Introduction
Autophagy is an evolutionarily catabolic process
that is activated when cells encounter stress, such as nutrient
starvation, energy-limiting conditions, endoplasmic reticulum
stress and reactive oxygen species. It involves the degradation of
unessential proteins or other components. Dysregulation of
autophagy contributes to multiple diseases, including cancer and
neurodegenerative diseases. In addition, autophagy is essential for
maintaining cellular homeostasis in eukaryotes through a mechanism
that can be described as a lysosome-dependent degradation of
intracellular components.
Under normal conditions, basal autophagy is critical
in regulating cell proliferation, apoptosis, differentiation,
development, hematopoiesis, inflammation and aging. Dysfunctional
autophagy is associated with various diseases, including cancer,
diabetes, myopathies, cell death, aging, and neurodegenerative,
cardiovascular, heart, lung, infectious and autoimmune diseases
(1-6). Mosaic deletion of Atg5 or
liver-specific deletion of Atg7 resulted in benign liver adenoma
and accumulation of sequestosome 1 (SQSTM1)/p62 in hepatocytes
(7). Loss of function for ATG5 has
also been found in patients with gastrointestinal cancer, and these
ATG5 dysfunctions may interfere with normal apoptosis and autophagy
during gastrointestinal cancer pathogenesis (8). Mutations of specific tumor suppressor
genes such as ultraviolet irradiation resistance-associated gene
(UVRAG) and Beclin 1 (BECN1) could also lead to reduced clearance
of intracellular damage components, thereby causing tumor
development (9). In addition, the
loss of UVRAG or BECN1 was associated with gastric, colon, breast
and prostate cancer, as well as hepatocellular and cervical
squamous cell carcinoma (10).
Leukemia is a hematological malignancy caused by
abnormal proliferation and differentiation of hematopoietic stem
cells. It is a clonal hematological tumor characterized by the
proliferation and accumulation of lymphoid or myeloid progenitor
cells in the entire bone marrow and the blocking of their
differentiation. Drug resistance remains an important cause of
treatment failure and high mortality in leukemia. Previous studies
focused on autophagy regulation and leukemia treatment demonstrated
the ability for autophagy to degrade fusion oncoproteins, including
promyelocytic leukemia-retinoic acid receptor α and breakpoint
cluster region-Abelson kinase, and then promote leukemic cell death
(11-14). Conditional deletion of Atg7 in
hematopoietic stem cells (HSCs) affected autophagy activity and led
to invasive myeloproliferation and death in mice (15). Low autophagy levels and high NOTCH
signaling levels in HSCs were evident in patients with acute
myeloid leukemia (AML). Low autophagy also upregulated the NOTCH
signaling pathway in mouse models (16). Akt-mTOR, a critical regulatory
pathway in autophagy, was also vital for the maintenance of HSCs.
It could maintain the balance between self-renewal and
differentiation of HSCs, and prevented the transformation of HSCs
into leukemia stem cells (17).
Unc51-like autophagy activating kinase 1 (ULK1) is a substrate of
caspase-3, and ULK1-induced autophagy suppressed leukemogenesis
caused by the fusion oncoprotein acute myeloid gene 1
(AML1)-myeloid transforming gene 8 (ETO) in AML (18).
Although autophagy is a cytoplasmic event and the
majority of studies have focused on protein-level signaling in the
cytoplasm, transcriptional and epigenetic regulation of various
autophagy components in the nucleus is also critical. In addition,
various histone modifications provide essential functions in the
autophagy process. As an increasing number of transcription factors
become widely studied in autophagy, including transcription factor
EB (TFEB) (19, 20), E2F transcription factor 1 (21,22),
and forkhead box proteins of class O subgroup family members
(23-26), the associated epigenetic mechanisms
involved in controlling chromatin recognition of transcription
factors are also receiving more research attention (27). The deubiquitinase ubiquitin
specific peptidase 44 affected autophagy by downregulating H2B
monoubiquitination under starvation conditions (28). G9a, a methyltransferase, directly
suppressed the transcriptional activation of key regulators of
autophagy genes by remodeling the chromatin landscape (29). In addition, G9a depletion in
Drosophila melanogaster suppressed autophagy by altering the
expression level of Atg8a in a methyltransferase-independent manner
(30). The methyltransferase
enhancer of zeste homolog 2 inhibited autophagy via activation of
the mTOR signaling pathway, which revealed a new link between
autophagy, epigenetic regulation and tumorigenesis in human
colorectal carcinoma (31). The
histone acetyltransferase hMOF (also called KAT8 or MYST1) and
NAD+-dependent deacetylase sirtuin 1 regulate the H4K16
acetylation levels of autophagy-related genes, which determine cell
survival (32). Arginine
methyltransferase 1 functions as a co-activator of TFEB and
regulates histone H3R17 methylation levels of autophagy-related and
lysosomal genes after nutrient starvation (20).
Cytogenetic deletions involving the long arm of
chromosome 5 are frequently observed in malignant bone marrow
diseases, including myelodysplasia and AML. The affected patients
exhibit drug resistance, rapid disease progression and short
survival. KDM3B is one of the genes prone to be lost on chromosome
5 (33). As a histone demethylase,
KDM3B has a Jumonji C domain, which is mutated or deleted in
numerous diseases and tumors, such as breast cancer (34) and myeloid leukemia (35). KDM3s (including KDM3A, KDM3B and
Jumonji Domain Containing 1C) regulate chromatin stability and
transcription in human colorectal cancer stem cells through
synergistic Wnt/β-catenin signaling, thus controlling their
tumorigenic potential (36). A
previous study showed that KDM3B played a key role in regulating
the cell cycle and proliferation of HepG2 cells (37). In addition, KDM3B plays critical
roles in prostate cancer genesis (38), ferroptosis (39), differentiation of functional
spermatogonia in mice (40) and
maintenance of genomic stability (41). Moreover, KDM3B has been
investigated in previous studies on hematopoiesis and leukemia,
since it acts as a tumor suppressor gene in AML (42,43)
and can regulate the expression of the leukemia oncogene LIM domain
only 2 (44). H4R3me2s and H3K9me2
could be demethylated by KDM3B to promote gene expression in the
development of hematopoietic stem and progenitor cells (45). Despite the currently available
knowledge about KDM3B in other aspects, the molecular mechanisms
underlying the role of KDM3B in regulating autophagy in leukemia
remain unclear.
The present study identified the molecular basis by
which KDM3B regulates the transcription of the autophagy-related
gene GABARAPL1 in the leukemia cell lines K562, THP1 and
U937. In addition, it was found that the expression of KDM3B
promoted the formation of autophagosomes in leukemia cells under
stimulations. The present findings reveal the mechanism that KDM3B
employs to regulate autophagy as an epigenetic regulator within
leukemia cells.
Materials and methods
Antibodies and reagents
The following antibodies were used in the present
study: Anti-light chain 3 β (LC3B) [Sigma-Aldrich; Merck KGaA; cat.
no. L7543; 1:1,000 for western blotting (WB) and 1:500 for
immunofluorescence (IF)], anti-SQSTM1 (Abcam; cat. no. ab109012;
1:5,000 for WB), anti-GABARAPL1 (Cell Signaling Technology, Inc.;
cat. no. 26632T; 1:1,000 for WB), anti-H3K9me2 (ABclonal Biotech
Co., Ltd.; cat. no. A2359; 1:1,000 for WB), anti-H4R3me2s (ABclonal
Biotech Co., Ltd.; cat. no. A3159; 1:1,000 for WB), anti-KDM3B
[Invitrogen; Thermo Fisher Scientific, Inc.; cat. no. PA5-17170;
1:1,000 for WB; and Cell Signaling Technology, Inc.; cat. no.
5377S; 1:200 for chromatin immunoprecipitation (ChIP)], anti-GAPDH
(Proteintech Group, Inc.; cat. no. 60004-1-Ig; 1:20,000 for WB),
horseradish peroxidase (HRP)-labeled secondary antibody conjugates
(ABclonal Biotech Co., Ltd.; cat. nos. AS003 and AS014) and
Cy3-labeled goat anti-rabbit IgG (H + L) (Beyotime Institute of
Biotechnology; cat. no. A0516; 1:300 for IF).
DAPI was purchased from Beijing Solarbio Science
& Technology Co., Ltd. (cat. no. S2110), while bafilomycin A1
(BafA1) was obtained from Sigma-Aldrich; Merck KGaA (cat. no.
196000; 100 nM; 2 h for autophagy flux), rapamycin from
MedChemExpress (cat. no. HY-10219; 5 µM for autophagy
induction) and puromycin from Beijing Solarbio Science &
Technology Co., Ltd. (cat. no. P8230; 2 µg/ml for cell
screening).
Plasmid constructs
The full-length human KDM3B (NM_016604) was
cloned into the GV367 vector (Shanghai GeneChem Co., Ltd.), and a
plasmid encoding Flag-tagged wild-type (WT) KDM3B was
constructed.
Cell culture, transfection and generation
of knockout (KO) cells
The human cell lines used in the present study were
obtained from the Leibniz-Institute DSMZ-German Collection of
Microorganisms and Cell Cultures. The cell types of the three cell
lines are as follow: K562 (chronic myeloid leukemia in blast
crisis), THP1 (acute monocytic leukemia) and U937 (acute monocytic
leukemia). All the three cell lines used in the present study have
been authenticated using the standard STR (short tandem repeats)
genotyping method. The three cell lines were cultured in RPMI-1640
medium (Cellmax Technologies; cat. no. CGM112.05), while 293T cells
(Leibniz-Institute DSMZ-German Collection of Microorganisms and
Cell Cultures; cat. no. ACC 635) were cultured in DMEM (Cellmax
Technologies; cat. no. CGM101.05). The aforementioned media
contained 10% fetal bovine serum (FBS) (Cellmax Technologies; cat.
no. SA211.02). Cells were incubated in a 5%
CO2-humidified incubator at 37°C.
