Introduction
Hepatocellular carcinoma (HCC) is the most common
type of malignant liver cancer with high rates of recurrence and
mortality (1). Several studies
reported that various genetic factors, such as gene mutations and
epigenetic modifications, as well as environmental factors, are
involved in HCC development (2).
Despite improvements in the diagnosis and treatment of HCC in
recent decades, HCC prognosis is poor and there are limited
effective treatments (3). Tumor
metastasis is a leading cause of poor prognosis for patients with
HCC (4); therefore, determining
the underlying biological mechanisms promoting HCC metastasis is
crucial for the development of diagnostic and therapeutic
interventions for HCC.
β-Klotho (KLB) is a single-pass transmembrane
protein and is predominantly expressed in the liver, pancreas and
white adipose tissues (5). The
extracellular region of KLB consists of two internal repeats that
share significant structural homology with family 1 glycosidase
members, but lack intrinsic enzymatic activity (5,6).
Human KLB is essential for the high-affinity binding of fibroblast
growth factor (FGF)-19 and FGF21 to their cognate FGF receptors
(FGFRs) (7). Upon food intake, the
gut secretes FGF19, which binds to the FGFR4/KLB complex in
hepatocytes to accelerate the metabolic response to feeding
(8). By contrast, the liver
secretes FGF21, the starvation hormone, which binds to the
FGFR1c/KLB complex in adipocytes to induce metabolic responses
under fasting conditions (9). It
has been indicated that aberrant expression of FGF19-FGFR4 is a
metastatic driver of HCC (10) and
that KLB amplified FGF19-FGFR4 signaling in HCC (11). However, the activation of FGF21-KLB
signaling and its role in HCC metastasis have remained to be fully
elucidated.
Epigenetic regulations, such as DNA methylation,
histone modification and microRNA regulation, are heritable and
stable changes in gene expression that do not alter the DNA
sequence (12,13). Epigenetic mechanisms have been
reported to have crucial roles in tumor metastasis, including HCC
metastasis (14). Histone
acetylation is a widely studied post-translational epigenetic
regulation that facilitates the dissociation of DNA and histone
octamers and allows transcription factor binding to specific sites
on the DNA to initiate gene transcription (15). Histone acetylation and
deacetylation are conducted by histone acetyltransferases and
histone deacetylases (HDACs), respectively (16). Previous studies demonstrated that
HDACs have a critical role in tumor metastasis; therefore,
epigenetic drugs may serve as effective therapeutic interventions
against liver cancers (17,18).
However, since epigenetic modifications occur in a cell or
gene-specific manner, identification of the key genes of
acetylation modifications is essential for the development of HCC
treatments.
The present study found that KLB expression was
increased in HCC tissues and was associated with HCC metastasis. In
addition, KLB knockdown with simultaneous FGF21 overexpression
promoted epithelial-mesenchymal transition (EMT) and HCC cell
motility. Furthermore, that HDAC3 was indicated to be a potential
deacetylase for KLB and treatment with HDAC3 inhibitor led to KLB
inactivation, resulting in the blockade of FGF21-KLB signaling,
further increasing the expression of EMT induction-related genes in
HCC cells. Taken together, the present results indicate that
aberrant acetylated modification of FGF21-KLB signaling contributes
to EMT and HCC metastasis. Therefore, FGF21-KLB signaling may serve
as a potential therapeutic target for HCC treatment.
Materials and methods
Cell culture and treatment
The human liver cancer cell lines LM3, HepG2,
PLC/PRF/5, Li-7 and Huh7 and the murine HCC cell line Hepa1-6 were
provided by the Hepatobiliary Institute of Nanjing University,
while the normal hepatocyte cell line MIHA was provided by the
Shanghai Institute of Cell Biology. Unless otherwise specified, all
the experiments were conducted using the Huh7 cell line. Short
tandem repeat profiling was conducted to authenticate all the cell
lines used in the present study. The cells were cultured in DMEM or
RPMI-1640 supplemented with 10% FBS (all from Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin,
and maintained at 37°C and 5% CO2 in a humidified
incubator. When the cell density reached 60–70%, the cells were
treated with HDAC3 inhibitors, RGFP966 (cat. no. S7229; Selleck
Chemicals) or Trichostatin A (TSA; cat. no. S1045; Selleck
Chemicals).
Clinical specimens
HCC tissues and the corresponding adjacent normal
tissues were provided by the Affiliated Drum Tower Hospital of
Nanjing University Medical School (Nanjing, China) with written
informed consent from the patients concerned. The present study was
approved by the local ethics committee. Western blot (WB) and
immunohistochemistry (IHC) assays were used to evaluate the protein
levels in the HCC tissues, as described previously (19). The following primary antibodies
were purchased from Abcam: anti-KLB (WB, 1;1,000 dilution; IHC,
1:200 dilution; cat. no. ab106794) and anti-β-catenin (WB, 1:1,000
dilution; IHC, 1:400 dilution; cat. no. ab223075).
