are also
highly expressed in head and neck squamous cell carcinoma (81.5%,
22/27) (85). Furthermore, GAGE-1
expression is upregulated in 43.3% of hepatocellular carcinoma
tissue (26/60) and GAGE-1/-2 are co-expressed in 26.8% of ovarian
cancer tissues (11/41) (86,87).
Activation of GAGEs was observed in MM, colorectal cancer, lung
cancer and papillary and follicular thyroid cancer (88-91).
Like that of most CTAs, GAGE expression is regulated by
epigenetics. In breast cancer, for instance, high levels of
promoter methylation of GAGE were detected by methylation-specific
PCR analysis and enrichment of H3K4me3 was observed to be
correlated with different expression levels of GAGEs (92).
Owing to its restricted expression in testis and
ovary, antibody against GAGE-1 was reported in patients with HCC
(23.33%), liver cirrhosis (13.1%) and hepatitis B (3.3%), as well
as normal human individuals (3.4%) (86). Taking serum from patients with
melanoma as the specific primary antibody, autoantibodies against
GAGE were detected in the serum of 4/72 patients, whereas none were
observed in 72 healthy controls (93).
Unlike MAGEs and SSXs, which have highly conserved
protein domains, GAGEs have no distinct secondary or tertiary
structure (94), indicating that
they are intrinsically disordered proteins despite the homology in
their amino acid sequences.
GAGE protein expression was identified in multiple
tumors, including neuroblastoma, esophageal carcinoma and stomach
cancer (80). Levels of GAGE
protein were associated with a poor differentiation level of
malignant thyroid diseases, indicating a role in tumorigenesis
(91). Cellular levels of the
apoptotic regulators interferon regulatory factor 1 and
nucleophosmin were regulated by GAGE expression, which contribute
to resistance to cytotoxic agents (95). Furthermore, GAGEs are also involved
in the development of radiation resistance through the regulation
of chromatin accessibility and DNA repair efficiency (96).
The XAGE family, consisting of at least 3 homologous
clusters (XAGE-1, -2 and -3), was identified after screening the
expressed sequence tag database (http://www.ncbi.nlm.nih.gov/ncicgap) for PAGE family
member 4 (PAGE-4) homologous genes. Members of the XAGE family are
clustered on chromosome X (Xp11.21-Xp11.3), where SSXs, GAGEs and
MAGE-D, -H, -I and -J were also mapped (97). XAGEs are mainly expressed in
placenta, testis and sarcoma tissues, except XAGE-3, which is
expressed only in placenta but not the testis or any tumor lesions
(97).
A total of four transcript variants, XAGE-1a, -1b,
-1c, -1d, have been identified in various types of tumor (98), including lung cancer (XAGE-1b and
XAGE-1d, 30.6%) (99),
hepatocellular carcinoma (XAGE-1b, 64.4%; XAGE-1c, 15.6%; XAGE-1d,
26.0%) (100,101), prostate cancer (XAGE-1, 35,2%)
(102) and Ewing's sarcoma
(XAGE-1, 33.3%) (97). Of note,
XAGE-1b is expressed in almost all melanomas (103).
ELISA of 278 patients with prostate cancer revealed
that antibodies against XAGE-1 were detectable in two stage-D2
patients, but not in healthy controls (102). Humoral response to XAGE-1b was
also confirmed in patients with NSCLC (104). T-cell response against XAGE-1b
was reported in lung adenocarcinoma tissues and T-cell and B-cell
epitopes of XAGE-1b protein were identified by Yazdi et al
(105).
All XAGE transcripts contain a relatively large
secondary open reading frame, which encodes putative proteins in
homology with XAGE-1 primary protein (97). Considering strong homology between
XAGEs, tumor vaccines targeting multiple XAGEs may become a novel
therapeutic strategy for generating efficient antitumor
effects.
The association between high levels of XAGE
expression and poor outcomes in patients with cancer implies that
XAGEs may have an important role in tumorigenesis and cancer
progression. Patients with HCC with positive XAGE-1 mRNA expression
had a relatively lower 2-year survival rate (101). In particular, XAGE-1b promoted
adenoid cystic carcinoma progression by regulating the cell cycle
(shortening the G0/G1 and prolonging the
G2/M phase) and enhanced resistance to apoptotic effects
induced by tumor necrosis factor-α (106).
The PAGE family is a GAGE-like gene family that was
identified by a combination of experimental expression analyses and
computerized database mining (107). The PAGE family consists of five
members, namely PAGE1-5, which share a significant homology in
amino acid sequence (108).
Different from the other four members, PAGE-4 is the most
well-studied, with significant expression in prostate cancer and
therapeutic potential.
By Northern blot, PAGE-1 RNA expression was revealed
in prostate cancer and uterine cancer tissues. Expression of PAGE-2
was detected in the colorectal cancer cell line Caco-2 and PAGE-4
is highly expressed in prostate cancer (109,110). The expression pattern of PAGEs is
not as restricted in neoplasms, such as prostate cancer and
colorectal cancer, as other CTA families mentioned above (107,111). PAGE-1 mRNA may also be found in
certain normal tissues, including testis, prostate, uterus and
placenta, which signifies that more attention should be paid to
develop PAGE-targeted immunotherapy, avoiding severe side effects
(117).
The expression of PAGEs is associated with the
demethylation status of CpG residues within regions proximal to the
transcription start sites. PAGE-2 expression may also be activated
by 5-aza-CdR treatment in colorectal cancer cell lines (111). As to immunogenicity, humoral
response against PAGE-3 was reported in 3.8 and 2.9% of patients
with NSCLC from two cohorts, but not in patients with benign lung
disease (40).
PAGEs are small proteins containing 102-146 amino
acids with high abundance of hydrophilic/charged residues and
certain hydrophobic residues, indicating that they are
intrinsically disordered proteins (112). As an ensemble of interconverting
conformations without rigid 3D structure, these proteins are
compared to 'dancing protein cloud' and are inclined to partially
form instantaneous secondary structures, which function as
potential ligand binding sites in succession (112-115).
PAGEs have important biological roles as cellular
transformation promoters and metastasis suppressors in cancer.
PAGE-4 is also highly expressed in high-grade prostatic
intraepithelial neoplasia and was considered a tumorigenic
precursor (116). Consistent with
that, upregulated PAGE-4 expression protects cancer cells from
oxidative stress through modulating the MAPK signaling pathway
(117). PAGE-4 interacts with and
potentiates proto-oncogene c-Jun transactivation through
conformational changes, indicating a new vulnerability of prostate
cancer (118).
NY-ESO-1 was cloned from a cDNA library of
esophageal cancer using recombinant cDNA library serological
analysis technology in 1997 (119). Structurally, it is a 180 amino
acid-long protein, containing epitopes of both cellular and humoral
responses in the glycine-rich N-terminal and hydrophobic C-terminal
regions (120).
As a well-studied CTA, NY-ESO-1 protein expression
has been identified in a variety of cancers, but not in normal
adult tissues except immune-privileged organs, such as the testis
and placenta (123). NY-ESO-1 has
been detected in myxoid liposarcoma (45/64, 70.3%) (124), SS (20/25, 80.0%) (123), osteosarcoma (3/9, 33.3%)
(125), esophageal cancer
(83/227, 36.6%) (126),
colorectal cancer (13/60, 21.7%) and breast cancer (37/97, 38.1%)
(127), In addition, upregulated
NY-ESO-1 was found in metastatic tumor sites and to be associated
with high risk of recurrence and a poor survival rate (128-131). So far, the expression pattern of
NY-ESO-1 has been fully illustrated by several excellent reviews
(121,132-134). Owing to the specific expression
pattern, therapeutic strategies targeting NY-ESO-1 have achieved
certain effects with limited off-target events (133), which will be discussed in the
next section. Like most CTAs, regulation of NY-ESO-1 expression in
tumors is also mediated by several epigenetic events, involving
tightly controlled sequential interaction of histone deacetylases,
histone methyltransferase, DNA methyltransferases and transcription
factors (121). There is a clear
correlation between high NY-ESO-1 antigen expression and the
hypomethylation status of promoters in various tumor cell types
(83).
NY-ESO-1 is one of the most immunogenic CTAs in
various cancer types. Humoral response to NY-ESO-1 was reported in
several cancers, including breast cancer (73%), ovarian cancer
(30%), melanoma (9.4%), adult T-cell leukemia (11.6%), lung cancer
(4-12.5%), thyroid cancer (36%), bladder cancer (12.5%) and
esophageal cancer (13%) (38,135). In terms of cellular response,
NY-ESO-1-specific T-cell response was detected in 10.64% of
patients with HCC (42). Patients
with melanoma who had NY-ESO-1 antibodies exhibited CD8+ T-cell
responses, and NY-ESO-1-specific T cells in patients with
neuroblastoma were reported to produce interferon-γ (136-138).
NY-ESO-1 expression in cancer tissues was indicated
to be associated with lymph node metastasis, higher differentiation
grade and advanced clinical stage, indicating an important role in
regulating tumor development and progression (121,134). In MM, NY-ESO-1 knockdown caused
impaired growth of MM cell lines and reduced osteolytic lesions,
and it upregulated the expression of E-cadherin, p21 and p53 in
vivo (139). Furthermore,
NY-ESO-1 expression in mesenchymal stem cells was downregulated
after differentiation, suggesting a role in cell differentiation
(38). Furthermore, a positive
correlation between NY-ESO-1 and forkhead box P3 levels was
reported in the TME of NSCLC (140).
Due to their restricted expression pattern in
tumors, CTAs are promising targets for therapeutic vaccines.
Certain CTA subfamilies, such as MAGEs, are more attractive
candidates for the development of cancer vaccines. Thus far,
numerous CTA-based tumor vaccines have been developed and are able
to induce antitumor response through direct administration as a DNA
vaccine, RNA vaccine or protein vaccine (Table I). In addition,
nanomaterial-derived delivery systems have caught the attention of
researchers, with higher delivery efficiency and induced robust
immune response. These vaccine platforms are presented in the
following sections (Fig. 2).
As mentioned previously, numerous CTAs are expressed
in various tumor tissues at different frequencies. However, not all
of them have been used to construct cancer vaccines due to the
irregular expression frequencies and relatively low immunogenicity
(141). MAGE, NY-ESO-1, SSX,
cancer-testis SP-1 (CTSP-1), TTK protein kinase (TTK1),
insulin-like growth factor II mRNA-binding protein 3 (IMP-3), sperm
lysozyme-like protein 1 (SLLP1), placenta enriched 1 (PLAC1),
lactate dehydrogenase C, sperm autoantigenic protein 17 (sp17) and
PRAME nuclear receptor transcriptional regulator (PRAME) are the
most widely-used CTAs in tumor vaccines at present, which have
demonstrated promising results in preclinical research (15,142-146). However, vaccines targeting SSX,
CTSP-1, SLLP1 and PLAC1 are barely used in clinical trials due to
limitations regarding safety, stability and effectiveness (147). According to data from www.clinicaltrials.gov, NY-ESO-1 and MAGEs are the
most widely-used CTAs to treat malignancies with high
immunogenicity (accounting for 37 and 36% of all clinical trials,
respectively). They may stimulate both cellular and humoral immune
responses with considerable safety and antigenicity (148). Vaccines targeting LY6K (also
known as URLC10), TTK1, IMP-3, PRAME and sp17 are also used in
several clinical trials to control tumor progression (www.clinicaltrials.gov).