For transfection, 293T cells were cultured in cell
plates using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.; cat. no. L3000015). For starvation
treatments, the cells were cultured in Earle's Balanced Salt
Solution (EBSS) (Beijing Solarbio Science & Technology Co.,
Ltd.; cat. no. H2020) for various durations.
To generate KDM3B KO cells, lentiviral
supernatant containing targeting sequence to KDM3B
(purchased from Shanghai GeneChem Co., Ltd.) was transfected into
target cells (K562, THP1, U937 and 293T). Briefly, KDM3B KO
cell lines were constructed using the CRISPR/CAS9 lentivirus
packaging technique. The targeting gRNA sequence was constructed
into lentivirus vector Lenti-sgRNA-CAS9 (cat. no. GV392; Shanghai
GeneChem Co., Ltd.), and the lentiviral supernatant was added to
the cells in medium containing 10% FBS, centrifuged at 1,000 × g
for 1 h at room temperature, and then incubated in a 5%
CO2-humidified incubator at 37°C. After 2 days of viral
infection, 2 µg/ml puromycin was added to the cell culture
medium to screen positive cells. After 2 weeks of screening,
multiple monoclonal cell colonies were selected using a 96-well
plate by the limited dilution method. Multiple cell lines with
different KO levels were obtained by monoclonal amplification
culture. When the cells proliferated to >1×106 cells,
the cells were collected for WB with an anti-KDM3B antibody. A
monoclonal cell colony with complete KDM3B KO was selected
for each cell line for subsequent experiments. The targeting
sequence was as follows: 5′-GTCCAATGGTGTTCTAGCCA-3′.
Total cellular protein extraction and
WB
Cells were collected and washed with pre-cold PBS.
Their proteins were extracted by adding RIPA buffer (Beijing
Solarbio Science & Technology Co., Ltd.; cat. no. R0020),
supplemented with protease inhibitors (Beyotime Institute of
Biotechnology; cat. no. P1010), incubated on ice for ≥30 min, and
vortexed for 20 sec every 10 min. The soluble fraction was the
isolated by centrifugation at 14,000 × g for 10 min at 4°C, and the
supernatant was transferred to a new tube.
For immunoblotting, total cellular proteins were
subjected to quantification by using BCA Protein Assay Kit. The
protein extracts were resolved by SDS-PAGE in 12% polyacrylamide
(20 µg protein loaded per lane) and transferred to a
0.45-µm PVDF membrane (MilliporeSigma; cat. no. R1MB61296).
Membranes were blocked with 5% non-fat dried milk in TBS containing
0.1% Tween-20 (Shandong Sparkjade Scientific Instruments Co., Ltd.;
cat. no. ED0009) for 1 h at room temperature and incubated with the
indicated primary antibodies overnight at 4°C, followed by
incubation with HRP-conjugated goat secondary antibodies (1:2,000)
for 1 h at room temperature. The bands were analyzed using ECL Plus
detecting reagents (Beijing Solarbio Science & Technology Co.,
Ltd.; cat. no. PE0010) and Amersham Imager 600 (Cytiva).
IF of cells in suspension
Suspended cells were collected by centrifugation at
1,000 × g for 5 min at room temperature. Next, the pellet was
resuspended with 100 µl culture medium. A single-cell
coating at a density of 5-6×105 cells was obtained on
the slide using the Thermo Shandon Cytospin 4 machine (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The cells were fixed with 4% paraformaldehyde
(Beyotime Institute of Biotechnology; cat. no. P0099) for 20 min at
room temperature. Next, the cells were permeabilized with 0.1%
Triton X-100 (Beijing Solarbio Science & Technology Co., Ltd.;
cat. no. T8200) in PBS for 5 min and blocked with 5% bovine serum
albumin (BSA) (Beijing Solarbio Science & Technology Co., Ltd.;
cat. no. A8010) in PBS for 1 h at room temperature. The samples
were incubated with anti-LC3B primary antibody in 1% BSA in PBS
overnight at 4°C. Next, the cells were washed with PBS and
incubated with a secondary antibody in 1% BSA in PBS for 1 h at
room temperature, followed by DAPI (10 µg/ml; Beijing
Solarbio Science & Technology Co., Ltd.; cat. no. S2110)
staining. The cells were then mounted using coverslips and
visualized under a confocal microscope (Leica Microsystems, Inc.;
cat. no. TCS SP8).
Electron microscopy
Cells were fixed in 0.2 M sodium cacodylate buffer
(pH 7.4) containing 2% paraformaldehyde and 2.5% glutaraldehyde
overnight at 4°C, washed with 0.1 M sodium cacodylate buffer for 3
times at 25°C, postfixed in 1% osmium tetroxide for 2 h at 4°C and
stained with 1% uranyl acetate overnight at 4°C. Following stepwise
ethanol and acetone dehydration, samples were infiltrated with Epon
resin overnight at 60°C. Finally, the samples were sectioned at a
thickness of 70 nm and observed by transmission electron microscopy
(TEM; Hitachi, Ltd.; cat. no. HT7700).
RNA-sequencing (RNA-seq) analyses
mRNA sequencing was performed using WT and
KDM3B KO K562 cells (Shanghai GeneChem Co., Ltd.). The cells
were collected after starvation in EBSS medium for 4 h before RNA
was extracted. A total of 1 µg RNA per sample was used as
input material for RNA sample preparations. Sequencing libraries
were generated using NEBNext® Ultra™ RNA Library Prep
Kit for Illumina® (New England BioLabs, Inc.) following
the manufacturer's recommendations, and index codes were added to
attribute sequences to each sample. The clustering of the
index-coded samples was performed on a cBot Cluster Generation
System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, Inc.)
according to the manufacturer's instructions. After cluster
generation, the library preparations were sequenced on an Illumina
Novaseq platform (Illumina, Inc.), and 150-bp paired-end reads were
generated. Clean data (clean reads) were obtained by removing reads
containing adapter, poly-N and general low-quality reads from raw
data. At the same time, the clean data were inspected for Q20, Q30
and GC content. The sequencing process itself has the possibility
of machine error, and the inspection of sequencing error rate
distribution can reflect the quality of sequencing data. If the
sequencing error rate is represented by e (46) and the base mass value of Illumina
is represented by Qphred, Qphred=-10log10(e). Q20 represents the
percentage of bases with a phred value higher than 20 in the
population, and Q30 represents the percentage of bases with a phred
value higher than 30 in the population. GC content represents the
percentage of G and C in the four bases of clean reads.
All downstream analyses were based on the resulting
high-quality clean data. Reference genome and gene model annotation
files were downloaded from genome websites directly. Indexing of
the reference genome was built using Hisat2 v2.0.5 software, and
paired-end clean reads were aligned to the reference genome.
Differential expression analysis of two conditions/groups was
performed using the DESeq2 package (47). The resulting P-values were adjusted
using Benjamini and Hochberg's approach for controlling the false
discovery rate. Genes with an adjusted P<0.05 found by DESeq2
were considered to be differentially expressed. Gene Ontology (GO)
enrichment analysis of differentially expressed genes was
implemented via the clusterProfiler R package, in which gene length
bias was corrected. GO terms with corrected P<0.05 were
considered as significantly enriched by differential expressed
genes. The clusterProfiler R package was used to evaluate the
statistical enrichment of differential expression genes in Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways.
RNA extraction, cDNA synthesis and
reverse transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted using TRIzol®
(Shandong Sparkjade Scientific Instruments Co., Ltd.), and cDNA was
obtained from 2 µg total RNA by RT using the Evo M-MLV cDNA
Synthesis kit (Jiangsu Accuracy Biotechnology Co., Ltd.; cat. no.
AG11706) according to the manufacturer's instructions. qPCR was
carried out by Applied Biosystems 7500 Fast Real-Time PCR System
(Thermo Fisher Scientific, Inc.) with SYBR Green Premix Pro TaqHS
qPCR Kit (Rox Plus) (Jiangsu Accuracy Biotechnology Co., Ltd.; cat.
no. AG11718). The thermocycling conditions were as follows: Initial
denaturation, 95°C for 30 sec; followed by 40 cycles of
denaturation (95°C for 10 sec); annealing (55°C for 20 sec) and
extension (72°C for 30 sec). The relative gene expression was
analysed using the 2−ΔΔCq method (48). ACTB was used as a control
for normalization. All reactions were performed as triplicates. The
following primers were used: ACTB forward,
5′-ATTGCCGACAGGATGCAGAA-3′ and reverse,
5′-ACATCTGCTGGAAGGTGGACAG-3′; and GABARAPL1 forward,
5′-GAATCCACCTGAGACC-3′ and reverse, 5′-GCCTTACACTGCCATA-3′.
Luciferase assays
For luciferase assays, the GABARAPL1 gene
promoter regions (from ~1 kb upstream of the transcription start
site to ~200 bp downstream) were amplified from genomic DNA and
inserted into the pGL3.0-basic vector (Promega Corporation) by two
restriction endonucleases, NheI and HindIII. For
plasmid transfection in 293T cells, Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.; cat. no. L3000015) was
used according to the manufacturer's instructions. Luciferase
activities were measured after 48 h of transfection using a
Dual-Luciferase Reporter Assay System (Promega Corporation; cat.
no. E2920) according to the manufacturer's instructions, and Sirius
L Tube Luminometer (Titertek-Berthold). Renilla luciferase
activity was used as a control for normalization. Experiments were
independently repeated ≥3 times. The following primers were used:
GABARAPL1-NheI forward, 5′-CTA GCT AGC GCT TGA TAC
TCT CTT TTC CA-3′ and GABARAPL1-HindIII reverse,
5′-CCC AAG CTT TGA CCC TGT CCC GCT CC-3′.