Bioinformatics analysis
Transcript levels of KLB in different cancer tissues
and their corresponding normal tissues were analyzed by the
ONCOMINE (https://www.oncomine.org/) and Tumor
IMmune Estimation Resource (TIMER) (https://cistrome.shinyapps.io/timer/) databases. The
transcriptomic data of KLB in normal and HCC samples were obtained
from the Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/tcga) and its expression in HCC
samples was further determined based on tumor grade, individual
cancer stage, TP53 mutation status and nodal metastasis status
using the UALCAN database. The Kaplan-Meier plotter (https://kmplot.com/) was used to analyze the
prognostic value of the KLB gene in patients with HCC. Gene
set evaluation analysis (GSVA) (https://www.aclbi.com/static/index.html#/) of TCGA
data was used to determine the potential biological function of
KLB.
Small interfering RNA (siRNA)
assay
siRNAs were used to alter the expression of KLB and
HDAC3 in the Huh7 cell line. The siRNAs with the highest silencing
efficiency were screened and used for gene function research. After
screening, the Huh7 cells (60–70% confluency in a 6-well plate)
were treated with KLB- or HDAC3-siRNAs (si-KLB1,
5′-CGCUAUAGGAAUACAAUGUTT-3′; si-KLB2, 5′-GCUUCAAGCAAUAAGGUUATT-3′;
and si-HDAC3, 5′-CACAAAUACGGAAAUUACUTT-3′) and the corresponding
control-siRNAs (TSINGKE Biological Technology), which were
encapsulated by the INTERFERin reagent (Polyplus-transfection SA).
The sequence of the control siRNA was not provided by the
supplier.
Lentivirus transductions
To inhibit KLB expression, the Huh7 cell line was
infected with lentiviral vectors carrying short hairpin RNA against
KLB (sh-KLB; target sequence, 5′-GCTTCAAGCAATAAGGTTA-3′). To
overexpress FGF21, the huh7 cell line was transfected with
lentiviral vectors carrying the FGF21 gene (GeneChem; sequences
provided in supplementary data). Empty lentiviral vectors were used
as internal controls.
WB and co-immunoprecipitation (co-IP)
assays
Proteins were extracted from the HCC tissues and
cell samples using RIPA buffer (KeyGEN BioTECH) with
phenylmethylsulfonyl fluoride, according to the manufacturer's
instructions, and the WB and co-IP assays were conducted as
described previously (20).
Anti-KLB (WB, 1:1,000 dilution; cat. no. ab106794), anti-E-cadherin
(WB, 1:2,000; cat. no. ab40772), anti-claudin-1 (WB, 1:2,000; cat.
no. ab211737), anti-MMP9 (WB, 1:2,000; cat. no. ab76003),
anti-β-catenin (WB, 1:1,000; cat. no. ab223075), anti-laminB1 (WB,
1:5,000; cat. no. ab108536), anti-β-actin (WB, 1:1,000; cat. no.
ab8226) and anti-GAPDH (WB, 1:10,000; cat. no. ab181602) were
purchased from Abcam; anti-vimentin (WB, 1:20,000; cat. no.
60330-1-Ig) and anti-HDAC3 (WB, 1:1,000; cat. no. 10255-1-AP) were
purchased from ProteinTech; and anti-acetylated lysine (WB,
1:1,000; cat. no. 9441) was purchased from Cell Signaling
Technology, Inc. The bicinchoninic acid assay was conducted to
determine the protein concentration. Each experiment was replicated
thrice. The results of the WB assay were processed by Image J
software (National Institutes of Health, version 1.8.0).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the HCC cells using
TRIzol™ (Thermo Fisher Scientific, Inc.) and subjected to RT-qPCR
analysis using the ABI PRISM 7500 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.), as described
previously (20). β-Actin was used
as the internal reference gene and the 2−ΔΔCq method was
used to evaluate the relative mRNA expression (21). The PCR primers were designed by
Realgene (Nanjing, China) and are listed in Table I.
 | Table I.Primers for quantitative real-time
PCR. |
Table I.
Primers for quantitative real-time
PCR.