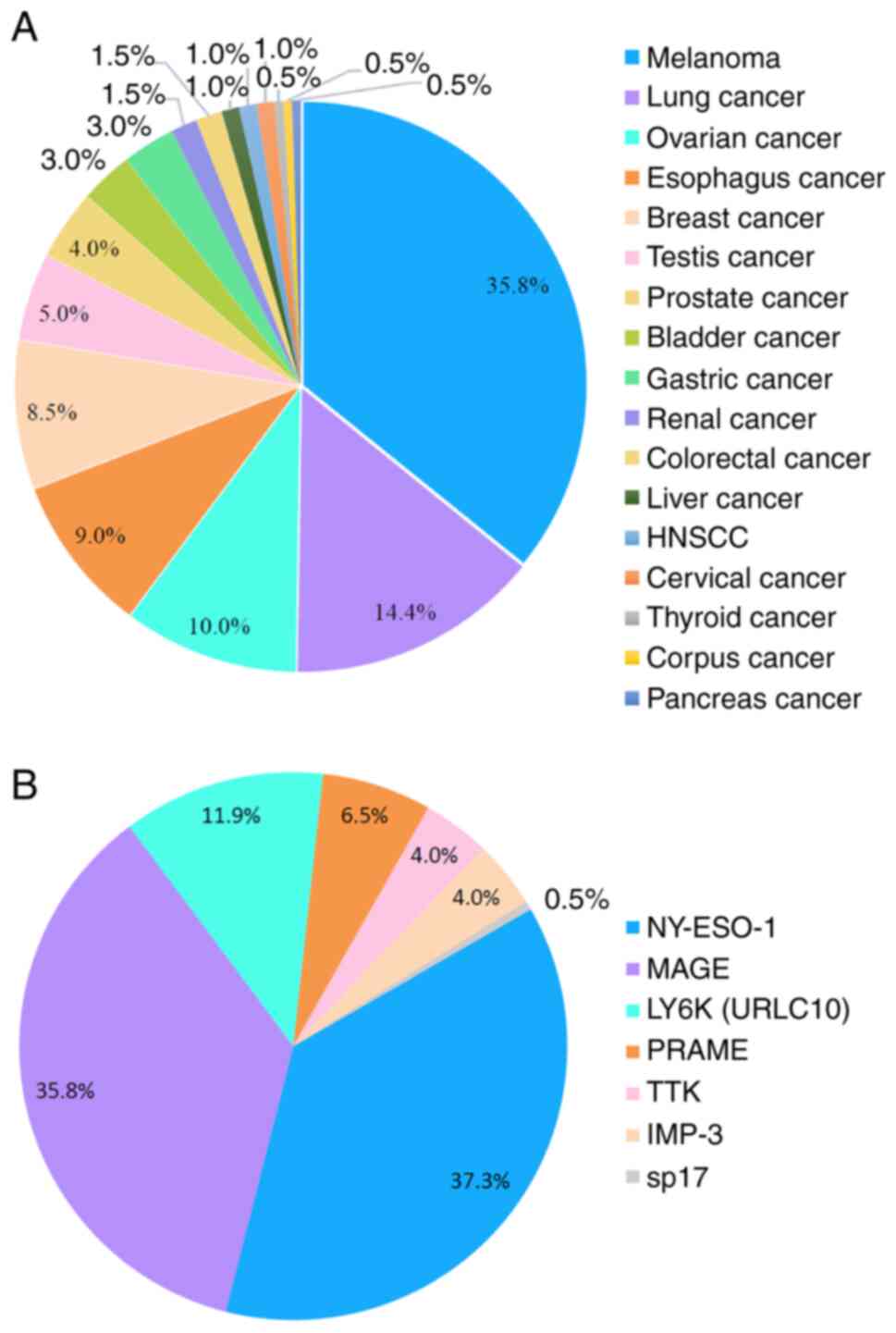
Melanoma, lung cancer and ovarian cancer have been
categorized as CTA-high-expressing malignancies, breast cancer and
prostate cancer as CTA-moderately-expressing malignancies, and
colorectal cancer, kidney cancer and pancreatic cancer as
CTA-low-expressing malignancies (83). Consistently, most CTA-based vaccine
trials were performed in patients with melanoma (72.36%),
particularly those targeting MAGEs and NY-ESO-1. According to data
from clinical trials, lung cancer is the second-most common
malignancy treated with CTA vaccines and therapeutic targets are
LY6K, TTK and IMP-3 (149). There
have been fewer clinical trials on the use of CTA vaccines for the
treatment of ovarian cancer than lung cancer and the commonly used
target is NY-ESO-1, similarly to the role of NY-ESO-1 in melanoma
(Fig. 3).
In addition to gene targets, the form of antigen
delivery also has an important impact on vaccination efficacy,
which includes DNA vaccines, RNA vaccines and peptide vaccines.
Among them, DNA vaccine is a well-studied type of vaccination with
the longest history. DNA vaccines may induce both cellular and
humoral responses and have several advantages, such as low
incidence of side effects, high stability, simplicity and repeated
administration (150). Innate
immune response may also be stimulated by DNA vaccine due to the
presence of CpG motifs and the double strand nucleotide (151). Taking melanoma as an example,
studies using multiple melanoma-associated antigens are ongoing,
combining them with molecular adjuvants to enhance the antitumor
effect (152).
mRNA vaccine represents a promising immunotherapy
approach with rapid development, safe administration and relatively
low-cost manufacture (153).
Compared to DNA vaccine, advantages of mRNA vaccine are as follows:
i) Protein expression rate and magnitude of mRNA are higher than
for DNA vaccines; ii) unlike DNA vaccines, there is no insertional
mutagenesis for mRNA vaccines, which do not integrate into the
genome (8); and iii) production of
mRNA vaccines is less time-consuming and they are less
comprehensive to manufacture than plasmid DNA (154,155). In general, mRNA vaccine has
attracted widespread interest for the treatment of both infectious
disease and malignancies (156).
Peptide vaccines are characterized by better safety
and tolerance without any serious adverse events in comparison to
traditional anti-tumor therapies and are considered a promising
vaccination approach, which directly delivers synthetic or natural
tumor-specific, -associated peptide to induce antitumor effects
(157,158). There are several advantages of
peptide-based therapeutic cancer vaccines, including convenient
production, low carcinogenic potential, cost-effective manufacture,
high chemical stability and insusceptibility to pathogen
contamination (156).
Peptide-based vaccine has become a major focus of cancer vaccine
study with promising clinical possibilities. Peptides used for
cancer vaccines usually consist of small peptides (generally 7-14
amino acids) with immunogenicity expressed on target cells. Peptide
vaccines have been tested in clinical trials for multiple cancers,
including esophageal cancer (159,160), melanoma (161), lung cancer (162,163), head and neck squamous cell
carcinoma (164) and pancreatic
cancer (165). Application and
challenges of CTA-based cancer vaccines
SSXs and MAGE-As are commonly used targets of DNA
vaccines to generate antigen-specific CD4+ and CD8+ T-cell
responses (166,167). Taking advantage of the homology
between MAGE-As, researchers designed and optimized a consensus
MAGE-A DNA vaccine to treat melanoma, which was able to cross-react
with numerous MAGE-A isoforms. Immunization with the MAGE-A vaccine
in mice induced robust CD8+ T-cell responses against multiple
isoforms (14/15), exhibited cytotoxic effects to significantly
inhibit tumor growth and prolonged mouse survival to a median of 50
days, which was 2-fold of that of the control group (168). SSXs are also widely used in
cancer vaccines to induce humoral and cellular responses (15,176,169,170). In a previous report, DNA vaccine
encoding SSX2 was reported to induce enhanced peptide-specific
immune responses and cytotoxic T cells were detectable in mice
immunized with modified SSX2 plasmid DNA vaccine (176). In addition to single-gene DNA
vaccines, fusion-gene DNA vaccines are being studied and have
demonstrated higher immunogenicity. In preclinical research, both
SSX2-MAGEA3 and MAGEA3-SSX2 DNA vaccines achieved improved
antitumor effects in the treatment of esophageal cancer compared to
either MAGEA3 or SSX2 DNA vaccine (15).
Safety concerns regarding DNA vaccines are usually
hypersensitivity reaction and mutation risk. In a phase I/II
clinical trial for prostate cancer, 50% (13/26) of patients
exhibited a delayed-type hypersensitivity reaction after treatment
with naked DNA of proteasome 20S subunit alpha and CD86 (171). In another phase I trial of erb-b2
receptor tyrosine kinase 2-postive breast cancer, mild to moderate
complications were reported in 82% of patients with injection site
reactions, 36% with fatigue and 33% with flu-like syndrome
(172). Furthermore, DNA vaccines
may integrate into the host genome and increase the risk of genomic
alteration; the production of anti-DNA autoantibodies may also
limit their application (173).
Despite the efficacy and safety demonstrated in
clinical trials, clinical translation of DNA vaccines targeting
CTAs remains limited, mainly by two factors: Immunosuppressive TME
and low immunogenicity profiles in human studies. According to a
previous report, optimized SSX2 DNA vaccination led to increased
expression of programmed cell death 1 (PD-1) and PD-1 ligand 1 on
CD8+ T cells and tumor cells, respectively, signifying the
importance of combined treatment with chemotherapy, radiation
therapy and immune checkpoint blockade (2,150,174). Conversely, strategies to improve
immunogenicity have been categorized into several aspects,
including antigen selection, vaccine construct optimization and
delivery method diversity (167).
For antigen selection, homology between CTA subfamily members
should be considered to avoid safety events and personalized
antigens are universally recommended. Vaccine construct design is
also responsible for DNA vaccine efficacy. Future design should pay
more attention to codon optimization, promoter selection and
plasmid vector backbone, as well as adjuvant selection (167).
Effective delivery methods have an important role
in DNA vaccination. In general, it is more complex to deliver DNA
vaccine than RNA vaccine due to its larger dimension and the
necessity for nuclear localization. Plasmids containing target
sequences are usually administered directly into the tumor site to
produce specific antigens, as well as by mucosal delivery and
intramuscular injection (171).
Electroporation and intradermal needle-free delivery system were
recently developed to enhance vaccination efficacy (175). Furthermore, rapid advancements in
biomaterials have facilitated improvements in the efficacy of DNA
vaccines (176). Nanoparticles
(liposomes or polymeric particles) are recommended to deliver DNA
vaccines to target cells, which significantly enhanced
encapsulation efficiency and stability, improved cellular uptake
and avoided toxicity (176,177). However, the hallmarks of DNA
vaccines are their ability to present native conformational
immunogens and prime both humoral and cellular immune responses
(167). DNA vaccines remain a
well-accepted strategy with their stability, scalability and low
cost for manufacture, and it is worthwhile to make efforts to
develop and investigate methods with improved delivery efficacy
(176).
A phase I/IIa clinical trial applied RNA vaccine
encoding five tumor-associated antigens (NY-ESO-1, MAGE-C1,
MAGE-C2, survivin and a trophoblast glycoprotein named CV9201) to
treat 46 patients with NSCLC. Administrated intradermally, CV9201
generated antigen-specific immune responses in 63% of patients, and
the median progression-free survival (PFS) and OS were prolonged to
5.0 [95% confidence interval (CI), 1.8-6.3] months and 10.8 (95%
CI, 8.1-16.7) months, respectively. The two- and three-year
survival rates were 26.7 and 20.7%, respectively. Furthermore, only
mild to moderate adverse events were observed in most patients
(16). Similar to DNA vaccines, a
combined treatment strategy was recommended to conquer the
immunosuppressive TME and to improve outcomes. mRNA-based vaccine
encoding six NSCLC antigens (NY-ESO-1, MAGE-C1, MAGE-C2, 5T4,
survivin and MUC-1) was utilized in a phase Ib clinical trial in
combination with local radiation. Monitoring data revealed
increased antigen-specific humoral immunity and cellular immunity
in 80 and 40% of patients, respectively. This vaccination has
achieved significantly prolonged PFS and OS at 2.87 (95% CI
1.43-4.27) months and 13.95 (95% CI 8.93-20.87) months,
respectively (178). Furthermore,
researchers recently reported an intravenous-administrated
liposomal RNA vaccine (RNA-LPX), which encoded four non-mutated,
tumor-associated antigens, including NY-ESO-1, MAGE-A3, tyrosinase
and transmembrane phosphatase with tensin homology. For 89 patients
with melanoma with treatment-refractory tumors (previous checkpoint
inhibitor treatment), vaccination induced a durable
antigen-specific cytotoxic T-cell response and a higher tumor
regression rate was achieved at 35% after combined treatment with
anti-PD-1 (179).