Chromatin immunoprecipitation (ChIP)-qPCR
analysis
For ChIP assay, a kit from Beyotime Institute of
Biotechnology (cat. no. P2078) was used. Briefly, cells were
collected and crosslinked with 1% formaldehyde for 10 min in PBS at
room temperature, followed by quenching for 5 min with 0.125 M
glycine. After centrifugation at 1,000 × g for 5 min at 4°C, the
cell pellets were lysed in SDS-lysis buffer, supplemented with 1 mM
PMSF (Beyotime Institute of Biotechnology, Inc.; cat. no. ST506),
and sonicated. The supernatant lysates were incubated with protein
A+G agarose/salmon sperm DNA (Beyotime Institute of Biotechnology,
Inc.; cat. no. P2078-1) for pre-clearing, and then subjected to IP
with IgG (Beyotime Institute of Biotechnology, Inc.; cat. no.
A7016) and the indicated antibodies. The immunoprecipitants were
washed with low-salt wash buffer, high-salt wash buffer, LiCl wash
buffer and Tris-EDTA buffer before being eluted in elution buffer
(1% SDS and 0.1 M NaHCO3). Next, the supernatant was incubated with
5 M NaCl for 4 h at 65°C to reverse crosslink, and digested with
0.5 M EDTA, 1 M Tris (pH 6.5) and proteinase K for 1 h at 45°C. DNA
was purified and then analyzed by RT-qPCR. The enrichment value of
bound DNA was calculated relative to the input. The following
primers were used in ChIP-qPCR assay: GABARAPL1-P1 region
forward, 5′-TGCCCTAATGCCCAATCTTC-3′ and reverse,
5′-TTGGCCAATGCAGAGCTGT-3′; and GABARAPL1-P2 region forward,
5′-TCTGCATTGGCCAAAGGGAT-3′ and reverse,
5′-TAGCTGCAGAAACGTCCGCT-3′.
Cell viability assay
Cell Counting Kit (CCK)-8 assay was used to detect
cell viability. K562, THP1 and U937 cells (WT and KDM3B KO)
were seeded into 96-well plates with 1×104 cells per
well. Cells were cultured for 48 h, and then 10 µl CCK-8
(Beijing Solarbio Science & Technology Co., Ltd.; cat. no.
CA1210) was added to each well and incubated for 4 h at 37°C. The
total volume in each well was 100 µl. The absorbance was
measured at 450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Statistical analysis
Statistical significance was performed using
unpaired Student's t-test, One-way analysis of variance (ANOVA) or
Two-way ANOVA with multiple comparisons test unless otherwise
stated. All statistical analyses were assessed using GraphPad Prism
9.0 (Dotmatics), unless otherwise stated. Data are presented as the
mean ± SD. In all analysis, P<0.05 was considered to indicate a
statistically significant difference. All experiments were
independently repeated at least three times.
The clinical data and gene expression profiles were
obtained from The Cancer Genome Atlas database (https://portal.gdc.cancer.gov). The survival rate and
expected survival time were used to measure disease prognosis.
Survival curves were generated using the Kaplan-Meier method
followed by the log-rank test. The correlation between KDM3B and
GABARAPL1 was analyzed by Spearman's correlation analysis. Cox and
logistic regression were used to analyze the relationship between
KDM3B, GABARAPL1 expression levels and AML clinical phenotypes.
Data were expressed as the median (interquartile range) or the mean
± SD for continuous variables.
Results
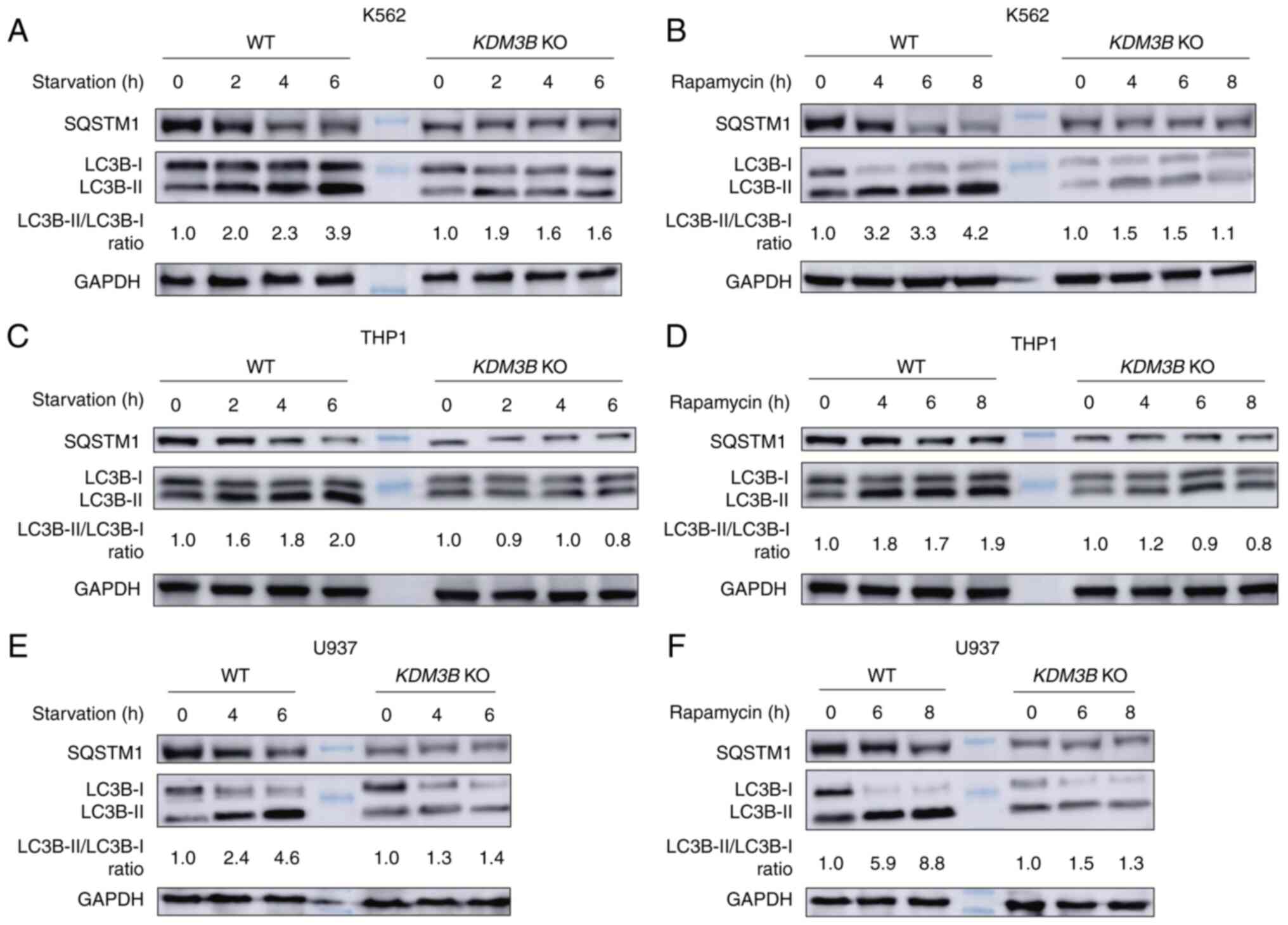
Loss of KDM3B decreases LC3B-II levels
and inhibits autophagy in different leukemia cells
To investigate the effect of KDM3B expression on
leukemia cell autophagy, the autophagic process was first analyzed
in K562, THP1 and U937 cells under starvation or rapamycin
stimulation by examining the conversion of LC3B-I to LC3B-II as a
standard marker of autophagic activity and the decrease of the
autophagic substrate SQSTM1. Western blot analysis showed that
LC3B-II levels were significantly increased upon starvation or
rapamycin induction in K562 cells, while the SQSTM1 levels showed
an opposite trend (Fig. S1A). A
similar trend was observed in THP1 and U937 cells (Fig. S1B and C).
To further explore the role of KDM3B in autophagy,
four different KDM3B KO stable cell lines (KDM3B KO
K562 cells, KDM3B KO THP1, KDM3B KO U937 and
KDM3B KO 293T cells) were constructed using CRISPR/Cas9
technology (Fig. S1D). WB
confirmed efficient KO in all cell lines (Fig. S1E).
CCK-8 assay was also used to measure cell viability
after KDM3B KO. As demonstrated in Fig. S1F, compared with the WT group, the
cell viability was significantly increased after KDM3B KO,
suggesting that KDM3B expression could inhibit cell
proliferation.
Next, the autophagic process was analyzed in WT and
KDM3B KO K562 cells upon starvation induction by WB. It was
observed that the LC3B-II levels were increased in WT K562 cells
compared with cells under normal culture conditions. This increase
was time dependent, becoming significant with increased starvation
time. The SQSTM1 levels exhibited an opposite trend. However,
KDM3B KO inhibited LC3B-II formation, while the LC3B-II and
SQSTM1 levels remained stable after starvation induction (Fig. 1A).
The autophagic process was also investigated in WT
and KDM3B KO K562 cells by rapamycin stimulation. WB
revealed the accumulation of LC3B-II and the degradation of SQSTM1
in WT K562 cells upon rapamycin stimulation compared with cells
under normal culture conditions. KDM3B KO inhibited LC3B-II
formation, whereas the LC3B-II or SQSTM1 levels did not differ
after rapamycin stimulation (Fig.
1B). Similar trends for LC3B-II and SQSTM1 levels were also
observed in THP1 and U937 cells (Fig.
1C-F). Collectively, these results suggested that KDM3B was a
critical regulator of autophagy.