| Gene name | Forward primer | Reverse primer |
|---|
| β-actin |
5′-ATTGCCGACAGGATGCAGAA-3′ |
5′-GCTGATCCACATCTGCTGGAA-3′ |
| KLB |
5′-TCTGTCATCCTGTCAGCACTT-3′ |
5′-CCAGTCCCAATACCCCAGAAAAA-3′ |
| AXIN2 |
5′-CAACACCAGGCGGAACGAA-3′ |
5′-GCCCAATAAGGAGTGTAAGGACT-3′ |
| MMP7 |
5′-GAGTGAGCTACAGTGGGAACA-3′ |
5′-CTATGACGCGGGAGTTTAACAT-3′ |
| C-MYC |
5′-GGCTCCTGGCAAAAGGTCA-3′ |
5′-CTGCGTAGTTGTGCTGATGT-3′ |
| TCF7 |
5′-CTGGCTTCTACTCCCTGACCT-3′ |
5′-ACCAGAACCTAGCATCAAGGA-3′ |
|
β-Catenin |
5′-AAAGCGGCTGTTAGTCACTGG-3′ |
5′-CGAGTCATTGCATACTGTCCAT-3′ |
|
E-cadherin |
5′-CGAGAGCTACACGTTCACGG-3′ |
5′-GGGTGTCGAGGGAAAAATAGG-3′ |
|
N-cadherin |
5′-TCAGGCGTCTGTAGAGGCTT-3′ |
5′-ATGCACATCCTTCGATAAGACTG-3′ |
| SLUG |
5′-CGAACTGGACACACATACAGTG-3′ |
5′-CTGAGGATCTCTGGTTGTGGT-3′ |
| ZEB1 |
5′-GATGATGAATGCGAGTCAGATGC-3′ |
5′-ACAGCAGTGTCTTGTTGTTGT-3′ |
| FGF21 |
5′-CTGTGGGTTTCTGTGCTGG-3′ |
5′-CCGGCTTCAAGGCTTTCAG-3′ |
Cell viability, EdU, apoptosis and
colony-formation assays
Cell viability, EdU and apoptosis assays were
conducted using a cell counting kit-8 (cat. no. C0037), the
BeyoClick™ EdU kit (cat. no. C0075S) and an Annexin V-PE kit
(C1065S; Beyotime Institute of Biotechnology), respectively,
following the manufacturer's instructions. For the colony-formation
assay, si-KLB-treated cells and negative control cells (n=500 each)
were incubated in a 12-well plate and cultured for 2 weeks.
Thereafter, the cells were fixed with methanol for 30 min and
stained with 0.1% crystal violet for 30 min at room temperature.
Finally, the colonies containing ≥50 cells were counted.
Wound-healing, cell-migration and
cell-invasion assays
Wound-healing, cell migration and cell invasion
assays were conducted as described previously (22). For the cell migration and invasion
assays, migrated or invaded cells were counted using a microscope
in six random fields. For the wound-healing assay, cell migration
was calculated as the percentage of wound closure.
Immunofluorescence (IF) assay
The IF assay was conducted as described previously
(20). In brief, treated cells
were seeded in 24-well plates and incubated for 24 h. Thereafter,
the cells were fixed using 4% paraformaldehyde and washed thrice
with PBS. The cells were then treated with anti-vimentin antibodies
(IF, 1:500 dilution; cat. no. 60330-1-Ig, ProteinTech), followed by
incubation with goat anti-mouse IgG H&L (Alexa
Fluor® 488; IF, 1:500 dilution; cat. no. ab150113;
Abcam). The cells were then observed under a fluorescent microscope
(Leica Microsystems GmbH) and the images were obtained.
Animal model
5×106 Huh7 cells were injected into male
BALB/c nude mice (GemPharmatech; age, 3–4 weeks; weight, 16–20 g;
n=6 per group) through the tail vein to generate the mouse lung
metastasis model. The mice were housed with 5 mice per cage on a
12-h light/dark cycle, controlled humidity (50–60%) and temperature
(24–26°C) and had free access to food and water (23). After 6 weeks, the mice were
anesthetized and euthanized by intraperitoneal injection of
pentobarbital sodium 150–200 mg/kg. The lungs were excised, imaged
and embedded in paraffin. Tumor metastases were analyzed by
hematoxylin and eosin staining according to standard procedures.
The Ethics Committee of the Affiliated Drum Tower Hospital of
Nanjing University Medical School (Nanjing, China) approved the
animal experiments.
Statistical analysis
GraphPad Prism v8.02 (Dotmatics) was used to conduct
data analyses and to calculate the P-value. Student t-test and
one-way analysis of variance (ANOVA) with Dunnett's post-hoc test
were used for comparisons between two or multiple groups,
respectively. P<0.05 was considered to indicate a statistically
significant difference.
Results
KLB expression is elevated in HCC and
associated with HCC metastasis
KLB, a vital component of the endocrine FGFR
complex, is involved in tumor progression in several cancers. To
obtain KLB expression profiles in human cancers, pan-cancer KLB
expression data were retrieved from the ONCOMINE and TIMER
databases. As presented in Fig. 1A and
B, KLB expression was lower in a majority of cancer tissues,
including breast cancer, colorectal cancer, lung cancer and
pancreatic cancer, compared with their corresponding normal
tissues. However, KLB expression was markedly increased in HCC
tissues compared to the corresponding normal tissues (Fig. 1B). Differential-expression analysis
of the TCGA-liver hepatocellular carcinoma data using paired and
unpaired t-tests revealed that KLB expression was significantly
increased in the HCC tissues compared with the normal tissues
(Fig. 1C and D). Immunoblotting
and IHC staining were conducted to assess the protein levels of KLB
in 10 human HCC tissues and their corresponding adjacent
non-cancerous tissues, to further confirm KLB expression in HCC.