In general, adverse events of mRNA vaccination are
mild to moderate, such as flu-like syndrome and injection site
reaction. Recently, safety concerns about mRNA vaccines were
raised, as a higher occurrence of adverse effects was observed,
particularly grade 3 adverse reactions, including anaphylactic
shock, myocarditis and pericarditis, cytokine release syndrome and
cerebral venous thrombosis (180,181). Furthermore, modified mRNA may
combine with serum proteins and form a vascular occlusion, which
has potential toxicity (182). In
a phase I/II clinical study (NCT03639714), patients with advanced
metastatic cancers were treated with mRNA vaccine encoding
neoantigen. Most of them were well-tolerated but one patient
experienced pyrexia, duodenitis and increased transaminases and
hyperthyroidism (183). mRNA
vaccine may be administered by various methods, such as
intradermal, intranasal, subcutaneous, intranodal, intratumoral,
intramuscular and intravenous injection (184). Thereafter, delivery to target
cells is achieved usually by lipid-based nanoparticles to protect
mRNA from degradation and may significantly improve the delivery
efficiency of mRNA vaccines (180). Viruses may also be designed to
deliver mRNA encoding peptides that are later displayed by tumor
cells and/or other cells (185).
Furthermore, peptide vectors and polymer vectors may also
facilitate the delivery of mRNA vaccines (186).
However, there are still several points to be fully
elucidated concerning the specific mechanism of mRNA vaccine
delivered to the immune system, and strategies to overcome the
instability of mRNA, as well as to improve the effectiveness of
most vaccines, require to be developed (148). Furthermore, the immunosuppressive
TME remains the most significant hurdle for mRNA vaccines to induce
a robust antitumor effect (8).
Future investigations should pay more attention to improving
antigen expression efficacy and duration, as well as to promoting
antigen presentation efficiency.
Similar to DNA vaccines, MAGEs and NY-ESO-1 are
also the most common targets of peptide anticancer vaccines. A
previous clinical trial reported significant tumor regression
observed in 28% of patients with melanoma (7/25) who received
peptide vaccine treatment targeting MAGE-3.A1, but no cytotoxic
T-lymphocyte (CTL) responses were detected. In particular, complete
tumor regression was observed in two patients who survived >2
years after the treatment (5).
Vaccination with human leukocyte antigen (HLA)-A2-binding NY-ESO-1
peptides generated detectable specific antibody in 41.7% (5/12) of
patients with metastatic tumors expressing NY-ESO-1, and
peptide-specific CD8+ T-cell reactions were detected in 4 of 7
NY-ESO-1 antibody-negative patients (136). In a phase II clinical trial using
peptide vaccine targeting three CTAs (TTK, LY6K, IMP-3), LY6K-,
IMP3- and TTK-specific CD8+ T-cell responses were observed in 63,
60 and 45% of patients with esophageal cancer, respectively. Of
note, the median survival time of HLA-A*2402-positive groups were
improved to 4.6 and 2.6 months, respectively (160). On the other hand, adjuvants are
usually applied to induce more efficient T-cell responses in
combination with CTA peptides. For instance, the immunostimulant
AS15 was administered with recombinant MAGE-A3 protein to enhance
the antitumor effect in 25 patients with resected stage IIB-IV
melanoma (NCT01425749), and durable antibody responses were
observed in all patients, as well as T-cell response in sentinel
immunized nodes. Either injected intramuscularly (Group A, n=13) or
intradermally/subcutaneously (Group B, n=12), MAGE-A3/AS15
vaccination achieved multifunctional CD4+ T-cell responses to
MAGE-A3 in 64% of patients (16/24) and prolonged OS to two years in
90% of patients (187).
In terms of adverse events of peptide vaccines,
erythema is the most frequently encountered, while rare events
include nausea, increased aspartate aminotransaminase, diarrhoea,
myalgia and fatigue. Peptides given alone do not elicit strong
immune responses in vivo due to quick degradation, absence
of danger signals required for APC activation and a lack of
costimulatory ability (188).
These limitations may be overcome by appropriate formulations.
Antigen peptides, adjuvants, as well as targeting sequences, may be
encapsulated into a single package that generates a strong T
cell-mediated response (188).
Poly (lactic-co-glycolic acid) (PLGA) and liposomes are the most
widely used drug delivery applications and have been studied for
numerous years with great biosafety and biodegradability (189). In a recent preclinical study,
PLGA nanoparticles were designed to deliver an immunogenic peptide
to enhance antigen delivery and presentation, and generated a
robust CD8+ CTL response against multiple myeloma in comparison
with free peptide (189).
Peptide vaccines are promising therapeutic
approaches with safety, good tolerance and effective immunization
(157). A general concern of
peptide-based vaccines is the relatively low frequency of CD8+ T
and CD4+ T-cell responses (190,191). Thus, challenges are still to be
overcome to develop peptide vaccines in the future, which include
identification of immunogenetic and neoepitopes, and stimulation of
more effective T-cell responses (158).
In spite of the frustrating results of tumor
vaccines in clinical trials, attempts never ceased to improve the
efficacy of vaccination cascades, including antigen identification,
antigen encapsulation, antigen delivery, antigen release and
antigen presentation (10).
Nanomaterials have attracted increasing attention due to their
potential to enhance the cancer vaccination cascade and facilitate
antitumor effect with less off-target events. Widely used
nanomaterials for developing tumor vaccines include polymeric
nanomaterials, endogenous nanocarriers, lipid-based nanoparticles
and biomimetic cell membrane-derived nanosystems (10,192). In general, nano vaccines are
produced via electrostatic interaction, covalent linking and
hydrophobic interaction to ensure efficient co-encapsulation of
antigens as well as adjuvants (193). The antigens and adjuvants
released from nano vaccine systems may effectively stimulate the
maturation of dendritic cells, which in turn induce the effector
T-cell response via cross-presentation and cytokine secretion.
Furthermore, cross-presentation, mediated by antibody-antigen
immune complex uptake via Fcγ receptors on APCs, also holds
significant potential to stimulate long-term antitumor cellular
immunity (194). Compared with
conventional vaccines, nanovaccines share advantages of increased
immunogenicity and co-delivery of multiple antigens, prolonged
biological activity, enhanced bioavailability, controlled antigen
release and protection of antigens from degradation (195,196).
Due to improved delivery efficiency, nanovaccines
have markedly expanded targeted antigens and facilitated the
development of individualized vaccines. However, CTA-targeted
nanovaccines are still to be fully investigated despite these
advantages mentioned above. In a preclinical trial, nanovaccines
loaded with MAGE-3 peptides have demonstrated great anti-tumor
activity in mice with transplanted gastric cancer and the tumor
inhibition rate was as high as 37.81% after treatment (197). In this research, peptide/chitosan
was conjugated with deoxycholic acid nanoparticles to encapsulate
MAGE-3 peptide, and vaccination resulted in the generation of
MAGE-3-specific CTLs and achieved significant tumor regression.
Nanoparticle assembled from pyruvate dehydrogenase E2 subunit
effectively delivered NY-ESO-1 and MAGE-A3 and achieved an additive
effect to induce a specific cell-mediated response resulting in
15-fold and 9-fold increases in cytotoxicity targeting cancer
cells, respectively (17).
Recently, Verma et al (13)
designed a self-assembled peptide-based nanotube entrapping a
MAGE-3-derived peptide (F-ΔF-M3) to increase the stability and
cellular uptake of M3. After immunization, CTL responses were
provoked in mice and led to a remarkable inhibition ratio of tumor
growth at 41% (13). In another
recently published phase I clinical trial, NY-ESO-1 expression in
dendritic cells was induced after delivery via a modified
lentivirus-based vector LV305 to treat 39 patients with sarcoma and
other types of solid tumor (melanoma, non-small cell lung cancer,
ovarian cancer and breast cancer). After intradermal injection,
disease control was achieved in 56.4% of all patients, and of note,
in 62.5% of patients with sarcoma. NY-ESO-1-specific CD4+ and/or
CD8+ T cells were generated in 52% of all patients (57% of sarcoma
patients), and median PFS reached both 4.6 months for all patients
(95% CI, 2.7-11.7 months) and patients with sarcoma (95% CI,
2.5-8.6 months) (198).
Due to repeat administration of cancer vaccines and
slow degradability of delivery materials, they may accumulate and
cause toxicity in the liver (199). While membrane vesicles are
usually considered ideal vectors to cargo vaccine components, the
complex contents in those vesicles may cause impaired glucose
tolerance and fasting hyperglycemia, and prolong in vivo
residence due to difficult metabolism (200). Genetically modified membrane
vesicles may also cause symptoms such as fatigue and fever, or even
continuous tumor development, which probably resulted from cargo in
membrane nanovesicles (201,202). Mesoporous silica nanoparticles
have been considered a classic delivery platform for cancer
vaccines with multiple advantages, but the inert Si-O-Si framework
may prevent degradation and lead to long-term biosafety issues
(203). Furthermore, the
multicomponent hybrid nanomaterials may cause complex
biodegradation and excretion problems, and potential toxicity is
another concern of metal ions and organic nanomaterials (204).
It has been well accepted that nanovaccines
represent a novel anticancer strategy. Given the broad activation
of CTAs in various cancer types and homology among subfamilies,
nanovaccine targeting one or more CTAs is expected to facilitate
antitumoral effects. However, nanovaccines have several drawbacks,
including fast clearance, and the mechanisms by which nanoparticles
are excreted from organisms after cellular uptake/targeting remain
largely elusive (179). In
addition, the effect of the physical properties of nanoparticles on
the biological interaction between the material and the human body
requires to be further studied to ensure the stability and
operability of the nanovaccine design process.
CTAs are a large protein family expressed in
malignant tumors and male testicular tissues, possessing certain
immunogenicity due to the blood-testis barrier, and may stimulate
humoral and cellular immunity in patients with malignancies.
Members of the same CTA subfamily usually share a similar
structure, co-expression pattern and biofunctions in tumors.
Exploration of the structural characteristics, biological functions
and immunogenicity of CTA families is helpful for developing
antitumor treatment strategies. The MAGE, SSX, GAGE, XAGE and PAGE
families are all X chromosome-linked CTA families and are activated
in various malignant tumors. Co-expression pattern and structural
homology shared between subfamily members render them optimal
targets for tumor diagnosis and treatment. So far, therapeutic
strategies targeting CTA have gained increasing attention to treat
tumors either used independently or in combination with other
treatments (148). CTAs are
usually used as cancer vaccine targets, yet clinical translation is
still limited in spite of promising results achieved at the
preclinical stage. The underlying reasons may be attributed to the
heterogenous expression in tumors and restricted expression of
certain CTAs in normal tissues. Thus, the co-expression pattern and
structural homology of CTA subfamily members should be taken into
account when designing CTA-based therapies.
Cancer vaccines represent a promising therapeutic
modality with several advantages. DNA, mRNA and peptide-based
cancer vaccines are commonly-used vaccination forms with their own
merits and drawbacks. During vaccination, low immunogenicity is
generally observed, leading to limited anticancer efficacy.