KDM3B promotes autophagosome
formation
To further investigate KDM3B functions in autophagy,
the present study explored whether KDM3B affects autophagosome
formation upon starvation induction. IF and confocal fluorescence
microscopy analyses identified the formation of a LC3B-positive
autophagosome upon starvation induction. First, the increase in
LC3B punctate cells was notably attenuated in KDM3B KO cells
compared with WT K562 cells (Fig.
2A). Second, TEM further showed an increase in the number of
autophagic vesicles in WT K562 cells but not in KDM3B KO
K562 cells (Fig. 2B). Under TEM,
the early stage of autophagosome formation was cup shaped and had a
double-membrane structure, which half enclosed the components that
needed to be degraded. The complete autophagosome was a bilayer
vesicle containing components that needed to be degraded.
Autophagosomes were generally 300-900 nm in size, with a mean size
of 500 nm. These results indicated that KDM3B played a role in
promoting autophagosome formation.
KDM3B-associated autophagy flux
To investigate the KDM3B-associated autophagy
process, the KDM3B-involved autophagy flux was evaluated via
KDM3B KO. Western blot analysis showed that KDM3B KO
inhibited LC3B-II formation in K562 upon starvation induction
(Figs. 1, and 3A and B). In addition, LC3B-II
accumulated during BafA1 treatment, and the autophagic substrate
SQSTM1 level exhibited a corresponding change with LC3B-II
(Fig. 3A). Consistent results were
observed in THP1 cells (Fig.
3B).
 | Figure 3Defects in autophagic flux caused by
the loss of KDM3B. (A and B) LC3B flux was analyzed in WT and
KDM3B KO K562 cells (A) or THP1 cells (B) in the absence or
presence of BafA1 (100 nM, 2 h). Cells were cultured and then
starved in EBSS (4 h) with or without BafA1. KDM3B KO
inhibited LC3B-II formation. BafA1 treatment resulted in the
accumulation of LC3B-II and SQSTM1. The LC3B-II/LC3B-I ratio is
indicated. (C) Autophagy flux was revealed by the mCherry-GFP-LC3
tandem reporter. The plasmid was transfected in WT or KDM3B
KO 293T cells. Representative images of the cells cultured in
normal, EBSS medium (4 h) or EBSS with BafA1 (100 nM) addition (2
h), respectively. Moreover, the formation of autophagosome (yellow:
mCherry-positive; GFP-positive) and autolysosome (red:
mCherry-positive; GFP-negative) was examined. Scale bar, 10
µm. (D) Statistical analysis of vesicles positive for both
GFP and mCherry (autophagosomes) and for mCherry (autolysosomes)
(>20 cells per experiment) in (C) Data (mean ± SD) were analyzed
with two-way ANOVA. ns, not significant;
****P<0.0001. KDM3B, histone lysine demethylase 3B;
WT, wild-type; KO, knockout; BafA1, bafilomycin A1; EBSS, Earle's
Balanced Salt Solution. |
Autophagy flux tests were performed using a tandem
fluorescent indicator, mCherry-GFP-LC3B, in KDM3B KO and WT
293T cells upon starvation induction. Since the green fluorescence
of the fusion protein is sensitive in lysosomes and quickly
quenched in autolysosomes, only red fluorescence could be detected
in autolysosomes. Therefore, results showing yellow or green puncta
indicate the presence of autophagosomes. Fluorescence analysis
using the aforementioned tandem fluorescent indicator system showed
that KDM3B KO significantly inhibited the formation of
autophagosomes compared with WT 293T cells upon starvation. Under
normal conditions, there was no significant difference between
KDM3B KO and WT 293T cells. However, further starvation and
BafA1 combined treatment showed a significant accumulation of
autophagosomes in WT 293T but not in KDM3B KO 293T cells
(Fig. 3C and D). These results
suggested that KDM3B promoted autophagosome formation and
functioned as a factor in autophagy flux.
KDM3B regulates the expression of
GABARAPL1, which is involved in the process of autophagy
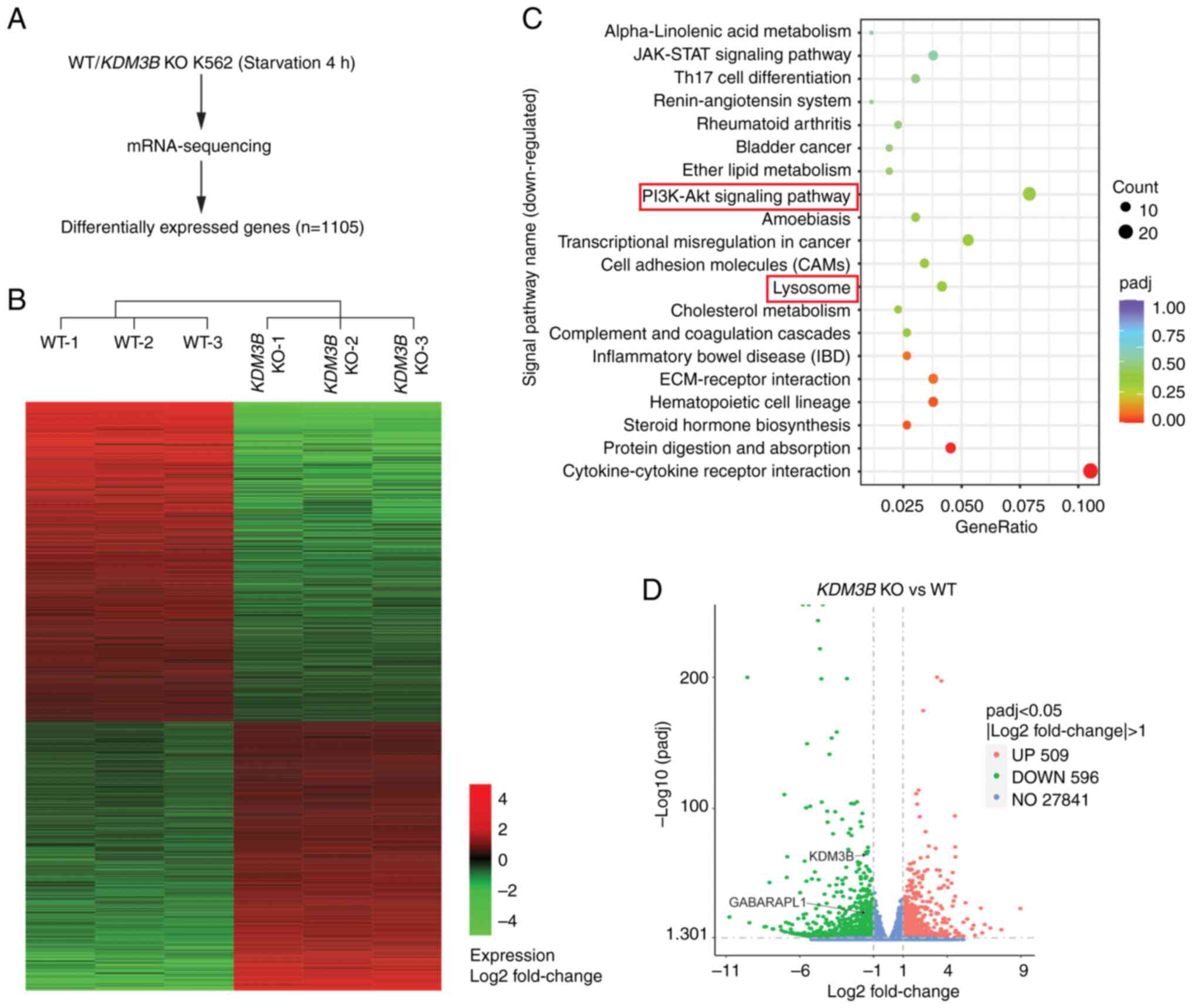
To explore the mechanism of KDM3B in the
transcriptional regulation of autophagy, RNA-seq was performed in
WT and KDM3B KO K562 cells after starvation treatment. A
total of 1,105 differentially expressed genes were found (Fig. 4A and B). KEGG analysis was
performed for all the downregulated genes after KDM3B KO in
K562 cells. The top 20 signaling pathways are shown in Fig. 4C. Among them, the PI3K-Akt and
lysosomal signaling pathways were closely associated the regulation
of autophagy.