The results revealed that KLB protein levels were markedly higher
in the HCC tissues (Fig. 1E),
which is consistent with previously published results (11). In addition, KLB expression was also
evaluated in various HCC cell lines (LM3, HepG2, PLC/PRF/5, Li-7
and Huh 7) and a normal hepatocyte cell line (MIHA), and the
results suggested that the mRNA and protein levels of KLB were
significantly upregulated in Huh7, HepG2 and Li-7 compared with
MIHA (Fig. 1F and G). Furthermore,
KLB expression was observed to gradually increase with tumor grade
(Fig. 1H). Altogether, these
findings confirmed that KLB expression is high in HCC tissues and
cell lines.
 | Figure 1.KLB expression is elevated in HCC and
associated with HCC metastasis. (A and B) Pan-cancer KLB expression
analysis using (A) ONCOMINE and (B) Tumor IMmune Estimation
Resource databases. (C and D) The transcriptomic data of KLB in HCC
tissues and adjacent normal tissues were obtained from the TCGA
database. Analysis of the KLB expression levels in HCC tissues and
adjacent normal tissues using (C) unpaired and (D) paired t-tests.
(E) Representative images of KLB expression obtained by western
blot analysis of HCC tissues. (F) Western blot and (G) quantitative
real-time PCR analyses indicated increased KLB expression in most
HCC cell lines. (H) Representative images of immunohistochemistry
staining of KLB in different stages of human HCC tissues compared
with the adjacent normal tissues (scale bars: Upper panel, 100 µm;
lower panel, 50 µm). (I) The non-alcohol-consuming HCC patients
were subjected to overall survival analysis based on KLB expression
level. (J) KLB expression in HCC according to tumor grade, disease
stage, TP53 mutation status and nodal metastasis status.
**P<0.01, ***P<0.001. ns, not significant; KLB, β-klotho;
HCC, hepatocellular carcinoma; N, normal tissue; C, cancer tissue;
HR, hazard ratio; TCGA, The Cancer Genome Atlas; ACC,
adrenocortical carcinoma; BLCA, Bladder Urothelial Carcinoma; BRCA,
Breast invasive carcinoma; CESC, Cervical squamous cell carcinoma
and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD,
colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell
lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme;
HNSC, head and neck squamous cell carcinoma; KICH, kidney
chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney
renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG,
brain lower grade glioma; LIHC, liver hepatocellular carcinoma;
LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma;
MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD,
pancreatic adenocarcinoma; PCPG, pheochromocytoma and
paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum
adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD,
stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA,
thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial
carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma. |
To determine the clinical significance of increased
KLB expression in HCC, a survival analysis was performed using
Kaplan-Meier plotter. It was indicated that, although KLB is
elevated in HCC, its high expression was associated with a
favorable prognosis in non-alcohol-consuming patients with HCC
(Fig. 1I), thus implying that KLB
may serve as a protective modulator in HCC. These results suggest
that high expression of KLB may be considered a prognostic
biomarker for a specific population of patients with HCC. Further
survival analysis of KLB gene expression in non-alcohol-consuming
HCC patients with and without vascular invasion revealed that KLB
was a specific prognostic marker for patients with HCC without
vascular invasion (Fig. S1A and
B). To further determine the expression of KLB in HCC
development, KLB expression was analyzed according to tumor grade,
disease stage, TP53 mutation status and lymph node metastasis
status. As presented in Fig. 1J,
KLB expression increased continuously with HCC development,
suggesting that KLB has an important role in HCC development and
metastasis.
Knockdown of KLB promotes HCC cell
migration and invasion
To elucidate the role of KLB in HCC progression, a
KLB-knockdown Huh7 cell line was generated (Fig. S1C). Consistent with the results of
a previous study (11), the EdU,
apoptosis, CCK-8 and colony-formation assays revealed that KLB
knockdown reduced cell proliferation and induced cell apoptosis in
Huh7 cells (Fig. 2A-D). These
findings demonstrate the involvement of KLB in the pathogenesis of
HCC and highlight the need to investigate its role in enhancing the
FGF19-FGFR4 signaling pathway. However, wound-healing migration,
Transwell migration and Matrigel invasion assays in Huh7 and
Hepa1-6 cell lines revealed that KLB knockdown enhanced HCC cell
motility (Fig. 2E-H). This may
partly explain why high expression of KLB is associated with a
favorable prognosis in patients with HCC without vascular
invasion.