Furthermore, the immunosuppressive TME also hinders the immune
response induced by a specific antigen. In view of this,
nanomaterial-derived delivery systems may be applied to realize
co-delivery of multiple antigens, as well as immune modulators to
reverse the immunosuppressive TME. Since antigen identification has
long been attributed to be the main reason for the low efficiency
of the cancer vaccination cascade, resulting in poor performance in
clinical trials, CTA subfamilies should be considered optimal
candidates for designing cancer vaccines. In particular, CTA-based
nanovaccines will become an attractive strategy for enhancing the
antitumor effects of cancer vaccines in the future. At the same
time, attention should also be paid to maintaining the balance
between the complexity and composition of the nanostructure and the
therapeutic effect, to minimize the toxicity of nanomaterials and
maximize the therapeutic efficacy.
Not applicable.
Conceptualization, FFC; methodology, SNR and ZYZ;
analysis and interpretation of data, MYL and DRW; writing-original
draft preparation, SNR and ZYZ; writing-review and editing, SNR;
visualization, RJG; design and funding acquisition, XDF and FFC.
All authors have read and agreed to the published version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the National Key Research and
Development Program (grant nos. 2021YFC2400600 and 2021YFC2400603),
the National Natural Science Foundation of China (grant no.
32271446), Program of Health Department of Jilin Province (grant
nos. 2022SCZ26, 2022JC075 and 2022LC121), Norman Bethune Program of
Jilin University (grant nos. 2022B21 and 2022B37), the China
Postdoctoral Science Foundation (grant no. 2022TQ0118) and the
Natural Science Foundation of Jilin Province (grant nos.
YDZJ202101ZYTS024 and YDZJ202301ZYTS427).
|
1
|
Gibbs ZA and Whitehurst AW: Emerging
contributions of cancer/testis antigens to neoplastic behaviors.
Trends Cancer. 4:701–712. 2018.
|
|
2
|
Saxena M, van der Burg SH, Melief CJM and
Bhardwaj N: Therapeutic cancer vaccines. Nat Rev Cancer.
21:360–378. 2021.
|
|
3
|
Florke Gee RR, Chen H, Lee AK, Daly CA,
Wilander BA, Fon Tacer K and Potts PR: Emerging roles of the MAGE
protein family in stress response pathways. J Biol Chem.
295:16121–16155. 2020.
|
|
4
|
Lian Y, Meng L, Ding P and Sang M:
Epigenetic regulation of MAGE family in human cancer
progression-DNA methylation, histone modification, and non-coding
RNAs. Clin Epigenetics. 10:1152018.
|
|
5
|
Marchand M, van Baren N, Weynants P,
Brichard V, Dréno B, Tessier MH, Rankin E, Parmiani G, Arienti F,
Humblet Y, et al: Tumor regressions observed in patients with
metastatic melanoma treated with an antigenic peptide encoded by
gene MAGE-3 and presented by HLA-A1. Int J Cancer. 80:219–230.
1999.
|
|
6
|
Parvizpour S, Razmara J, Pourseif MM and
Omidi Y: In silico design of a triple-negative breast cancer
vaccine by targeting cancer testis antigens. Bioimpacts. 9:45–56.
2019.
|
|
7
|
Vansteenkiste JF, Cho BC, Vanakesa T, De
Pas T, Zielinski M, Kim MS, Jassem J, Yoshimura M, Dahabreh J,
Nakayama H, et al: Efficacy of the MAGE-A3 cancer immunotherapeutic
as adjuvant therapy in patients with resected MAGE-A3-positive
non-small-cell lung cancer (MAGRIT): A randomised, double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 17:822–835.
2016.
|
|
8
|
Miao L, Zhang Y and Huang L: mRNA vaccine
for cancer immunotherapy. Mol Cancer. 20:412021.
|
|
9
|
Sahin U and Türeci O: Personalized
vaccines for cancer immunotherapy. Science. 359:1355–1360.
2018.
|
|
10
|
Chen F, Wang Y, Gao J, Saeed M, Li T, Wang
W and Yu H: Nanobiomaterial-based vaccination immunotherapy of
cancer. Biomaterials. 270:1207092021.
|
|
11
|
Polla Ravi S, Shamiya Y, Chakraborty A,
Elias C and Paul A: Biomaterials, biological molecules, and
polymers in developing vaccines. Trends Pharmacol Sci. 42:813–828.
2021.
|
|
12
|
Xiao L, Huang Y, Yang Y, Miao Z, Zhu J,
Zhong M, Feng C, Tang W, Zhou J, Wang L, et al: Biomimetic
cytomembrane nanovaccines prevent breast cancer development in the
long term. Nanoscale. 13:3594–3601. 2021.
|
|
13
|
Verma P, Biswas S, Yadav N, Khatri A,
Siddiqui H, Panda JJ, Rawat BS, Tailor P and Chauhan VS: Delivery
of a cancer-testis antigen-derived peptide using conformationally
restricted dipeptide-based self-assembled nanotubes. Mol Pharm.
18:3832–3842. 2021.
|
|
14
|
Huang W, Zhang Q, Li W, Yuan M, Zhou J,
Hua L, Chen Y, Ye C and Ma Y: Development of novel nanoantibiotics
using an outer membrane vesicle-based drug efflux mechanism. J
Control Release. 317:1–22. 2020.
|
|
15
|
Jian W, Li X, Kang J, Lei Y, Bai Y and Xue
Y: Antitumor effect of recombinant Mycobacterium smegmatis
expressing MAGEA3 and SSX2 fusion proteins. Exp Ther Med.
16:2160–2166. 2018.
|
|
16
|
Sebastian M, Schröder A, Scheel B, Hong
HS, Muth A, von Boehmer L, Zippelius A, Mayer F, Reck M,
Atanackovic D, et al: A phase I/IIa study of the mRNA-based cancer
immunotherapy CV9201 in patients with stage IIIB/IV non-small cell
lung cancer. Cancer Immunol Immunother. 68:799–812. 2019.
|
|
17
|
Neek M, Tucker JA, Kim TI, Molino NM,
Nelson EL and Wang SW: Co-delivery of human cancer-testis antigens
with adjuvant in protein nanoparticles induces higher cell-mediated
immune responses. Biomaterials. 156:194–203. 2018.
|
|
18
|
van der Bruggen P, Traversari C, Chomez P,
Lurquin C, De Plaen E, Van den Eynde B, Knuth A and Boon T: A gene
encoding an antigen recognized by cytolytic T lymphocytes on a
human melanoma. J Immunol. 178:2617–2621. 2007.
|
|
19
|
van der Bruggen P, Traversari C, Chomez P,
Lurquin C, De Plaen E, Van den Eynde B, Knuth A and Boon T: A gene
encoding an antigen recognized by cytolytic T lymphocytes on a
human melanoma. Science. 254:1643–1647. 1991.
|
|
20
|
Chomez P, De Backer O, Bertrand M, De
Plaen E, Boon T and Lucas S: An overview of the MAGE gene family
with the identification of all human members of the family. Cancer
Res. 61:5544–5551. 2001.
|
|
21
|
Barker PA and Salehi A: The MAGE proteins:
Emerging roles in cell cycle progression, apoptosis, and
neurogenetic disease. J Neurosci Res. 67:705–712. 2002.
|
|
22
|
Lee AK and Potts PR: A comprehensive guide
to the MAGE family of ubiquitin ligases. J Mol Biol. 429:1114–1142.
2017.
|
|
23
|
Simpson AJG, Caballero OL, Jungbluth A,
Chen YT and Old LJ: Cancer/testis antigens, gametogenesis and
cancer. Nat Rev Cancer. 5:615–625. 2005.
|
|
24
|
De Plaen E, Arden K, Traversari C, Gaforio
JJ, Szikora JP, De Smet C, Brasseur F, van der Bruggen P, Lethé B,
Lurquin C, et al: Structure, chromosomal localization, and
expression of 12 genes of the MAGE family. Immunogenetics.
40:360–369. 1994.
|
|
25
|
Rogner UC, Wilke K, Steck E, Korn B and
Poustka A: The melanoma antigen gene (MAGE) family is clustered in
the chromosomal band Xq28. Genomics. 29:725–731. 1995.
|
|
26
|
Li S, Shi X, Li J and Zhou X:
Pathogenicity of the MAGE family. Oncol Lett. 22:8442021.
|
|
27
|
van den Elsen GA, Tobben L, Ahmed AI,
Verkes RJ, Kramers C, Marijnissen RM, Olde Rikkert MG and van der
Marck MA: Effects of tetrahydrocannabinol on balance and gait in
patients with dementia: A randomised controlled crossover trial. J
Psychopharmacol. 31:184–191. 2017.
|
|
28
|
Kerkar SP, Wang ZF, Lasota J, Park T,
Patel K, Groh E, Rosenberg SA and Miettinen MM: MAGE-A is more
highly expressed than NY-ESO-1 in a systematic immunohistochemical
analysis of 3668 cases. J Immunother. 39:181–187. 2016.
|
|
29
|
Fon Tacer K, Montoya MC, Oatley MJ, Lord
T, Oatley JM, Klein J, Ravichandran R, Tillman H, Kim M, Connelly
JP, et al: MAGE cancer-testis antigens protect the mammalian
germline under environmental stress. Sci Adv. 5:eaav48322019.
|
|
30
|
Hao YH, Doyle JM, Ramanathan S, Gomez TS,
Jia D, Xu M, Chen ZJ, Billadeau DD, Rosen MK and Potts PR:
Regulation of WASH-dependent actin polymerization and protein
trafficking by ubiquitination. Cell. 152:1051–1064. 2013.
|
|
31
|
Liu S, Sang M, Xu Y, Gu L, Liu F and Shan
B: Expression of MAGE-A1, -A9, -A11 in laryngeal squamous cell
carcinoma and their prognostic significance: A retrospective
clinical study. Acta Otolaryngol. 136:506–513. 2016.
|
|
32
|
Hou SY, Sang MX, Geng CZ, Liu WH, Lü WH,
Xu YY and Shan BE: Expressions of MAGE-A9 and MAGE-A11 in breast
cancer and their expression mechanism. Arch Med Res. 45:44–51.
2014.
|
|
33
|
Guo L, Sang M, Liu Q, Fan X, Zhang X and
Shan B: The expression and clinical significance of
melanoma-associated antigen-A1, -A3 and -A11 in glioma. Oncol Lett.
6:55–62. 2013.
|
|
34
|
De Smet C, Loriot A and Boon T:
Promoter-dependent mechanism leading to selective hypomethylation
within the 5' region of gene MAGE-A1 in tumor cells. Mol Cell Biol.
24:4781–4790. 2004.
|
|
35
|
Wischnewski F, Pantel K and Schwarzenbach
H: Promoter demethylation and histone acetylation mediate gene
expression of MAGE-A1, -A2, -A3, and -A12 in human cancer cells.
Mol Cancer Res. 4:339–349. 2006.
|
|
36
|
Laiseca JE, Ladelfa MF, Cotignola J, Peche
LY, Pascucci FA, Castaño BA, Galigniana MD, Schneider C and Monte
M: Functional interaction between co-expressed MAGE-A proteins.
PLoS One. 12:e01783702017.
|
|
37
|
Mahmoud AM: Cancer testis antigens as
immunogenic and oncogenic targets in breast cancer. Immunotherapy.
10:769–778. 2018.
|
|
38
|
Salmaninejad A, Zamani MR, Pourvahedi M,
Golchehre Z, Hosseini Bereshneh A and Rezaei N: Cancer/testis
antigens: Expression, regulation, tumor invasion, and use in
immunotherapy of cancers. Immunol Invest. 45:619–640. 2016.
|
|
39
|
Õunap K, Kurg K, Võsa L, Maiväli Ü, Teras
M, Planken A, Ustav M and Kurg R: Antibody response against
cancer-testis antigens MAGEA4 and MAGEA10 in patients with
melanoma. Oncol Lett. 16:211–218. 2018.
|
|
40
|
Djureinovic D, Dodig-Crnković T, Hellström
C, Holgersson G, Bergqvist M, Mattsson JSM, Pontén F, Ståhle E,
Schwenk JM and Micke P: Detection of autoantibodies against
cancer-testis antigens in non-small cell lung cancer. Lung Cancer.