GABARAPL1, an autophagy-related gene, was
identified among all the downregulated genes through analysis using
volcano plots (Fig. 4D). The KDM3B
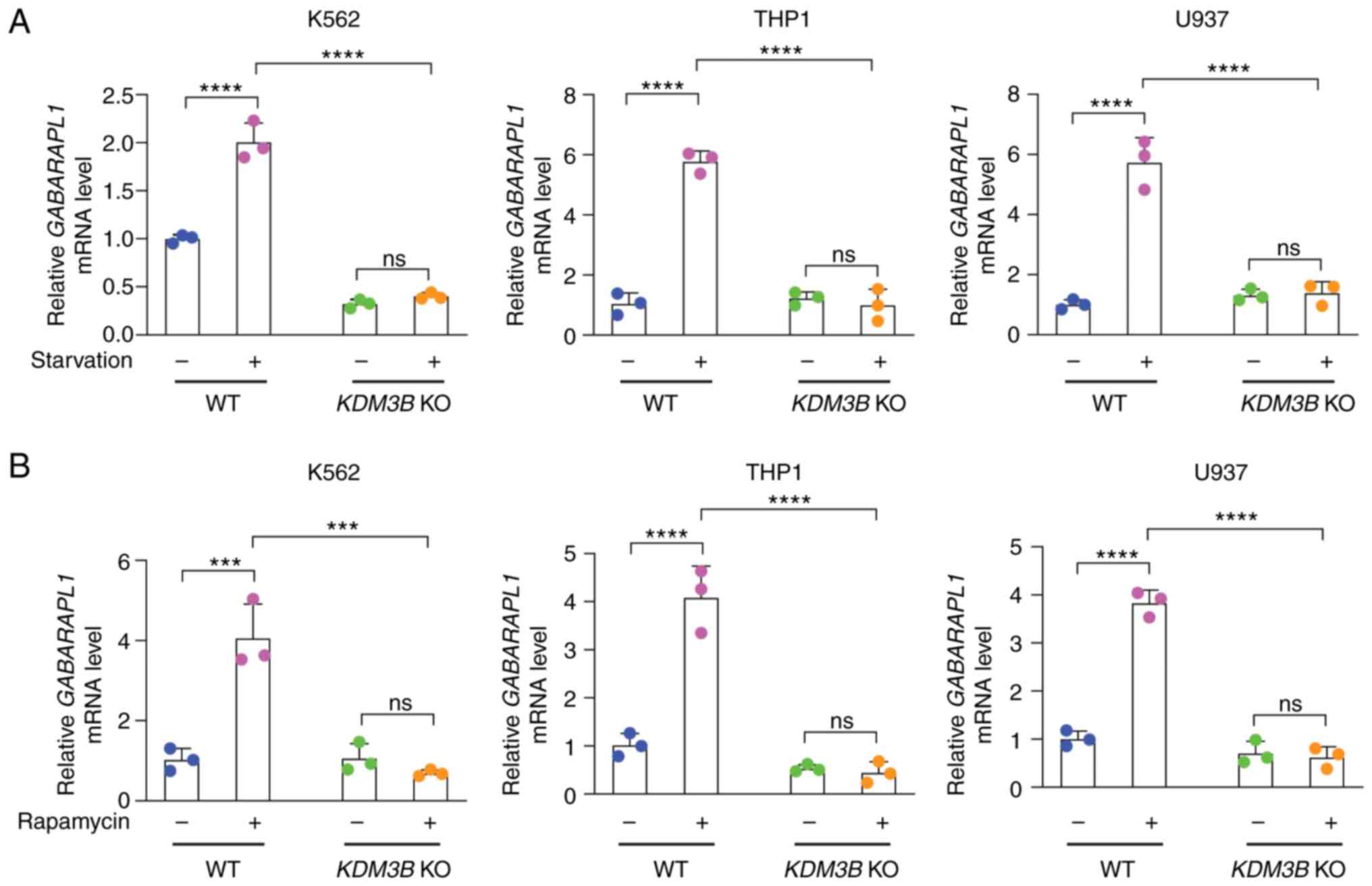
dependency of the GABARAPL1 gene was validated by RT-qPCR.
Three cell lines, K562, THP1 and U937 cells (WT and KDM3B
KO), were cultured under normal and EBSS conditions (Fig. 5A) or subjected to rapamycin
treatment (Fig. 5B), and the mRNA
expression of GABARAPL1 was quantified through qPCR. The
results showed that the mRNA expression of GABARAPL1 was
increased in WT cell lines upon starvation or rapamycin induction
compared with cell lines under normal culture conditions. However,
the mRNA expression of GABARAPL1 did not increase in
KDM3B KO cell lines, which was consistent with the results
of RNA-seq. This result suggested that KDM3B may be involved in the
regulation of the GABARAPL1 gene, which participates in the
autophagic process.
KDM3B is associated with the promoter of
the GABARAPL1 gene
The present study focused on GABARAPL1 for
additional analysis. To examine the KDM3B-dependent epigenetic
regulation of GABARAPL1, two possible promoter regions for
KDM3B association were mapped in the ChIP experiment (Fig. 6A). WT K562 cells were starved in an
EBSS medium for 4 h and then used for ChIP assay. Agarose gel
electrophoresis showed a significant binding of KDM3B to the −1,119
to −360 region (P1 region) of the GABARAPL1 promoter
compared with the −374 to +155 region (P2 region) (Fig. 6B). The efficiency of IP was
evaluated by WB. As demonstrated in Fig. 6B, the data suggested that the added
anti-KDM3B antibody successfully precipitated the KDM3B protein in
cells. The ChIP-qPCR experiment also confirmed the recruitment of
KDM3B to the −1,119/−360-GABARAPL1-promoter (Fig. 6C).
 | Figure 6Depletion of KDM3B alters KDM3B
promoter binding of GABARAPL1. (A) Schematic diagram of
primer relative positions (P1 region and P2 region) on the
GABARAPL1 gene promoter used for (B) and (C). (B) ChIP
assay. Upper, sonicated chromatin from WT K562 cells starved in
EBSS (4 h) was immunoprecipitated with anti-KDM3B or preimmune IgG
(control). Agarose gel electrophoresis showed that a fragment
corresponding to the -1119 to -360 region (P1 region) of
GABARAPL1 promoter was amplified using the
immunoprecipitated DNA as a template. Below: Western blot analysis
of KDM3B in the immunoprecipitated samples, followed by
immunoblotting with the anti-KDM3B antibody. A total protein of 20
µg was added into each well for western blot analysis. (C)
Quantitative PCR of ChIP analysis revealed that KDM3B can bind to
the GABARAPL1 gene promoter (P1 region) in vivo. The
data were normalized by input. (D) Luciferase assays. WT,
KDM3B KO or KDM3B-rescued 293T cells were transiently
transfected with pGL3.0-GABARAPL1 promoter. Luciferase
activities were measured followed by EBSS medium for 4 h. Data
(mean ± SD) were analyzed with one-way ANOVA. ns, not significant;
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. KDM3B,
histone lysine demethylase 3B; GABARAPL1, GABA type A
receptor-associated protein like 1; ChIP, chromatin
immunoprecipitation; WT, wild-type; KO, knockout; EBSS, Earle's
Balanced Salt Solution. |
Next, a luciferase assay was performed to confirm
the KDM3B-mediated regulation of GABARAPL1 at the
transcriptional level. A luciferase vector containing the promoter
region of GABARAPL1 was transiently overexpressed in WT and
KDM3B KO 293T cells (Fig. 6D). A
significant increase in luciferase signal was observed in WT 293T
cells compared with KDM3B KO 293T cells, indicating that
certain functional regulatory elements by KDM3B existed in this
region. The data showed that KDM3B expression could regulate the
transcription of GABARAPL1. In summary, these results
indicated that KDM3B-dependent chromatin modifications occurred in
GABARAPL1, which was involved in the autophagic process.
Therefore, KDM3B could bind to the promoter of GABARAPL1 and
activate its transcription.
Re-expression of WT KDM3B rescues
GABARAPL1 expression and autophagy
To clarify if KDM3B is necessary for autophagy, WT
KDM3B (Flag-tagged) was re-expressed in KDM3B KO 293T cells.
WB showed that the LC3B-II levels were increased in WT 293T cells
upon starvation induction compared with normal culture conditions.
The SQSTM1 levels exhibited the opposite trend. However,
KDM3B KO inhibited LC3B-II formation, and the LC3B-II or
SQSTM1 levels did not differ during starvation induction. When
KDM3B expression was rescued in KDM3B KO 293T cells,
prominent LC3B-II formation reappeared under starvation conditions,
and SQSTM1 exhibited an opposite trend (Fig. S2A).
RT-qPCR was used to confirm the mRNA expression of
GABARAPL1 in 293T cells. The results demonstrated that the
GABARAPL1 mRNA expression was increased in WT 293T cells
upon starvation induction compared with normal culture conditions,
but not in KDM3B KO 293T cells. When KDM3B expression was
rescued in KDM3B KO 293T cells, it could promote
GABARAPL1 expression (Fig.
S2B). Finally, WT or KDM3B KO 293T and K562 cells were
used to evaluate the GABARAPL1 protein expression levels. Western
blot analysis showed that the GABARAPL1 levels were increased in WT
cells after stimulation compared with normal culture conditions.
KDM3B KO inhibited GABARAPL1 expression (Fig. S2C and D). GABARAPL1 levels
recovered when KDM3B expression was rescued in KDM3B KO 293T
cells (Fig. S2C). Re-expression
of WT KDM3B in KDM3B KO 293T cells also rescued the
luciferase activity (Fig. 6D).
Collectively, these data indicated that KDM3B was a crucial
molecule in autophagy and functioned by increasing the expression
of GABARAPL1.
Identification of the correlation between
KDM3B and GABARAPL1
The occurrence of leukemia is closely related to the
dysfunction of autophagy. In addition to exploring the role of
KDM3B in autophagy in AML cell lines, the information of patients'
samples from the online public database (https://portal.gdc.cancer.gov) was also analyzed to
reveal the expression changes of KDM3B and GABARAPL1 in normal
controls and patients with AML, and the relationship between
expression levels and patient survival was analyzed. According to
the data analysis, increased KDM3B and GABARAPL1 gene expressions
were both associated with favorable outcomes of the patients
(Fig. S3A and B). Lower KDM3B and
GABARAPL1 expression levels were determined in patients with AML
compared with normal controls (Fig.
S3C). Furthermore, a positive correlation between KDM3B and
GABARAPL1 expression was identified in AML patients (Fig. S3D). In addition, multiple
regression analyses (Fig. S4)
were performed to detect the association between KDM3B, GABARAPL1
expression levels and certain AML clinical variables. The results
indicated that, like the majority of other clinical phenotypes, the
KDM3B (HR= 0.711, P= 0.102) and GABARAPL1 (HR= 0.944, P=0.591)
expression levels could not independently predict AML prognosis.
These results suggested that KDM3B and GABARAPL1 may interact with
other clinical variables and be beneficial to the prognosis of AML.
Hence, logistic regression analysis (Fig. S5) was performed and the results
suggested that a low expression of KDM3B correlated with FAB
classifications (OR=0.423, P=0.013). GABARAPL1 expression
correlated not only with FAB classifications (OR=3.421,
P<0.001), but also with BM blasts (OR=0.262, P<0.001). But
neither was associated with certain common gene mutations,
including nucleophosmin 1 (NPM1), and fms related receptor tyrosine
kinase 3 (FLT3).