 | Figure 2.Knockdown of KLB promotes HCC cell
migration and invasion. (A) EdU (scale bar, 50 µm), (B) flow
cytometry, (C) cell counting kit-8 and (D) colony-formation assays
(scale bar, 15 mm diameter) of Huh7 cells after KLB knockdown. (E
and F) Transwell migration and invasion assays of Huh7 cells after
48 h of si-KLB treatment. (E) Representative images (scale bar, 100
µm) of Transwell migration and invasion assays of Huh7 cells after
KLB knockdown. (F) Representative images (scale bar, 100 µm) of
Transwell migration and invasion assays of Hepa1-6 cells after KLB
knockdown. (G) Wound closure (wound-healing assay) was monitored at
0, 24 and 48 h after wounding (scale bar, 100 µm) in KLB knockdown
Huh7 cells. (H) Wound closure (wound healing assay) was monitored
at 0, 24 and 48 h after wounding (scale bar, 100 µm) in KLB
knockdown Hepa1-6 cells. *P<0.05, **P<0.01, ***P<0.001.
ns, not significant; KLB, β-klotho; HCC, hepatocellular carcinoma;
NC, negative control; si-KLB, small interfering RNA targeting KLB;
OD, optical density. |
KLB is identified as a novel upstream
regulator of β-catenin signaling
GSVA was performed to further explore the underlying
biological function of KLB in HCC and the results demonstrated that
KLB is closely associated with β-catenin signaling (Fig. 3A). In addition, it was found that
KLB knockdown significantly inhibited the mRNA and protein
expression of several critical genes in the β-catenin signaling
pathway, including β-catenin, C-MYC, MMP7, transcription factor
(TCF)-7 and phosphorylated glycogen synthase kinase
3β-S9 in Huh7 cells; however, the expression of nuclear
β-catenin was not obvious (Fig. 3B and
C). These results further verified the accuracy of the
enrichment analysis, thus suggesting that KLB is possibly involved
in the β-catenin-mediated signaling activation in HCC. IHC staining
of the HCC tissues and their corresponding normal tissues revealed
a high correlation between KLB and β-catenin gene (CTNNB1)
expression, consistent with the results of the gene expression
profiling interactive analysis (Fig.
3D and E).
 | Figure 3.KLB is identified as a novel upstream
regulator of β-catenin signaling. (A) GSVA revealed that KLB
expression was closely associated with β-catenin signaling. (B)
Quantitative real-time PCR and (C) western blot assays indicated
that KLB knockdown inhibited the expression of β-catenin
signaling-related genes. (D) Immunohistochemistry analysis (scale
bars, 50 µm) and (E) gene expression profiling interactive analysis
of hepatocellular carcinoma tissues and their corresponding normal
tissues showed a strong correlation between KLB and
CTNNB1 expression. *P<0.05, **P<0.01. ns, not
significant; KLB, β-klotho; GSVA, Gene Set Variation Analysis; NC,
negative control; si-KLB, small interfering RNA targeting KLB;
CTNNB1, β-catenin; GSK, glycogen synthase kinase; TCF-7,
transcription factor-7. |
FGF21-KLB signaling inhibits HCC
metastasis via the β-catenin signaling pathway
Aberrant FGF19-FGFR4-KLB signaling is a metastatic
driver for HCC (10), while the
function of FGF21-KLB signaling in HCC metastasis remains to be
clarified. Therefore, to explore the role of FGF21-KLB signaling in
regulating HCC metastasis, Huh7 cells were transduced with
lentivirus carrying the FGF21 gene to induce stable
expression of FGF21 (Fig. S1D).
Several studies demonstrated that β-catenin expression increased,
while E-cadherin expression decreased in diabetic FGF21-knockout
mice (24). In the present study,
it was found that enhanced FGF21 expression inhibited the
transcription of AXIN2, CTNNB1, C-MYC and TCF7.
However, KLB knockdown in FGF21-overexpressing cells (Fig. S1E and F) caused increased
expression of CTNNB1 and TCF7 (Fig. 4A and B), suggesting that FGF21 is
an upstream mediator of the β-catenin signaling pathway and that
its regulation is dependent on KLB expression. Thereafter, it was
examined whether FGF21-KLB induced HCC metastasis via the EMT and
it was found that FGF21 overexpression enhanced the protein level
of E-cadherin (essential for the establishment of stable adherent
junctions), which is downregulated during EMT (Fig. 4D). Subsequently, the mRNA levels of
E-cadherin and several EMT inducers, including
N-cadherin, Slug and zinc finger E-box binding homeobox 1
(ZEB1), were measured in both FGF21-overexpressing and
sh-KLB-treated FGF21-overexpressing cells (Fig. 4C). Compared with
FGF21-overexpressing cells, sh-KLB-treated FGF21-overexpressing
cells had reduced mRNA levels of E-cadherin and increased
mRNA levels of Slug and ZEB1 (key transcriptional
repressors of E-cadherin). Furthermore, compared with the
FGF21-overexpressing cells, claudin-1 [a repressor of
E-cadherin (25)] was increased,
while zonula occludens (ZO)−1 (a member of the ZO
proteins that constitute tight junctions) was decreased in the
sh-KLB-treated FGF21-overexpressing cells (Fig. 4E). Meanwhile, IF staining revealed
an upregulation of vimentin expression in the sh-KLB-treated
FGF21-overexpressing cells compared with the FGF21-overexpressing
cells (Fig. 4F). Furthermore, the
wound-healing migration, Transwell migration and Matrigel invasion
assays revealed that the sh-KLB-treated FGF21-overexpressing cells
had an enhanced migration and invasion ability compared with the
FGF21-overexpressing cells (Fig.