125:157–163. 2018.
|
|
41
|
Mischo A, Kubuschok B, Ertan K, Preuss KD,
Romeike B, Regitz E, Schormann C, de Bruijn D, Wadle A, Neumann F,
et al: Prospective study on the expression of cancer testis genes
and antibody responses in 100 consecutive patients with primary
breast cancer. Int J Cancer. 118:696–703. 2006.
|
|
42
|
Zang C, Zhao Y, Qin L, Liu G, Sun J, Li K,
Zhao Y, Sheng S, Zhang H, He N, et al: Distinct tumour
antigen-specific T-cell immune response profiles at different
hepatocellular carcinoma stages. BMC Cancer. 21:10072021.
|
|
43
|
Connerotte T, Van Pel A, Godelaine D,
Tartour E, Schuler-Thurner B, Lucas S, Thielemans K, Schuler G and
Coulie PG: Functions of Anti-MAGE T-cells induced in melanoma
patients under different vaccination modalities. Cancer Res.
68:3931–3940. 2008.
|
|
44
|
Huang LQ, Brasseur F, Serrano A, De Plaen
E, van der Bruggen P, Boon T and Van Pel A: Cytolytic T lymphocytes
recognize an antigen encoded by MAGE-A10 on a human melanoma. J
Immunol. 162:6849–6854. 1999.
|
|
45
|
Gure AO, Chua R, Williamson B, Gonen M,
Ferrera CA, Gnjatic S, Ritter G, Simpson AJ, Chen YT, Old LJ and
Altorki NK: Cancer-testis genes are coordinately expressed and are
markers of poor outcome in non-small cell lung cancer. Clin Cancer
Res. 11:8055–8062. 2005.
|
|
46
|
Zhang S, Zhai X, Wang G, Feng J, Zhu H, Xu
L, Mao G and Huang J: High expression of MAGE-A9 in tumor and
stromal cells of non-small cell lung cancer was correlated with
patient poor survival. Int J Clin Exp Patho. 8:541–550. 2015.
|
|
47
|
Qi Y, Cao KX, Xing FC, Zhang CY, Huang Q,
Wu K, Wen FB, Zhao S and Li X: High expression of MAGE-A9 is
associated with unfavorable survival in esophageal squamous cell
carcinoma. Oncol Lett. 14:3415–3420. 2017.
|
|
48
|
Sang M, Wang L, Ding C, Zhou X, Wang B,
Wang L, Lian Y and Shan B: Melanoma-associated antigen genes-an
update. Cancer Lett. 302:85–90. 2011.
|
|
49
|
Doyle JM, Gao J, Wang J, Yang M and Potts
PR: MAGE-RING protein complexes comprise a family of E3 ubiquitin
ligases. Mol Cell. 39:963–974. 2010.
|
|
50
|
Xiao TZ, Suh Y and Longley BJ: MAGE
proteins regulate KRAB zinc finger transcription factors and KAP1
E3 ligase activity. Arch Biochem Biophys. 563:136–144. 2014.
|
|
51
|
Su S, Chen X, Geng J, Minges JT, Grossman
G and Wilson EM: Melanoma antigen-A11 regulates
substrate-specificity of Skp2-mediated protein degradation. Mol
Cell Endocrinol. 439:1–9. 2017.
|
|
52
|
Cui J, Wang L, Zhong W, Chen Z, Chen J,
Yang H and Liu G: Development and validation of epigenetic
signature predict survival for patients with laryngeal squamous
cell carcinoma. DNA Cell Biol. 40:247–264. 2021.
|
|
53
|
Cui J, Chen Y, Ou Y, Liu G, Wen Q, Zhu W,
Liang L, Chen Z, Yang H, Wang L and Wei M: Cancer germline antigen
gene MAGEB2 promotes cell invasion and correlates with immune
microenvironment and immunotherapeutic efficiency in laryngeal
cancer. Clin Immunol. 240:1090452022.
|
|
54
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K,
Cecconi F, Choi AMK, et al: Autophagy in major human diseases. EMBO
J. 40:e1088632021.
|
|
55
|
Ravichandran R, Kodali K, Peng J and Potts
PR: Regulation of MAGE-A3/6 by the CRL4-DCAF12 ubiquitin ligase and
nutrient availability. EMBO Rep. 20:e473522019.
|
|
56
|
Güre AO, Wei IJ, Old LJ and Chen YT: The
SSX gene family: Characterization of 9 complete genes. Int J
Cancer. 101:448–453. 2002.
|
|
57
|
Crew AJ, Clark J, Fisher C, Gill S, Grimer
R, Chand A, Shipley J, Gusterson BA and Cooper CS: Fusion of SYT to
two genes, SSX1 and SSX2, encoding proteins with homology to the
Kruppel-associated box in human synovial sarcoma. EMBO J.
14:2333–2340. 1995.
|
|
58
|
Skytting B, Nilsson G, Brodin B, Xie Y,
Lundeberg J, Uhlén M and Larsson O: A novel fusion gene, SYT-SSX4,
in synovial sarcoma. J Natl Cancer Inst. 91:974–975. 1999.
|
|
59
|
Feng X, Huang YL, Zhang Z, Wang N, Yao Q,
Pang LJ, Li F and Qi Y: The role of SYT-SSX fusion gene in
tumorigenesis of synovial sarcoma. Pathol Res Pract.
222:1534162021.
|
|
60
|
Fligman I, Lonardo F, Jhanwar SC, Gerald
WL, Woodruff J and Ladanyi M: Molecular diagnosis of synovial
sarcoma and characterization of a variant SYT-SSX2 fusion
transcript. Am J Pathol. 147:1592–1599. 1995.
|
|
61
|
dos Santos NR, Torensma R, de Vries TJ,
Schreurs MW, de Bruijn DR, Kater-Baats E, Ruiter DJ, Adema GJ, van
Muijen GN and van Kessel AG: Heterogeneous expression of the SSX
cancer/testis antigens in human melanoma lesions and cell lines.
Cancer Res. 60:1654–1662. 2000.
|
|
62
|
Lim FL, Soulez M, Koczan D, Thiesen HJ and
Knight JC: A KRAB-related domain and a novel transcription
repression domain in proteins encoded by SSX genes that are
disrupted in human sarcomas. Oncogene. 17:2013–2018. 1998.
|
|
63
|
dos Santos NR, de Bruijn DR, Kater-Baats
E, Otte AP and van Kessel AG: Delineation of the protein domains
responsible for SYT, SSX, and SYT-SSX nuclear localization. Exp
Cell Res. 256:192–202. 2000.
|
|
64
|
Cronwright G, Le Blanc K, Götherström C,
Darcy P, Ehnman M and Brodin B: Cancer/testis antigen expression in
human mesenchymal stem cells: Down-regulation of SSX impairs cell
migration and matrix metalloproteinase 2 expression. Cancer Res.
65:2207–2215. 2005.
|
|
65
|
Anderson WJ, Maclean FM, Acosta AM and
Hirsch MS: Expression of the C-terminal region of the SSX protein
is a useful diagnostic biomarker for spermatocytic tumour.
Histopathology. 79:700–707. 2021.
|
|
66
|
Johansen S and Gjerstorff MF: Interaction
between polycomb and SSX proteins in pericentromeric
heterochromatin function and its implication in cancer. Cells.
9:2262020.
|
|
67
|
Wei R, Dean DC, Thanindratarn P, Hornicek
FJ, Guo W and Duan ZF: Cancer testis antigens in sarcoma:
Expression, function and immunotherapeutic application. Cancer
Lett. 479:54–60. 2020.
|
|
68
|
Türeci O, Chen YT, Sahin U, Güre AO, Zwick
C, Villena C, Tsang S, Seitz G, Old LJ and Pfreundschuh M:
Expression of SSX genes in human tumors. Int J Cancer. 77:19–23.
1998.
|
|
69
|
Jones PA and Gonzalgo ML: Altered DNA
methylation and genome instability: A new pathway to cancer? Proc
Natl Acad Sci USA. 94:2103–2105. 1997.
|
|
70
|
Atanackovic D, Arfsten J, Cao Y, Gnjatic
S, Schnieders F, Bartels K, Schilling G, Faltz C, Wolschke C,
Dierlamm J, et al: Cancer-testis antigens are commonly expressed in
multiple myeloma and induce systemic immunity following allogeneic
stem cell transplantation. Blood. 109:1103–1112. 2007.
|
|
71
|
Neumann F, Kubuschok B, Ertan K, Schormann
C, Stevanovic S, Preuss KD, Schmidt W and Pfreundschuh M: A peptide
epitope derived from the cancer testis antigen HOM-MEL-40/SSX2
capable of inducing CD4+ and CD8+ T-cell as well as B-cell
responses. Cancer Immunol Immunother. 60:1333–1346. 2011.
|
|
72
|
Hasegawa K, Koizumi F, Noguchi Y, Hongo A,
Mizutani Y, Kodama J, Hiramatsu Y and Nakayama E: SSX expression in
gynecological cancers and antibody response in patients. Cancer
Immun. 4:162004.
|
|
73
|
Cheever MA, Allison JP, Ferris AS, Finn
OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL,
Weiner LM and Matrisian LM: The prioritization of cancer antigens:
A national cancer institute pilot project for the acceleration of
translational research. Clin Cancer Res. 15:5323–5337. 2009.
|
|
74
|
McBride MJ, Pulice JL, Beird HC, Ingram
DR, D'Avino AR, Shern JF, Charville GW, Hornick JL, Nakayama RT,
Garcia-Rivera EM, et al: The SS18-SSX fusion oncoprotein hijacks
BAF complex targeting and function to drive synovial sarcoma.
Cancer Cell. 33:1128–1141.e7. 2018.
|
|
75
|
Banito A, Li X, Laporte AN, Roe JS,
Sanchez-Vega F, Huang CH, Dancsok AR, Hatzi K, Chen CC,
Tschaharganeh DF, et al: The SS18-SSX oncoprotein hijacks
KDM2B-PRC1 1 to drive synovial sarcoma. Cancer Cell. 33:527–541.e8.
2018.
|
|
76
|
Déjardin J: Switching between epigenetic
states at pericentromeric heterochromatin. Trends Genet.
31:661–672. 2015.
|
|
77
|
Schwartz YB, Kahn TG, Nix DA, Li XY,
Bourgon R, Biggin M and Pirrotta V: Genome-wide analysis of
polycomb targets in drosophila melanogaster. Nat Genet. 38:700–705.
2006.
|
|
78
|
Barco R, Garcia CB and Eid JE: The
synovial sarcoma-associated SYT-SSX2 oncogene antagonizes the
polycomb complex protein Bmi1. PLoS One. 4:e50602009.
|
|
79
|
Wang J, Wang H, Hou W, Liu H, Zou Y, Zhang
H, Hou L, McNutt MA and Zhang B: Subnuclear distribution of SSX
regulates its function. Mol Cell Biochem. 381:17–29. 2013.
|
|
80
|
Gjerstorff MF and Ditzel HJ: An overview
of the GAGE cancer/testis antigen family with the inclusion of
newly identified members. Tissue Antigens. 71:187–192. 2008.
|
|
81
|
Gjerstorff MF, Johansen LE, Nielsen O,
Kock K and Ditzel HJ: Restriction of GAGE protein expression to
subpopulations of cancer cells is independent of genotype and may
limit the use of GAGE proteins as targets for cancer immunotherapy.
Br J Cancer. 94:1864–1873. 2006.
|
|
82
|
Gjerstorff MF, Kock K, Nielsen O and
Ditzel HJ: MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen
expression during human gonadal development. Hum Reprod.