Discussion
Autophagy is a relatively conserved multistep
degradation process of intracellular components. The activation of
the ULK1 complex and PI3K complex 1 (PtdIns3K-C1) causes vesicle
nucleation and initiates autophagosome formation (49,50).
GABARAPL1 plays a vital role in the initiation of autophagy. It
participates in the re-localization of ULK1 to the phagophore, and
regulates the activation and phosphorylation of ULK1 (51). The assembled ULK1 complex can also
recruit and activate PtdIns3K-C1 together with GABARAPL1 (52,53).
GABARAPL1 is more likely to be involved in closure than elongation
during the extension and closure stages from the phagophore. By
contrast, LC3B is more involved in the extension stage (54). Therefore, LC3 deficiency leads to
the formation of smaller autophagosomes, while GABARAP deficiency
results in larger autophagosomes. The final step of autophagy is
the fusion of the autophagosome and lysosome, which includes the
transport of the autophagosome to the lysosome through the
microtubule skeleton before the fusing process. Previous studies
showed that LC3B was more prone to autophagosome re-localization
(55,56), while GABARAPL1 was more involved in
the fusion process and could also help to increase the number of
available lysosomes to ensure the regular progress of autophagy
(57,58). Notably, a previous study has shown
that LC3B can assist with the fusion process in cells lacking
GABARAPLs (59).
Although there are partial functional compensations
between GABARAP and LC3 subfamily members, the focus of each
member's function is different. Controversies exist about their
expression patterns and roles in different cancer types. Certain
studies have found that the expression of Gabarapl1 in rat brains
is significantly higher than that of other Atg8 family members
(60,61). This observation suggests that
Gabarapl1 may have a role in regulating central nervous system
functions, such as the regulatory association between autophagy and
numerous neurodegenerative diseases. In addition, Gabarapl1 is
highly expressed in the hypothalamus and is regulated by estrogen.
The hypothalamus contains the signaling pathway regulating male and
female reproduction, revealing another potential function of
Gabarapl1 in addition to autophagy regulation. Given the importance
of these members, targeted studies on their expression and
regulation patterns are of great importance in identifying
effective treatments for different types of cancer.
Among the types of leukemia within the focus of the
present study, published studies on KDM3B and GABARAPL1 are
relatively rare. A previous study showed that microRNA (miR)-15a-5p
was highly expressed in patients with chemoresistant AML, and
miR-15a-5p downregulated the expression of four autophagy-related
genes, namely ATG9a, ATG14, GABARAPL1 and sphingomyelin
phosphodiesterase 1, which further inhibited autophagy and
eliminated the therapeutic effect of daunorubicin-induced autophagy
(62). KDM3B, which was initially
considered a tumor suppressor gene, is located in the region of
chromosome 5q31, which is often absent in abnormal bone marrow
development and AML (33).
Analysis of patients with AML showed that KDM3B underexpression was
associated with poor prognosis, while KDM3B downregulation promoted
the proliferation of NB4 cells. After treatment of NB4 cells with
all-trans retinoic acid, KDM3B promoted promyelocytic
leukemia/retinoic acid receptor α fusion protein degradation,
thereby promoting NB4 differentiation into mature granulocytes
(63). However, it has also been
reported that KDM3B is upregulated in patients with acute
lymphoblastic leukemia, and inhibits the all-trans retinoic
acid-induced differentiation of HL-60 cells (44).
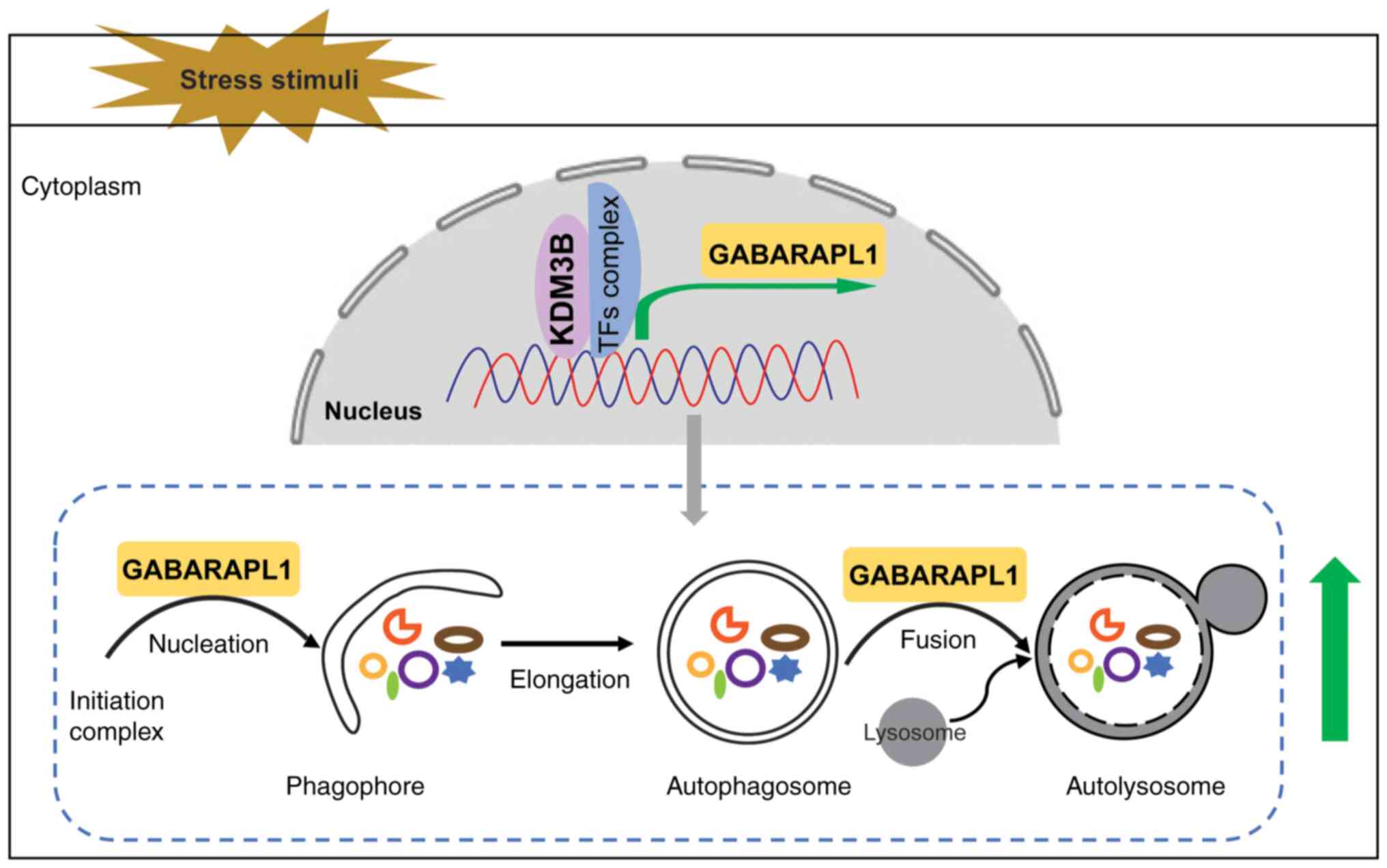
The present study showed that genetic depletion of
the histone lysine demethylase KDM3B attenuated the level of
LC3B-II, suppressed autophagosome formation and increased the
accumulation of SQSTM1. Furthermore, the expression of KDM3B
ensured autophagy flux under stress conditions. The current data
suggested that KDM3B regulated the expression of GABARAPL1, which
was involved in autophagy at the transcriptional level via
associating with the target gene promoter, leading to increased
transcription of GABARAPL1 during the autophagic process (Fig. 7). Subsequently, GABARAPL1
participated in different stages of autophagy, including the
recruitment and activation of the complex at the initiation stage,
and the fusion process at the later stage. The present RNA-seq
results showed the effect of KDM3B KO on autophagy in the leukemia
cell line K562 under autophagy induction conditions. Among all the
downregulated genes, multiple candidates associated with autophagy
were found. However, after further validation of the candidates
using RT-qPCR (data not shown), GABARAPL1 was finally identified as
a promising gene related to autophagy. In a previous study, RNA-seq
was also conducted on HepG2 cells with KDM3B KO (64) but no GABARAPL1 was found. This
difference may be due to different sequencing methods or sample
sources, as different cancer cell types have different patterns in
gene expression. To more comprehensively analyze the influence of
KDM3B KO on downstream target genes, candidate genes identified by
other studies should be further verified in leukemia cells in
subsequent experiments.
The present study also focused on screening
signaling pathways that may be related to autophagy. After KDM3B
KO, the PI3K-Akt and lysosome-related signaling pathways were found
in the top 20 downregulated pathways closely associated with
autophagy in cells. This finding further supported the notion that
KDM3B may be involved in the regulation of autophagy. The
aforementioned two pathways played important roles in the initial
formation stage and late maturation stage of autophagy,
respectively. GABARAPL1 also played a vital role in the formation
stage of autophagosomes and the fusion stage with lysosomes. These
results indicated that KDM3B regulated GABARAPL1 expression through
transcription, affecting signaling pathways and autophagy. More
detailed regulatory mechanisms need to be verified in the
future.