4G,H). To confirm the effects of FGF21-KLB on the
tumorigenicity of HCC in vivo, a lung metastasis model was
established. Consistent with previous results, sh-KLB-treated
FGF21-overexpressing cells had an enhanced in vivo
metastatic ability compared with the FGF21-overexpressing cells
(Fig. 4I and J). Taken together,
these results suggest that FGF21 suppresses HCC cell metastasis
through the β-catenin signaling pathway, while KLB knockdown
promotes HCC cell motility under FGF21 overexpression.
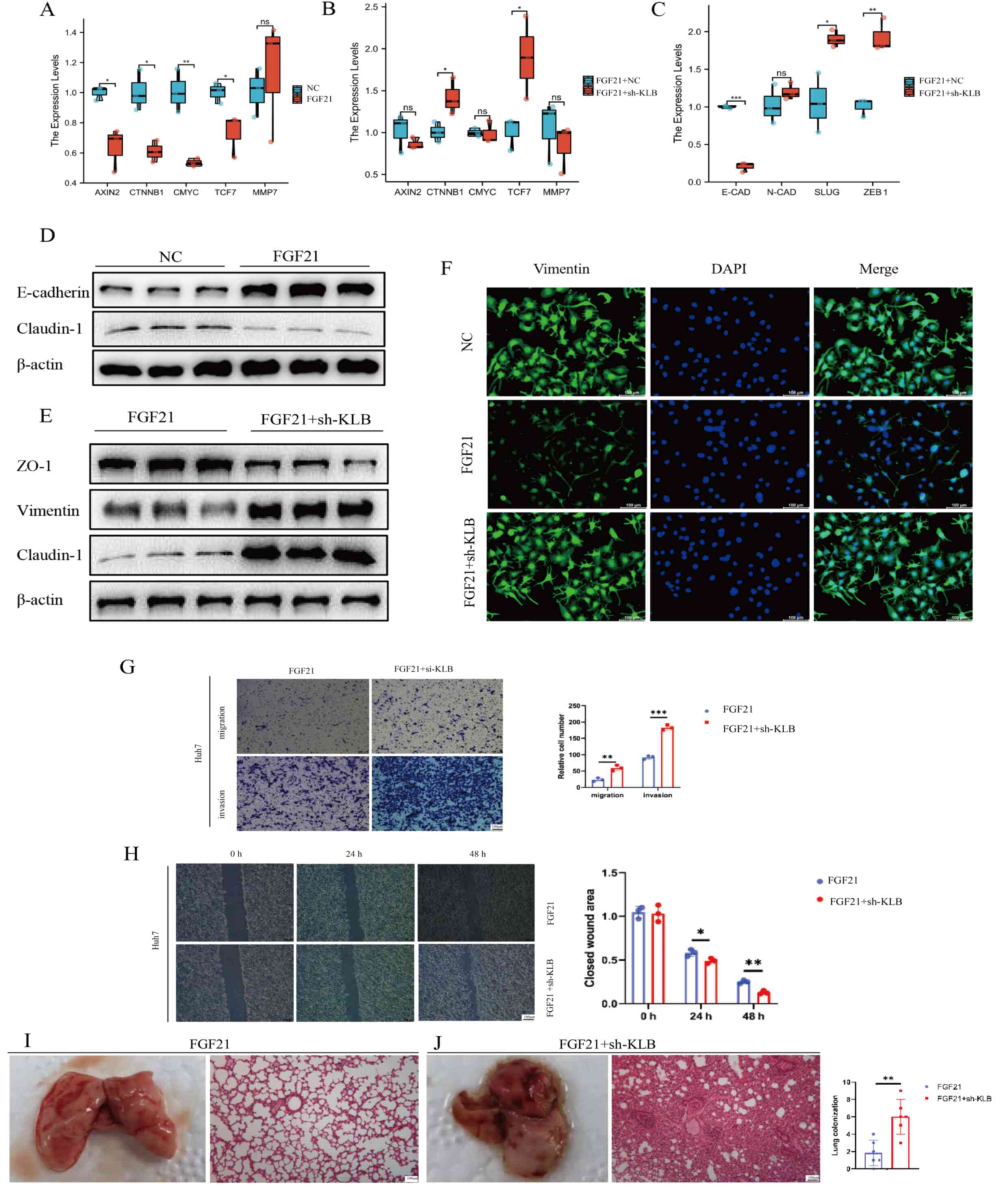 | Figure 4.FGF21-KLB signaling inhibited
hepatocellular carcinoma (HCC) metastasis in vitro and in
vivo via the β-catenin signaling pathway. (A and B) The mRNA
levels of AXIN2, CTNNB1, c-MYC, TCF7 and MMP7 in the
indicated cells. (C) The mRNA levels of E-cadherin, N-cadherin,
Slug and ZEB1 in the indicated cells. (D) The protein
levels of E-cadherin and claudin-1 in the indicated cells. (E) The
protein levels of ZO-1, vimentin and claudin-1 in the indicated
cells. (F) Immunofluorescence assay to detect vimentin expression
in the indicated cells (scale bar, 100 µm). (G) Representative
images (scale bar, 100 µm) of Transwell migration and invasion
assays of the indicated cells. (H) Wound-healing assay of the
indicated cells. Wound closure was monitored at 0, 24 and 48 h
after wounding (scale bar, 100 µm). (I and J) Representative HE
images from the lung metastasis models established by injection of
(I) FGF21-overexpressing and (J) sh-KLB-treated
FGF21-overexpressing cells (scale bar, 100 µm). *P<0.05,
**P<0.01, ***P<0.001. ns, not significant. FGF21, fibroblast
growth factor 21; KLB, β-klotho; CTNNB1, β-catenin; NC, negative
control; sh-KLB, short hairpin RNA targeting KLB; TCF-7,
transcription factor-7; CAD, cadherin; ZO-1, zonula occludens 1;
ZEB1, zinc finger E-box binding homeobox 1. |
HDAC3 inhibitor-mediated acetylated
modification of KLB facilitates the activation of HCC
metastasis-promoting genes
The methylation level of the KLB promoter
decreased gradually with an increase in tumor grade and disease
stage and was associated with TP53 mutation status and lymph node
metastasis status (Fig. S1H). It
was hypothesized that the epigenetic modification of KLB may