22:953–960. 2007.
|
|
83
|
Gordeeva O: Cancer-testis antigens: Unique
cancer stem cell biomarkers and targets for cancer therapy. Semin
Cancer Biol. 53:75–89. 2018.
|
|
84
|
Tabatabaei Yazdi SA, Safaei M, Gholamin M,
Abdollahi A, Nili F, Jabbari Nooghabi M, Anvari K and Mojarrad M:
Expression and prognostic significance of cancer/testis antigens,
MAGE-E1, GAGE, and SOX-6, in glioblastoma: An immunohistochemistry
evaluation. Iran J Pathol. 16:128–136. 2021.
|
|
85
|
Götte K, Usener D, Riedel F, Hörmann K,
Schadendorf D and Eichmüller S: Tumor-associated antigens as
possible targets for immune therapy in head and neck cancer:
Comparative mRNA expression analysis of RAGE and GAGE genes. Acta
Otolaryngol. 122:546–552. 2002.
|
|
86
|
Chao NX, Li LZ, Luo GR, Zhong WG, Huang
RS, Fan R and Zhao FL: Cancer-testis antigen GAGE-1 expression and
serum immunoreactivity in hepatocellular carcinoma. Niger J Clin
Pract. 21:1361–1367. 2018.
|
|
87
|
Zhang SQ, Zhou XL, Yu H and Yu YH:
Expression of tumor-specific antigen MAGE, GAGE and BAGE in ovarian
cancer tissues and cell lines. BMC Cancer. 10:1632010.
|
|
88
|
Kutilin DS: Regulation of gene expression
of cancer/testis antigens in colorectal cancer patients. Mol Biol.
54:520–534. 2020.
|
|
89
|
Zhang R, Ma L, Li W, Zhou S and Xu S:
Diagnostic value of multiple tumor-associated autoantibodies in
lung cancer. Onco Targets Ther. 12:457–469. 2019.
|
|
90
|
Ghafouri-Fard S, Seifi-Alan M, Shamsi R
and Esfandiary A: Immunotherapy in multiple myeloma using
cancer-testis antigens. Iran J Cancer Prev. 8:e37552015.
|
|
91
|
Melo DH, Mamede RCM, Neder L, Silva WA Jr,
Barros-Filho MC, Kowalski LP, Pinto CAL, Zago MA, Figueiredo DLA
and Jungbluth AA: Expression of cancer/testis antigens MAGE-A,
MAGE-C1, GAGE and CTAG1B in benign and malignant thyroid diseases.
Oncol Lett. 14:6485–6496. 2017.
|
|
92
|
Sun F, Chan E, Wu Z, Yang X, Marquez VE
and Yu Q: Combinatorial pharmacologic approaches target
EZH2-mediated gene repression in breast cancer cells. Mol Cancer
Ther. 8:3191–3202. 2009.
|
|
93
|
Bazhin AV, Wiedemann N, Schnölzer M,
Schadendorf D and Eichmüller SB: Expression of GAGE family proteins
in malignant melanoma. Cancer Lett. 251:258–267. 2007.
|
|
94
|
Gjerstorff MF, Rösner HI, Pedersen CB,
Greve KB, Schmidt S, Wilson KL, Mollenhauer J, Besir H, Poulsen FM,
Møllegaard NE and Ditzel HJ: GAGE cancer-germline antigens are
recruited to the nuclear envelope by germ cell-less (GCL). PLoS
One. 7:e458192012.
|
|
95
|
Kular RK, Yehiely F, Kotlo KU, Cilensek
ZM, Bedi R and Deiss LP: GAGE, an antiapoptotic protein binds and
modulates the expression of nucleophosmin/B23 and interferon
regulatory factor 1. J Interferon Cytokine Res. 29:645–655.
2009.
|
|
96
|
Nin DS, Wujanto C, Tan TZ, Lim D, Damen
JMA, Wu KY, Dai ZM, Lee ZW, Idres SB, Leong YH, et al: GAGE
mediates radio resistance in cervical cancers via the regulation of
chromatin accessibility. Cell Rep. 36:1096212021.
|
|
97
|
Zendman AJW, Van Kraats AA, Weidle UH,
Ruiter DJ and Van Muijen GN: The XAGE family of
cancer/testis-associated genes: alignment and expression profile in
normal tissues, melanoma lesions and Ewing's sarcoma. Int J Cancer.
99:361–369. 2002.
|
|
98
|
Xie C and Wang GM: XAGE-1b cancer/testis
antigen is a potential target for immunotherapy in prostate cancer.
Urol Int. 94:354–362. 2015.
|
|
99
|
Nakagawa K, Noguchi Y, Uenaka A, Sato S,
Okumura H, Tanaka M, Shimono M, Ali Eldib AM, Ono T, Ohara N, et
al: XAGE-1 expression in non-small cell lung cancer and antibody
response in patients. Clin Cancer Res. 11:5496–5503. 2005.
|
|
100
|
Pan Z, Tang B, Hou Z, Zhang J, Liu H, Yang
Y, Huang G, Yang Y and Zhou W: XAGE-1b expression is associated
with the diagnosis and early recurrence of hepatocellular
carcinoma. Mol Clin Oncol. 2:1155–1159. 2014.
|
|
101
|
Gong L, Peng J, Cui Z, Chen P, Han H,
Zhang D and Leng X: Hepatocellular carcinoma patients highly and
specifically expressing XAGE-1 exhibit prolonged survival. Oncol
Lett. 1:1083–1088. 2010.
|
|
102
|
Koizumi F, Noguchi Y, Saika T, Nakagawa K,
Sato S, Eldib AM, Nasu Y, Kumon H and Nakayama E: XAGE-1 mRNA
expression in prostate cancer and antibody response in patients.
Microbiol Immunol. 49:471–476. 2005.
|
|
103
|
Mori M, Funakoshi T, Kameyama K, Kawakami
Y, Sato E, Nakayama E, Amagai M and Tanese K: Lack of XAGE-1b and
NY-ESO-1 in metastatic lymph nodes may predict the potential
survival of stage III melanoma patients. J Dermatol. 44:671–680.
2017.
|
|
104
|
Tarek MM, Shafei AE, Ali MA and Mansour
MM: Computational prediction of vaccine potential epitopes and
3-dimensional structure of XAGE-1b for non-small cell lung cancer
immunotherapy. Biomed J. 41:118–128. 2018.
|
|
105
|
Talebian Yazdi M, Loof NM, Franken KL,
Taube C, Oostendorp J, Hiemstra PS, Welters MJ and van der Burg SH:
Local and systemic XAGE-1b-specific immunity in patients with lung
adenocarcinoma. Cancer Immunol Immunother. 64:1109–1121. 2015.
|
|
106
|
Zhou B, Li T, Liu Y and Zhu N: Preliminary
study on XAGE-1b gene and its mechanism for promoting tumor cell
growth. Biomed Rep. 1:567–572. 2013.
|
|
107
|
Brinkmann U, Vasmatzis G, Lee B,
Yerushalmi N, Essand M and Pastan I: PAGE-1, an X chromosome-linked
GAGE-like gene that is expressed in normal and neoplastic prostate,
testis, and uterus. Proc Natl Acad Sci USA. 95:10757–10762.
1998.
|
|
108
|
Kulkarni P, Dunker AK, Weninger K and
Orban J: Prostate-associated gene 4 (PAGE4), an intrinsically
disordered cancer/testis antigen, is a novel therapeutic target for
prostate cancer. Asian J Androl. 18:695–703. 2016.
|
|
109
|
Suyama T, Shiraishi T, Zeng Y, Yu W,
Parekh N, Vessella RL, Luo J, Getzenberg RH and Kulkarni P:
Expression of cancer/testis antigens in prostate cancer is
associated with disease progression. Prostate. 70:1778–1787.
2010.
|
|
110
|
Zeng Y, Gao D, Kim JJ, Shiraishi T, Terada
N, Kakehi Y, Kong C, Getzenberg RH and Kulkarni P:
Prostate-associated gene 4 (PAGE4) protects cells against stress by
elevating p21 and suppressing reactive oxygen species production.
Am J Clin Exp Urol. 1:39–52. 2013.
|
|
111
|
Yilmaz-Ozcan S, Sade A, Kucukkaraduman B,
Kaygusuz Y, Senses KM, Banerjee S and Gure AO: Epigenetic
mechanisms underlying the dynamic expression of cancer-testis
genes, PAGE2, -2B and SPANX-B, during mesenchymal-to-epithelial
transition. PLoS One. 9:e1079052014.
|
|
112
|
Hellman M, Tossavainen H, Rappu P, Heino J
and Permi P: Characterization of intrinsically disordered prostate
associated gene (PAGE5) at single residue resolution by NMR
spectroscopy. PLoS One. 6:e266332011.
|
|
113
|
Salgia R, Jolly MK, Dorff T, Lau C,
Weninger K, Orban J and Kulkarni P: Prostate-associated gene 4
(PAGE4): Leveraging the conformational dynamics of a dancing
protein cloud as a therapeutic target. J Clin Med. 7:1562018.
|
|
114
|
Uversky VN: Dancing protein clouds: The
strange biology and chaotic physics of intrinsically disordered
proteins. J Biol Chem. 291:6681–6688. 2016.
|
|
115
|
Monika FJ, Simon I, Friedrich P and Tompa
P: Preformed structural elements feature in partner recognition by
intrinsically unstructured proteins. Biophys J. 88:560a2005.
|
|
116
|
Sampson N, Ruiz C, Zenzmaier C, Bubendorf
L and Berger P: PAGE4 positivity is associated with attenuated AR
signaling and predicts patient survival in hormone-naive prostate
cancer. Am J Pathol. 181:1443–1454. 2012.
|
|
117
|
Lv C, Fu S, Dong Q, Yu Z, Zhang G, Kong C,
Fu C and Zeng Y: PAGE4 promotes prostate cancer cells survive under
oxidative stress through modulating MAPK/JNK/ERK pathway. J Exp
Clin Cancer Res. 38:242019.
|
|
118
|
Rajagopalan K, Qiu R, Mooney SM, Rao S,
Shiraishi T, Sacho E, Huang H, Shapiro E, Weninger KR and Kulkarni
P: The stress-response protein prostate-associated gene 4,
interacts with c-Jun and potentiates its transactivation. Biochim
Biophys Acta. 1842:154–163. 2014.
|
|
119
|
Tavakoli Koudehi A, Mahjoubi B, Mirzaei R,
Shabani S and Mahjoubi F: AKAP4, SPAG9 and NY-ESO-1 in Iranian
colorectal cancer patients as probable diagnostic and prognostic
biomarkers. Asian Pac J Cancer Prev. 19:463–469. 2018.
|
|
120
|
Chen YT, Scanlan MJ, Sahin U, Türeci O,
Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M and Old
LJ: A testicular antigen aberrantly expressed in human cancers
detected by autologous antibody screening. Proc Natl Acad Sci USA.
94:1914–1918. 1997.
|
|
121
|
Raza A, Merhi M, Inchakalody VP,
Krishnankutty R, Relecom A, Uddin S and Dermime S: Unleashing the
immune response to NY-ESO-1 cancer testis antigen as a potential
target for cancer immunotherapy. J Transl Med. 18:1402020.
|
|
122
|
Smith SM and Iwenofu OH: NY-ESO-1: A
promising cancer testis antigen for sarcoma immunotherapy and
diagnosis. Chin Clin Oncol. 7:442018.
|
|
123
|
Pollack SM: The potential of the CMB305
vaccine regimen to target NY-ESO-1 and improve outcomes for
synovial sarcoma and myxoid/round cell liposarcoma patients. Expert
Rev Vaccines. 17:107–114. 2018.
|
|
124
|
Jo U, Roh J, Song MJ, Cho KJ, Kim W and
Song JS: NY-ESO-1 as a diagnostic and prognostic marker for myxoid
liposarcoma. Am J Transl Res. 14:1268–1278. 2022.
|
|
125
|
Hashimoto K, Nishimura S, Ito T, Oka N,
Kakinoki R and Akagi M: Clinicopathological assessment of
cancer/testis antigens NY-ESO-1 and MAGE-A4 in osteosarcoma. Eur J
Histochem. 66:33772022.
|
|
126
|
Nagata Y, Kageyama S, Ishikawa T, Kokura
S, Okayama T, Abe T, Murakami M, Otsuka K, Ariyoshi T, Kojima T, et
al: Prognostic significance of NY-ESO-1 antigen and PIGR expression
in esophageal tumors of CHP-NY-ESO-1-vaccinated patients as
adjuvant therapy. Cancer Immunol Immunother. 71:2743–2755.