In addition, the current WB results showed that the
H3K9me2 and H4R3me2s levels were decreased in WT cells after
stimulation compared with normal culture conditions. KDM3B KO
inhibited such observations. Therefore, it was hypothesized that
KDM3B affected the histone methylation level, changed the density
and accessibility of chromosomes, and facilitated the binding of
the transcriptional complex. However, this hypothesis needs to be
further verified through targeted experiments. A previous study
found that GABARAPL1 expression in breast cancer was affected by
DNA methylation, histone deacetylation and the transcription factor
(cAMP response element binding protein-1 (CREB-1) (65). Although KDM3B is an important
histone demethylase regulating the expression of GABARAPL1, whether
it accomplishes this through interaction with transcription factors
such as CREB-1 requires further investigation. The present results
revealed that GABARAPL1 expression was reduced after KDM3B KO. The
current RNA-seq results revealed that the upregulated signaling
pathways after KDM3B knockdown included the Wnt signaling pathway
(data not shown). A previous study indicated that GABARAPL1 could
act as a tumor suppressor and negatively regulate the Wnt/β-catenin
signaling pathway through autophagy degradation of Dishevelled
(66), which is consistent with
the present results. Wnt signaling plays a vital role in cell
polarity and development in embryonic stem cells. It has been
reported that KDM3A/B directly interacts with β-catenin in colon
cancer cells and transcriptionally regulates downstream target
genes in the Wnt/β-catenin signaling pathway. It affects H3/H4
histone acetylation levels, and regulates the occurrence and
development of colon cancer (36).
The current study did not produce sufficient experimental evidence
to conclude whether KDM3B can affect the Wnt signaling pathway by
regulating GABARAPL1 or autophagy, or via other mechanisms. Further
studies are necessary to yield more supporting evidence and
potentially propose a new avenue for treating leukemia and other
cancer types. According to the results of the present
bioinformatics analysis, KDM3B was positively correlated with
GABARAPL1 expression in AML. The expression levels of KDM3B and
GABARAPL1 in AML were lower than normal controls, but neither of
them can exist as independent prognostic factors. Therefore, more
related factors need to be discovered in future studies to provide
more references for the treatment of the disease.
In conclusion, the current study revealed that KDM3B
regulated GABARAPL1 as an epigenetic regulator involved in
autophagy in leukemia cells under stimulation. The data suggested
that KDM3B, as a new candidate regulator for autophagy, increased
autophagy in leukemia cells. The present findings elucidated the
mechanism that KDM3B employed to regulate autophagy as an
epigenetic regulator within leukemia cells, and may shed light on
developing epigenetic therapeutics for leukemia and different types
of cancer. One of the limitations of the present study is the lack
of clinical data from AML patients; further study using clinical
samples from AML patients will be undertaken in the upcoming
project. Future studies are warranted to uncover the association
between cell survival and autophagy caused by KDM3B in leukemia.
Understanding these mechanisms could reveal novel insights for
exploring the association between autophagy, cell survival, and
KDM3B epigenetic regulation in leukemia and other types of
cancer.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS designed and performed the research, analyzed the
data and wrote the manuscript. JQZ performed cell cultures and
related western blots. HHW performed molecular experiments. HYW and
YL helped in the analysis of data. ZBH supervised the project and
revised the manuscript. HYW and YL confirm the authenticity of all
the raw data. All the authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank professor Xin Xu
(Weifang Medical University) for critical reading of the manuscript
and Dr Junfeng Shi (Affiliated Hospital of Weifang Medical
University) for helping with RNA-seq analysis.
Funding
The present study was supported by the Shandong Provincial
Natural Science Foundation of China (grant nos. ZR2021QH001 and
ZR2020KC016) and the Weifang Science and Technology Bureau (grant
no. 2020YQFK013).
References
|
1
|
Leidal AM, Levine B and Debnath J:
Autophagy and the cell biology of age-related disease. Nat Cell
Biol. 20:1338–1348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ariosa AR and Klionsky DJ: Autophagy core
machinery: Overcoming spatial barriers in neurons. J Mol Med
(Berl). 94:1217–1227. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang Y and Klionsky DJ: Autophagy and
disease: Unanswered questions. Cell Death Differ. 27:858–871. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gatica D, Chiong M, Lavandero S and
Klionsky DJ: Molecular mechanisms of autophagy in the
cardiovascular system. Circ Res. 116:456–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kimmelman AC: The dynamic nature of
autophagy in cancer. Genes Dev. 25:1999–2010. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Condello M, Pellegrini E, Caraglia M and
Meschini S: Targeting autophagy to overcome human diseases. Int J
Mol Sci. 20:7252019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takamura A, Komatsu M, Hara T, Sakamoto A,
Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K and Mizushima N:
Autophagy-deficient mice develop multiple liver tumors. Genes Dev.
25:795–800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Capparelli C, Guido C, Whitaker-Menezes D,
Bonuccelli G, Balliet R, Pestell TG, Goldberg AF, Pestell RG,
Howell A, Sneddon S, et al: Autophagy and senescence in
cancer-associated fibroblasts metabolically supports tumor growth
and metastasis via glycolysis and ketone production. Cell Cycle.
11:2285–2302. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He S, Zhao Z, Yang Y, O'Connell D, Zhang
X, Oh S, Ma B, Lee JH, Zhang T, Varghese B, et al: Truncating
mutation in the autophagy gene UVRAG confers oncogenic properties
and chemosensitivity in colorectal cancers. Nat Commun. 6:78392015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takahashi Y, Coppola D, Matsushita N,
Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, et
al: Bif-1 interacts with Beclin 1 through UVRAG and regulates
autophagy and tumorigenesis. Nat Cell Biol. 9:1142–1151. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elzinga BM, Nyhan MJ, Crowley LC,
O'Donovan TR, Cahill MR and McKenna SL: Induction of autophagy by
Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates
Bcr-Abl protein. Am J Hematol. 88:455–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goussetis DJ, Gounaris E, Wu EJ, Vakana E,
Sharma B, Bogyo M, Altman JK and Platanias LC: Autophagic
degradation of the BCR-ABL oncoprotein and generation of
antileukemic responses by arsenic trioxide. Blood. 120:3555–3562.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao
Y, Yu Y, Xie M, Yin X, Livesey KM and Tang D: Autophagy regulates
myeloid cell differentiation by p62/SQSTM1-mediated degradation of
PML-RARalpha oncoprotein. Autophagy. 7:401–411. 2011. View Article : Google Scholar :
|
|
14
|
Isakson P, Bjoras M, Boe SO and Simonsen
A: Autophagy contributes to therapy-induced degradation of the
PML/RARA oncoprotein. Blood. 116:2324–2331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mortensen M, Soilleux EJ, Djordjevic G,
Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J,
Knight S, Jacobsen SE, et al: The autophagy protein Atg7 is
essential for hematopoietic stem cell maintenance. J Exp Med.
208:455–467. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao Y, Cai J, Zhang S, Yuan N, Fang Y,
Wang Z, Li X, Cao D, Xu F, Lin W, et al: Autophagy sustains
hematopoiesis through targeting notch. Stem Cells Dev.
24:2660–2673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu F, Chen Z, Liu J and Hou Y: The
Akt-mTOR network at the interface of hematopoietic stem cell
homeostasis. Exp Hematol. 103:15–23. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Man N, Tan Y, Sun XJ, Liu F, Cheng G,
Greenblatt SM, Martinez C, Karl DL, Ando K, Sun M, et al: Caspase-3
controls AML1-ETO-driven leukemogenesis via autophagy modulation in
a ULK1-dependent manner. Blood. 129:2782–2792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Settembre C, Di Malta C, Polito VA, Garcia
Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D,
Colella P, et al: TFEB links autophagy to lysosomal biogenesis.
Science. 332:1429–1433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko
HJ, Kweon MN, Won KJ and Baek SH: AMPK-SKP2-CARM1 signalling
cascade in transcriptional regulation of autophagy. Nature.
534:553–557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Polager S, Ofir M and Ginsberg D: E2F1
regulates autophagy and the transcription of autophagy genes.
Oncogene. 27:4860–4864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang K, Liu JD, Deng G, Ou ZY, Li SF, Xu
XL, Zhang MJ, Peng XQ and Chen FH: LncSIK1 enhanced the sensitivity
of AML cells to retinoic acid by the E2F1/autophagy pathway. Cell
Prolif. 55:e131852022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng Z: The FoxO-Autophagy Axis in Health
and Disease. Trends Endocrinol Metab. 30:658–671. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Milan G, Romanello V, Pescatore F, Armani
A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, et
al: Regulation of autophagy and the ubiquitin-proteasome system by
the FoxO transcriptional network during muscle atrophy. Nat Commun.
6:66702015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JW, Nam H, Kim LE, Jeon Y, Min H, Ha
S, Lee Y, Kim SY, Lee SJ, Kim EK and Yu SW: TLR4 (toll-like
receptor 4) activation suppresses autophagy through inhibition of
FOXO3 and impairs phagocytic capacity of microglia. Autophagy.
15:753–770. 2019. View Article : Google Scholar :
|
|
26
|
Zhang J, Ng S, Wang J, Zhou J, Tan SH,
Yang N, Lin Q, Xia D and Shen HM: Histone deacetylase inhibitors
induce autophagy through FOXO1-dependent pathways. Autophagy.
11:629–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baek SH and Kim KI: Epigenetic Control of
Autophagy: Nuclear Events Gain More Attention. Mol Cell.
65:781–785. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen S, Jing Y, Kang X, Yang L, Wang DL,
Zhang W, Zhang L, Chen P, Chang JF, Yang XM and Sun FL: Histone H2B
monoubiquitination is a critical epigenetic switch for the
regulation of autophagy. Nucleic Acids Res. 45:1144–1158.
2017.PubMed/NCBI
|
|
29
|
Artal-Martinez de Narvajas A, Gomez TS,
Zhang JS, Mann AO, Taoda Y, Gorman JA, Herreros-Villanueva M, Gress
TM, Ellenrieder V, Bujanda L, et al: Epigenetic regulation of
autophagy by the methyltransferase G9a. Mol Cell Biol.