contribute to its high expression in HCC. A previous study
demonstrated that HDAC3 catalyzes the acetylation of Klotho, a
paralog of KLB (26); however, its
role in KLB modification has not yet been determined. Therefore, to
determine the regulatory effects of epigenetic changes on KLB
expression, HCC cells were treated with TSA (a broad-spectrum HDAC
inhibitor) and RGFP966 (an HDAC3-specific inhibitor). The results
demonstrated that KLB expression decreased in a dose-dependent
manner in TSA and RGFP966-treated HCC cells (Fig. 5A and B). Furthermore, it was found
that the KLB promoter was indeed acetylated and that its
acetylation was significantly increased after treatment with TSA
and RGFP966, suggesting that KLB is acetylated in HCC (Fig. 5C and D).
 | Figure 5.HDAC3 inhibitor-mediated acetylated
modification of KLB facilitates the activation of HCC
metastasis-promoting genes. (A and B) KLB expression decreased
after treatment with (A) TSA (broad-spectrum HDAC inhibitor) and
(B) RGFP966 (HDAC3-specific inhibitor) in a dose-dependent manner.
(C and D) KLB acetylation levels increased after treatment with (C)
TSA and (D) RGFP966 in a dose-dependent manner. The acetylation
status was determined by western blot assay using anti-AcK
antibodies. (E) Immunoprecipitation assay of the indicated cell
extracts using anti-KLB antibodies and IgG (control). Acetylated
KLB was detected by western blot assay using AcK. (F) The protein
levels of KLB, MMP9 and vimentin were detected in the indicated
cells. (G) The protein levels of ZO-1 and vimentin were detected in
the indicated cells. (H) Wound-healing assay demonstrated the
effect of RGFP966 on HCC cell migration with simultaneous FGF21
overexpression (scale bar, 100 µm). **P<0.01. FGF21, fibroblast
growth factor 21; HDAC3, histone deacetylase 3; AcK, acetylated
lysine; HCC, hepatocellular carcinoma; KLB, β-klotho; TSA,
trichostatin A; ZO-1, zonula occludens 1; si-HDAC3, small
interfering RNA targeting HDAC3; Ctrl, control; NC, negative
control; IP, immunoprecipitation; IB, immunoblot. |
Thereafter, a co-IP assay was performed to determine
whether HDAC3 is able to regulate the acetylation status of KLB.