2022.
|
|
127
|
Čeprnja T, Mrklić I, Perić Balja M,
Marušić Z, Blažićević V, Spagnoli GC, Juretić A, Čapkun V, Tečić
Vuger A, Vrdoljak E and Tomić S: Prognostic significance of
lymphocyte infiltrate localization in triple-negative breast
cancer. J Pers Med. 12:9412022.
|
|
128
|
Liu MY, Su H, Huang HL and Chen JQ: Cancer
stem-like cells with increased expression of NY-ESO-1 initiate
breast cancer metastasis. Oncol Lett. 18:3664–3672. 2019.
|
|
129
|
van Rhee F, Szmania SM, Zhan F, Gupta SK,
Pomtree M, Lin P, Batchu RB, Moreno A, Spagnoli G, Shaughnessy J
and Tricot G: NY-ESO-1 is highly expressed in poor-prognosis
multiple myeloma and induces spontaneous humoral and cellular
immune responses. Blood. 105:3939–3944. 2005.
|
|
130
|
Iura K, Kohashi K, Hotokebuchi Y, Ishii T,
Maekawa A, Yamada Y, Yamamoto H, Iwamoto Y and Oda Y: Cancer-testis
antigens PRAME and NY-ESO-1 correlate with tumour grade and poor
prognosis in myxoid liposarcoma. J Pathol Clin Res. 1:144–159.
2015.
|
|
131
|
Giavina-Bianchi M, Giavina-Bianchi P,
Sotto MN, Muzikansky A, Kalil J, Festa-Neto C and Duncan LM:
Increased NY-ESO-1 expression and reduced infiltrating CD3+ T cells
in cutaneous melanoma. J Immunol Res. 2015:7613782015.
|
|
132
|
Wang H, Chen D, Wang R, Quan W, Xia D, Mei
J, Xu J and Liu C: NY-ESO-1 expression in solid tumors predicts
prognosis: A systematic review and meta-analysis. Medicine
(Baltimore). 98:e179902019.
|
|
133
|
Gnjatic S, Nishikawa H, Jungbluth AA, Güre
AO, Ritter G, Jäger E, Knuth A, Chen YT and Old LJ: NY-ESO-1:
review of an immunogenic tumor antigen. Adv Cancer Res. 95:1–30.
2006.
|
|
134
|
Thomas R, Al-Khadairi G, Roelands J, et
al: NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives.
Frontiers in immunology. 9:9472018.
|
|
135
|
Astaneh M, Dashti S and Esfahani ZT:
Humoral immune responses against cancer-testis antigens in human
malignancies. Hum Antibodies. 27:237–240. 2019.
|
|
136
|
Jäger E, Gnjatic S, Nagata Y, Stockert E,
Jäger D, Karbach J, Neumann A, Rieckenberg J, Chen YT, Ritter G, et
al: Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and
antibody responses in peptide-vaccinated patients with NY-ESO-1+
cancers. Proc Natl Acad Sci USA. 97:12198–12203. 2000.
|
|
137
|
Jäger E, Nagata Y, Gnjatic S, Wada H,
Stockert E, Karbach J, Dunbar PR, Lee SY, Jungbluth A, Jäger D, et
al: Monitoring CD8 T cell responses to NY-ESO-1: Correlation of
humoral and cellular immune responses. Proc Natl Acad Sci USA.
97:4760–4765. 2000.
|
|
138
|
Barrow C, Browning J, MacGregor D, Davis
ID, Sturrock S, Jungbluth AA and Cebon J: Tumor antigen expression
in melanoma varies according to antigen and stage. Clin Cancer Res.
12:764–771. 2006.
|
|
139
|
Li F, Zhao F, Li M, Pan M, Shi F, Xu H,
Zheng D, Wang L and Dou J: Decreasing New York esophageal squamous
cell carcinoma 1 expression inhibits multiple myeloma growth and
osteolytic lesions. J Cell Physiol. 235:2183–2194. 2020.
|
|
140
|
Wang H, Xia Y, Yu J, Guan H, Wu Z, Ban D
and Wang M: Expression of New York esophageal squamous cell
carcinoma 1 and its association with Foxp3 and
indoleamine-2,3-dioxygenase in microenvironment of nonsmall cell
lung cancer. HLA. 94:39–48. 2019.
|
|
141
|
Ko TY, Kim JI and Lee SH: Relationship
between cancer stem cell marker CD133 and cancer germline antigen
genes in NCI-H292 lung cancer cells. Korean J Thorac Cardiovasc
Surg. 53:22–27. 2020.
|
|
142
|
Gong W, Hoffmann JM, Stock S, Wang L, Liu
Y, Schubert ML, Neuber B, Hückelhoven-Krauss A, Gern U, Schmitt A,
et al: Comparison of IL-2 vs IL-7/IL-15 for the generation of
NY-ESO-1-specific T cells. Cancer Immunol Immunother. 68:1195–1209.
2019.
|
|
143
|
Hirayama M, Tomita Y, Yuno A, Tsukamoto H,
Senju S, Imamura Y, Sayem MA, Irie A, Yoshitake Y, Fukuma D, et al:
An oncofetal antigen, IMP-3-derived long peptides induce immune
responses of both helper T cells and CTLs. Oncoimmunology.
5:e11233682016.
|
|
144
|
Hayashi R, Nagato T, Kumai T, Ohara K,
Ohara M, Ohkuri T, Hirata-Nozaki Y, Harabuchi S, Kosaka A, Nagata
M, et al: Expression of placenta-specific 1 and its potential for
eliciting anti-tumor helper T-cell responses in head and neck
squamous cell carcinoma. Oncoimmunology. 10:18565452020.
|
|
145
|
Minhas V, Kumar R, Moitra T, Singh R,
Panda AK and Gupta SK: Immunogenicity and contraceptive efficacy of
recombinant fusion protein encompassing Sp17 spermatozoa-specific
protein and GnRH: Relevance of adjuvants and microparticles based
delivery to minimize number of injections. Am J Reprod Immunol.
83:e132182020.
|
|
146
|
Taheri-Anganeh M, Savardashtaki A, Vafadar
A, Movahedpour A, Shabaninejad Z, Maleksabet A, Amiri A, Ghasemi Y
and Irajie C: In silico design and evaluation of PRAME+FliCΔD2D3 as
a new breast cancer vaccine candidate. Iran J Med Sci. 46:52–60.
2021.
|
|
147
|
Matteo M, Greco P, Levi Setti PE, Morenghi
E, De Rosario F, Massenzio F, Albani E, Totaro P and Liso A:
Preliminary evidence for high anti-PLAC1 antibody levels in
infertile patients with repeated unexplained implantation failure.
Placenta. 34:335–339. 2013.
|
|
148
|
Fan C, Qu H, Wang X, Sobhani N, Wang L,
Liu S, Xiong W, Zeng Z and Li Y: Cancer/testis antigens: From
serology to mRNA cancer vaccine. Semin Cancer Biol. 76:218–231.
2021.
|
|
149
|
Kono K, Mizukami Y, Daigo Y, Takano A,
Masuda K, Yoshida K, Tsunoda T, Kawaguchi Y, Nakamura Y and Fujii
H: Vaccination with multiple peptides derived from novel
cancer-testis antigens can induce specific T-cell responses and
clinical responses in advanced esophageal cancer. Cancer Sci.
100:1502–1509. 2009.
|
|
150
|
Lopes A, Vandermeulen G and Préat V:
Cancer DNA vaccines: Current preclinical and clinical developments
and future perspectives. J Exp Clin Cancer Res. 38:1462019.
|
|
151
|
Herrada AA, Rojas-Colonelli N,
González-Figueroa P, Roco J, Oyarce C, Ligtenberg MA and Lladser A:
Harnessing DNA-induced immune responses for improving cancer
vaccines. Hum Vaccin Immunother. 8:1682–1693. 2012.
|
|
152
|
Rezaei T, Davoudian E, Khalili S, Amini M,
Hejazi M, de la Guardia M and Mokhtarzadeh A: Strategies in DNA
vaccine for melanoma cancer. Pigment Cell Melanoma Res. 34:869–891.
2021.
|
|
153
|
Wu Y, Sang M, Liu F, Zhang J, Li W, Li Z,
Gu L, Zheng Y, Li J and Shan B: Epigenetic modulation combined with
PD-1/PD-L1 blockade enhances immunotherapy based on MAGE-A11
antigen-specific CD8+T cells against esophageal carcinoma.
Carcinogenesis. 41:894–903. 2020.
|
|
154
|
Jahanafrooz Z, Baradaran B, Mosafer J,
Hashemzaei M, Rezaei T, Mokhtarzadeh A and Hamblin MR: Comparison
of DNA and mRNA vaccines against cancer. Drug Discov Today.
25:552–560. 2020.
|
|
155
|
Heine A, Juranek S and Brossart P:
Clinical and immunological effects of mRNA vaccines in malignant
diseases. Mol Cancer. 20:522021.
|
|
156
|
Sahin U, Muik A, Derhovanessian E, Vogler
I, Kranz LM, Vormehr M, Baum A, Pascal K, Quandt J, Maurus D, et
al: COVID-19 vaccine BNT162b1 elicits human antibody and
TH1 T cell responses. Nature. 586:594–599. 2020.
|
|
157
|
Liu W, Tang H, Li L, Wang X, Yu Z and Li
J: Peptide-based therapeutic cancer vaccine: Current trends in
clinical application. Cell Prolif. 54:e130252021.
|
|
158
|
Nelde A, Rammensee HG and Walz JS: The
peptide vaccine of the future. Mol Cell Proteomics.
20:1000222021.
|
|
159
|
Iinuma H, Fukushima R, Inaba T, Tamura J,
Inoue T, Ogawa E, Horikawa M, Ikeda Y, Matsutani N, Takeda K, et
al: Phase I clinical study of multiple epitope peptide vaccine
combined with chemoradiation therapy in esophageal cancer patients.
J Transl Med. 12:842014.
|
|
160
|
Kono K, Iinuma H, Akutsu Y, Tanaka H,
Hayashi N, Uchikado Y, Noguchi T, Fujii H, Okinaka K, Fukushima R,
et al: Multicenter, phase II clinical trial of cancer vaccination
for advanced esophageal cancer with three peptides derived from
novel cancer-testis antigens. J Transl Med. 10:1412012.
|
|
161
|
Schwartzentruber DJ, Lawson DH, Richards
JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K,
Pockaj B, et al: gp100 peptide vaccine and interleukin-2 in
patients with advanced melanoma. N Engl J Med. 364:2119–2127.
2011.
|
|
162
|
Suzuki H, Fukuhara M, Yamaura T, Mutoh S,
Okabe N, Yaginuma H, Hasegawa T, Yonechi A, Osugi J, Hoshino M, et
al: Multiple therapeutic peptide vaccines consisting of combined
novel cancer testis antigens and anti-angiogenic peptides for
patients with non-small cell lung cancer. J Transl Med.
11:972013.
|
|
163
|
Kotsakis A, Papadimitraki E, Vetsika EK,
Aggouraki D, Dermitzaki EK, Hatzidaki D, Kentepozidis N, Mavroudis
D and Georgoulias V: A phase II trial evaluating the clinical and
immunologic response of HLA-A2(+) non-small cell lung cancer
patients vaccinated with an hTERT cryptic peptide. Lung Cancer.