33:3983–3993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
An PNT, Shimaji K, Tanaka R, Yoshida H,
Kimura H, Fukusaki E and Yamaguchi M: Epigenetic regulation of
starvation-induced autophagy in Drosophila by histone
methyltransferase G9a. Sci Rep. 7:73432017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li
T, Hattori N, Wang D, Du Y, Song B, et al: Epigenetic regulation of
autophagy by the methyltransferase EZH2 through an MTOR-dependent
pathway. Autophagy. 11:2309–2322. 2015. View Article : Google Scholar
|
|
32
|
Fullgrabe J, Lynch-Day MA, Heldring N, Li
W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ and
Joseph B: The histone H4 lysine 16 acetyltransferase hMOF regulates
the outcome of autophagy. Nature. 500:468–471. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu Z, Gomes I, Horrigan SK, Kravarusic J,
Mar B, Arbieva Z, Chyna B, Fulton N, Edassery S, Raza A and
Westbrook CA: A novel nuclear protein, 5qNCA (LOC51780) is a
candidate for the myeloid leukemia tumor suppressor gene on
chromosome 5 band q31. Oncogene. 20:6946–6954. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mar BG, Bullinger L, Basu E, Schlis K,
Silverman LB, Dohner K and Armstrong SA: Sequencing
histone-modifying enzymes identifies UTX mutations in acute
lymphoblastic leukemia. Leukemia. 26:1881–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
MacKinnon RN, Kannourakis G, Wall M and
Campbell LJ: A cryptic deletion in 5q31.2 provides further evidence
for a minimally deleted region in myelodysplastic syndromes. Cancer
Genet. 204:187–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork
K, Ramadoss S, Ding X, Li X and Wang CY: KDM3 epigenetically
controls tumorigenic potentials of human colorectal cancer stem
cells through Wnt/β-catenin signalling. Nat Commun. 8:151462017.
View Article : Google Scholar
|
|
37
|
An MJ, Kim DH, Kim CH, Kim M, Rhee S, Seo
SB and Kim JW: Histone demethylase KDM3B regulates the
transcriptional network of cell-cycle genes in hepatocarcinoma
HepG2 cells. Biochem Biophys Res Commun. 508:576–582. 2019.
View Article : Google Scholar
|
|
38
|
Sarac H, Morova T, Pires E, McCullagh J,
Kaplan A, Cingoz A, Bagci-Onder T, Onder T, Kawamura A and Lack NA:
Systematic characterization of chromatin modifying enzymes
identifies KDM3B as a critical regulator in castration resistant
prostate cancer. Oncogene. 39:2187–2201. 2020. View Article : Google Scholar :
|
|
39
|
Wang Y, Zhao Y, Wang H, Zhang C, Wang M,
Yang Y, Xu X and Hu Z: Histone demethylase KDM3B protects against
ferroptosis by upregulating SLC7A11. FEBS Open Bio. 10:637–643.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kuroki S, Maeda R, Yano M, Kitano S,
Miyachi H, Fukuda M, Shinkai Y and Tachibana M: H3K9 Demethylases
JMJD1A and JMJD1B Control Prospermatogonia to Spermatogonia
Transition in Mouse Germline. Stem Cell Reports. 15:424–438. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saavedra F, Gurard-Levin ZA,
Rojas-Villalobos C, Vassias I, Quatrini R, Almouzni G and Loyola A:
JMJD1B, a novel player in histone H3 and H4 processing to ensure
genome stability. Epigenetics Chromatin. 13:62020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu X, Nagel S, Quentmeier H, Wang Z,
Pommerenke C, Dirks WG, Macleod RAF, Drexler HG and Hu Z: KDM3B
shows tumor-suppressive activity and transcriptionally regulates
HOXA1 through retinoic acid response elements in acute myeloid
leukemia. Leuk Lymphoma. 59:204–213. 2018. View Article : Google Scholar
|
|
43
|
Xu X, Wang L, Hu L, Dirks WG, Zhao Y, Wei
Z, Chen D, Li Z, Wang Z, Han Y, et al: Small molecular modulators
of JMJD1C preferentially inhibit growth of leukemia cells. Int J
Cancer. 146:400–412. 2020. View Article : Google Scholar
|
|
44
|
Kim JY, Kim KB, Eom GH, Choe N, Kee HJ,
Son HJ, Oh ST, Kim DW, Pak JH, Baek HJ, et al: KDM3B is the H3K9
demethylase involved in transcriptional activation of lmo2 in
leukemia. Mol Cell Biol. 32:2917–2933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li S, Ali S, Duan X, Liu S, Du J, Liu C,
Dai H, Zhou M, Zhou L, Yang L, et al: JMJD1B demethylates H4R3me2s
and H3K9me2 to facilitate gene expression for development of
hematopoietic stem and progenitor cells. Cell Rep. 23:389–403.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Goldstein LD, Cao Y, Pau G, Lawrence M, Wu
TD, Seshagiri S and Gentleman R: Prediction and Quantification of
Splice Events from RNA-Seq Data. PLoS One. 11:e01561322016.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biology. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
49
|
Hosokawa N, Sasaki T, Iemura S, Natsume T,
Hara T and Mizushima N: Atg101, a novel mammalian autophagy protein
interacting with Atg13. Autophagy. 5:973–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Itakura E, Kishi C, Inoue K and Mizushima
N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase
complexes with mammalian Atg14 and UVRAG. Mol Biol Cell.
19:5360–5372. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ryu HY, Kim LE, Jeong H, Yeo BK, Lee JW,
Nam H, Ha S, An HK, Park H, Jung S, et al: GSK3B induces autophagy
by phosphorylating ULK1. Exp Mol Med. 53:369–383. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matsunaga K, Morita E, Saitoh T, Akira S,
Ktistakis NT, Izumi T, Noda T and Yoshimori T: Autophagy requires
endoplasmic reticulum targeting of the PI3-kinase complex via
Atg14L. J Cell Biol. 190:511–521. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Birgisdottir AB, Mouilleron S, Bhujabal Z,
Wirth M, Sjottem E, Evjen G, Zhang W, Lee R, O'Reilly N, Tooze SA,
et al: Members of the autophagy class III phosphatidylinositol
3-kinase complex I interact with GABARAP and GABARAPL1 via LIR
motifs. Autophagy. 15:1333–1355. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Weidberg H, Shvets E, Shpilka T, Shimron
F, Shinder V and Elazar Z: LC3 and GATE-16/GABARAP subfamilies are
both essential yet act differently in autophagosome biogenesis.
EMBO J. 29:1792–1802. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pankiv S, Alemu EA, Brech A, Bruun JA,
Lamark T, Overvatn A, Bjorkoy G and Johansen T: FYCO1 is a Rab7
effector that binds to LC3 and PI3P to mediate microtubule plus
end-directed vesicle transport. J Cell Biol. 188:253–269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fu MM, Nirschl JJ and Holzbaur ELF: LC3
binding to the scaffolding protein JIP1 regulates processive
dynein-driven transport of autophagosomes. Dev Cell. 29:577–590.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nguyen TN, Padman BS, Usher J, Oorschot V,
Ramm G and Lazarou M: Atg8 family LC3/GABARAP proteins are crucial
for autophagosome-lysosome fusion but not autophagosome formation
during PINK1/Parkin mitophagy and starvation. J Cell Biol.
215:857–874. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ebner P, Poetsch I, Deszcz L, Hoffmann T,
Zuber J and Ikeda F: The IAP family member BRUCE regulates
autophagosome-lysosome fusion. Nat Commun. 9:5992018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang X, Wang L, Lak B, Li J, Jokitalo E
and Wang Y: GRASP55 Senses Glucose Deprivation through
O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev Cell.
45:245–61.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mansuy-Schlick V, Tolle F, Delage-Mourroux
R, Fraichard A, Risold PY and Jouvenot M: Specific distribution of
gabarap, gec1/gabarap Like 1, gate16/gabarap Like 2, lc3 messenger
RNAs in rat brain areas by quantitative real-time PCR. Brain Res.
1073-1074:83–87. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tolle F, Risold PY, Mansuy-Schlick V,
Rossi E, Boyer-Guittaut M, Fraichard A and Jouvenot M: Specific
regional distribution of gec1 mRNAs in adult rat central nervous
system. Brain Res. 1210:103–115. PubMed/NCBI
|
|
62
|
Bollaert E, Claus M, Vandewalle V, Lenglez
S, Essaghir A, Demoulin JB and Havelange V: MiR-15a-5p confers
chemoresistance in acute myeloid leukemia by inhibiting autophagy
induced by daunorubicin. Int J Mol Sci. 22:51532021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang X, Fan H, Xu C, Jiang G, Wang H and
Zhang J: KDM3B suppresses APL progression by restricting chromatin
accessibility and facilitating the ATRA-mediated degradation of
PML/RARalpha. Cancer Cell Int. 19:2562019. View Article : Google Scholar
|
|
64
|
Jung H and Seo SB: Histone lysine
demethylase 3B (KDM3B) regulates the propagation of autophagy via
transcriptional activation of autophagy-related genes. PLoS One.
15:e02364032020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hervouet E, Claude-Taupin A, Gauthier T,
Perez V, Fraichard A, Adami P, Despouy G, Monnien F, Algros MP,
Jouvenot M, et al: The autophagy GABARAPL1 gene is epigenetically
regulated in breast cancer models. BMC Cancer. 15:7292015.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang Y, Wang F, Han L, Wu Y, Li S, Yang
X, Wang Y, Ren F, Zhai Y, Wang D, et al: GABARAPL1 negatively
regulates Wnt/β-catenin signaling by mediating Dvl2 degradation
through the autophagy pathway. Cell Physiol Biochem. 27:503–512.
2011. View Article : Google Scholar
|