86:59–66. 2014.
|
|
164
|
Yoshitake Y, Fukuma D, Yuno A, Hirayama M,
Nakayama H, Tanaka T, Nagata M, Takamune Y, Kawahara K, Nakagawa Y,
et al: Phase II clinical trial of multiple peptide vaccination for
advanced head and neck cancer patients revealed induction of immune
responses and improved OS. Clin Cancer Res. 21:312–321. 2015.
|
|
165
|
Okuyama R, Aruga A, Hatori T, Takeda K and
Yamamoto M: Immunological responses to a multi-peptide vaccine
targeting cancer-testis antigens and VEGFRs in advanced pancreatic
cancer patients. Oncoimmunology. 2:e270102013.
|
|
166
|
Smith HA, Rekoske BT and McNeel DG: DNA
vaccines encoding altered peptide ligands for SSX2 enhance
epitope-specific CD8+ T-cell immune responses. Vaccine.
32:1707–1715. 2014.
|
|
167
|
Li L and Petrovsky N: Molecular mechanisms
for enhanced DNA vaccine immunogenicity. Expert Rev Vaccines.
15:313–329. 2016.
|
|
168
|
Duperret EK, Liu S, Paik M, Trautz A,
Stoltz R, Liu X, Ze K, Perales-Puchalt A, Reed C, Yan J, et al: A
designer cross-reactive DNA immunotherapeutic vaccine that targets
multiple MAGE-A family members simultaneously for cancer therapy.
Clin Cancer Res. 24:6015–6027. 2018.
|
|
169
|
Smith HA, Cronk RJ, Lang JM and McNeel DG:
Expression and immunotherapeutic targeting of the SSX family of
cancer-testis antigens in prostate cancer. Cancer Res.
71:6785–6795. 2011.
|
|
170
|
Smith HA and McNeel DG: Vaccines targeting
the cancer-testis antigen SSX-2 elicit HLA-A2 epitope-specific
cytolytic T cells. J Immunother. 34:569–580. 2011.
|
|
171
|
Martínez-Puente DH, Pérez-Trujillo JJ,
Zavala-Flores LM, García-García A, Villanueva-Olivo A,
Rodríguez-Rocha H, Valdés J, Saucedo-Cárdenas O, Montes de Oca-Luna
R and Loera-Arias MJ: Plasmid DNA for therapeutic applications in
cancer. Pharmaceutics. 14:18612022.
|
|
172
|
Disis MLN, Guthrie KA, Liu Y, Coveler AL,
Higgins DM, Childs JS, Dang Y and Salazar LG: Safety and outcomes
of a plasmid DNA vaccine encoding the ERBB2 intracellular domain in
patients with advanced-Stage ERBB2-positive breast cancer: A phase
1 nonrandomized clinical trial. JAMA Oncol. 9:71–78. 2023.
|
|
173
|
Huang X, Zhang G, Tang TY, Gao X and Liang
TB: Personalized pancreatic cancer therapy: From the perspective of
mRNA vaccine. Mil Med Res. 9:532022.
|
|
174
|
Rekoske BT, Smith HA, Olson BM, Maricque
BB and McNeel DG: PD-1 or PD-L1 blockade restores antitumor
efficacy following SSX2 epitope-modified DNA vaccine immunization.
Cancer Immunol Res. 3:946–955. 2015.
|
|
175
|
Sobhani N, Scaggiante B, Morris R, Chai D,
Catalano M, Tardiel-Cyril DR, Neeli P, Roviello G, Mondani G and Li
Y: Therapeutic cancer vaccines: From biological mechanisms and
engineering to ongoing clinical trials. Cancer Treat Rev.
109:1024292022.
|
|
176
|
Zhang R, Billingsley MM and Mitchell MJ:
Biomaterials for vaccine-based cancer immunotherapy. J Control
Release. 292:256–276. 2018.
|
|
177
|
Zhang C, Ma Y, Zhang J, Kuo JC, Zhang Z,
Xie H, Zhu J and Liu T: Modification of lipid-based nanoparticles:
An efficient delivery system for nucleic acid-based immunotherapy.
Molecules. 27:19432022.
|
|
178
|
Papachristofilou A, Hipp MM, Klinkhardt U,
Früh M, Sebastian M, Weiss C, Pless M, Cathomas R, Hilbe W, Pall G,
et al: Phase Ib evaluation of a self-adjuvanted protamine
formulated mRNA-based active cancer immunotherapy, BI1361849
(CV9202), combined with local radiation treatment in patients with
stage IV non-small cell lung cancer. J Immunother Cancer.
7:382019.
|
|
179
|
Sahin U, Oehm P, Derhovanessian E,
Jabulowsky RA, Vormehr M, Gold M, Maurus D, Schwarck-Kokarakis D,
Kuhn AN, Omokoko T, et al: An RNA vaccine drives immunity in
checkpoint-inhibitor-treated melanoma. Nature. 585:107–112.
2020.
|
|
180
|
Van Hoecke L, Verbeke R, Dewitte H,
Lentacker I, Vermaelen K, Breckpot K and Van Lint S: mRNA in cancer
immunotherapy: Beyond a source of antigen. Mol Cancer.
20:482021.
|
|
181
|
Chen J, Chen J and Xu Q: Current
developments and challenges of mRNA vaccines. Annu Rev Biomed Eng.
24:85–109. 2022.
|
|
182
|
He Q, Gao H, Tan D, Zhang H and Wang JZ:
mRNA cancer vaccines: Advances, trends and challenges. Acta Pharm
Sin B. 12:2969–2989. 2022.
|
|
183
|
Palmer CD, Rappaport AR, Davis MJ, Hart
MG, Scallan CD, Hong SJ, Gitlin L, Kraemer LD, Kounlavouth S, Yang
A, et al: Individualized, heterologous chimpanzee adenovirus and
self-amplifying mRNA neoantigen vaccine for advanced metastatic
solid tumors: Phase 1 trial interim results. Nat Med. 28:1619–1629.
2022.
|
|
184
|
Lorentzen CL, Haanen JB, Met Ö and Svane
IM: Clinical advances and ongoing trials on mRNA vaccines for
cancer treatment. Lancet Oncol. 23:e450–e458. 2022.
|
|
185
|
Roy DG, Geoffroy K, Marguerie M, Khan ST,
Martin NT, Kmiecik J, Bobbala D, Aitken AS, de Souza CT, Stephenson
KB, et al: Adjuvant oncolytic virotherapy for personalized
anti-cancer vaccination. Nat Commun. 12:26262021.
|
|
186
|
Duan LJ, Wang Q, Zhang C, Yang DX and
Zhang XY: Potentialities and challenges of mRNA vaccine in cancer
immunotherapy. Front Immunol. 13:236472022.
|
|
187
|
Slingluff CL Jr, Petroni GR, Olson WC,
Smolkin ME, Chianese-Bullock KA, Mauldin IS, Smith KT, Deacon DH,
Varhegyi NE, Donnelly SB, et al: A randomized pilot trial testing
the safety and immunologic effects of a MAGE-A3 protein plus AS15
immunostimulant administered into muscle or into
dermal/subcutaneous sites. Cancer Immunol Immunother. 65:25–36.
2016.
|
|
188
|
Abd-Aziz N and Poh CL: Development of
peptide-based vaccines for cancer. J Oncol. 2022:97493632022.
|
|
189
|
Bae J, Parayath N, Ma W, Amiji M, Munshi N
and Anderson KC: BCMA peptide-engineered nanoparticles enhance
induction and function of antigen-specific CD8+
cytotoxic T lymphocytes against multiple myeloma: Clinical
applications. Leukemia. 34:19712020.
|
|
190
|
Kruit WH, Suciu S, Dreno B, Mortier L,
Robert C, Chiarion-Sileni V, Maio M, Testori A, Dorval T, Grob JJ,
et al: Selection of immunostimulant AS15 for active immunization
with MAGE-A3 protein: Results of a randomized phase II study of the
European organisation for research and treatment of cancer melanoma
group in metastatic melanoma. J Clin Oncol. 31:2413–2420. 2013.
|
|
191
|
Goepfert PA, Tomaras GD, Horton H,
Montefiori D, Ferrari G, Deers M, Voss G, Koutsoukos M, Pedneault
L, Vandepapeliere P, et al: Durable HIV-1 antibody and T-cell
responses elicited by an adjuvanted multi-protein recombinant
vaccine in uninfected human volunteers. Vaccine. 25:510–518.
2007.
|
|
192
|
Du G and Sun X: Engineering
nanoparticulate vaccines for enhancing antigen cross-presentation.
Curr Opin Biotechnol. 66:113–122. 2020.
|
|
193
|
Warrier VU, Makandar AI, Garg M, Sethi G,
Kant R, Pal JK, Yuba E and Gupta RK: Engineering anti-cancer
nanovaccine based on antigen cross-presentation. Biosci Rep.
39:BSR201932202019.
|
|
194
|
Miyamoto A, Honjo T, Masui M, Kinoshita R,
Kumon H, Kakimi K and Futami J: Engineering cancer/testis antigens
with reversible S-cationization to evaluate antigen spreading.
Front Oncol. 12:8693932022.
|
|
195
|
Zhang Y, Lin S, Wang XY and Zhu G:
Nanovaccines for cancer immunotherapy. Wiley Interdiscip Rev
Nanomed Nanobiotechnol. 11:e15592019.
|
|
196
|
Wen R, Umeano AC, Kou Y, Xu J and Farooqi
AA: Nanoparticle systems for cancer vaccine. Nanomedicine (Lond).
14:627–648. 2019.
|
|
197
|
Yang J, Li ZH, Zhou JJ, Chen RF, Cheng LZ,
Zhou QB and Yang LQ: Preparation and antitumor effects of
nanovaccines with MAGE-3 peptides in transplanted gastric cancer in
mice. Chin J Cancer. 29:359–364. 2010.
|
|
198
|
Somaiah N, Block MS, Kim JW, Shapiro GI,
Do KT, Hwu P, Eder JP, Jones RL, Lu H, Ter Meulen JH, et al:
First-in-class, first-in-human study evaluating LV305, a
dendritic-cell tropic lentiviral vector, in sarcoma and other solid
tumors expressing NY-ESO-1. Clin Cancer Res. 25:5808–5817.
2019.
|
|
199
|
Deng Z, Tian Y, Song J, An G and Yang P:
mRNA vaccines: The dawn of a new era of cancer immunotherapy. Front
Immunol. 13:8871252022.
|
|
200
|
Chen W, Wu Y, Deng J, Yang Z, Chen J, Tan
Q, Guo M and Jin Y: Phospholipid-membrane-based nanovesicles acting
as vaccines for tumor immunotherapy: Classification, mechanisms and
applications. Pharmaceutics. 14:24462022.
|
|
201
|
Wadman M: Public needs to prep for vaccine
side effects. Science. 370:10222020.
|
|
202
|
Kudo K, Miki Y, Carreras J, Nakayama S,
Nakamoto Y, Ito M, Nagashima E, Yamamoto K, Higuchi H, Morita SY,
et al: Secreted phospholipase A2 modifies extracellular
vesicles and accelerates B cell lymphoma. Cell Metab.
34:615–633.e8. 2022.
|
|
203
|
Cheng Y, Jiao X, Fan W, Yang Z, Wen Y and
Chen X: Controllable synthesis of versatile mesoporous organosilica
nanoparticles as precision cancer theranostics. Biomaterials.
256:1201912020.
|
|
204
|
Li J, Lu W, Yang Y, Xiang R, Ling Y, Yu C
and Zhou Y: Hybrid nanomaterials for cancer immunotherapy. Adv Sci
(Weinh). 10:e22049322023.
|