The si-HDAC3-transfected Huh7 cells (Fig. S1G) were subjected to IP analysis
using anti-KLB, followed by WB analysis with anti-acetylated-lysine
antibodies, to detect the levels of acetylated KLB. The results
revealed that KLB acetylation was significantly increased in the
si-HDAC3-treated group compared with the untreated group (Fig. 5E). Subsequently, to determine the
functional significance of HDAC3-mediated acetylation of KLB, the
effects of decreased HDAC3 on KLB-knockdown and KLB-overexpressing
cells were analyzed. As presented in Fig. 5F, treatment with either si-KLB or
HDAC3 inhibitor increased the expression of vimentin and MMP9,
while their combined application resulted in increased vimentin and
MMP9 expression. However, these effects were reversed by KLB
overexpression. Furthermore, the HDAC3 inhibitor induced a
significant decrease in ZO-1 expression and an increase in vimentin
expression, which was able to be inhibited by FGF21 overexpression
(Fig. 5G). In addition, the
wound-healing experiments demonstrated that the addition of HDAC3
inhibitor significantly enhanced HCC cell migration under FGF21
overexpression (Fig. 5H). Based on
the above results, it was hypothesized that HDAC3 inhibitor
inhibits KLB expression by aberrant acetylated modification,
resulting in the blockade of FGF21-KLB signaling, thereby
increasing the expression of EMT-inducing genes.
Discussion
Recurrence and distant metastasis are common in
patients with advanced HCC and their management poses a significant
challenge (27). Aberrant
activation of the receptor tyrosine kinase (RTK) signaling has been
found in HCC. In addition, the clinical benefits of sorafenib
further demonstrate the efficacy of targeting multiple RTK pathways
in HCC (28). However, another
study found that sorafenib treatment accelerates HCC metastasis
(29). While the US Food and Drug
Administration has recently approved several novel drugs for HCC
treatment, the majority of these are multikinase inhibitors,
similar to sorafenib, and are thus less likely to inhibit HCC
metastasis. Therefore, a comprehensive understanding of the
repertoire of pathways that are frequently and selectively
upregulated in HCC may facilitate the identification of novel
therapeutic targets for HCC treatment.
β-Catenin is elevated in HCC tissues and its
mutations are frequently identified in HCC, suggesting that
β-catenin may be critical for HCC development and progression
(30). It has been widely reported
that aberrant activation of the β-catenin signaling pathway is
closely associated with tumor invasion and metastasis and involved
in tumor microenvironment formation (31). Although the use of β-catenin
pathway-targeting molecules is unlikely to produce a viable
antitumor effect, identification of the factors upstream of the
β-catenin pathway may help in the identification of novel
therapeutic targets for HCC treatment. In the present study, KLB
was identified as a novel upstream regulator of β-catenin. However,
the underlying mechanisms by which KLB and β-catenin promote HCC
metastasis require further investigation.
FGF21, a stress-induced hormone, interacts with
FGFR-1 and KLB on the target cells via endocrine, paracrine and
autocrine pathways, and subsequently exerts its biological effects
by regulating energy balance and glucose-lipid balance (32). FGF21 is primarily expressed in
hepatocytes, adipose tissue and pancreas, and sufficient FGF21
directly reduces hepatic lipid accumulation in an
insulin-independent manner, and inhibits lipolysis in white adipose
tissues, further reducing circulating free fatty acid levels
(33). Administration of FGF21 in
rodents and non-human primates led to the alleviation of several
obesity-related metabolic complications, including reduced fat mass
and hyperglycemia, insulin resistance, dyslipidemia, cardiovascular
disease and nonalcoholic steatohepatitis (NASH), by reducing
oxidative stress and lipid peroxidation (34). Furthermore, FGF21 prevents the
development of advanced pathologies, such as pancreatic ductal
adenocarcinoma and HCC (35). A
study from 2006 found that overexpression of FGF21 in hepatocytes
led to a delay in the development of chemically-induced liver
tumors (36); however, the
underlying mechanisms were not known at the time. Recent studies
have confirmed that low FGF21 levels may activate Toll-like
receptor 4-interleukin-17A signaling pathway in hepatocytes to
promote NASH-HCC transformation, revealing the crucial role of
FGF21 in liver cancer (37).
Furthermore, high serum FGF21 levels are associated with worse
survival in patients with HCC, suggesting its use as a novel
metabolism-related prognostic biomarker for HCC (38). Therefore, the FGF21 signaling
pathway may serve as a potential prognostic factor, as well as a
therapeutic target for HCC. The application of several FGF21
analogs has led to significant improvements in dyslipidemia, liver
fat fraction and serum markers of liver fibrosis in patients with
NASH in the preclinical stages (32). However, FGF21 has limited
indications for clinical use due to its poor pharmacokinetic and
biophysical properties. Therefore, discerning the pathways
associated with FGF21-mediated regulation of HCC will facilitate
the development of effective HCC treatments. FGF21 is considered an
emerging therapeutic target for NASH and related metabolic diseases
and may provide a new perspective on the role of high KLB
expression in the prognosis of non-alcohol-consuming patients with
HCC. In the present study, the role of FGF21-KLB signaling in the
regulation of HCC metastasis was revealed from an epigenetic
standpoint. Mechanistically, FGF21 suppressed HCC metastasis by
inhibiting β-catenin signaling-mediated EMT, while
acetylation-driven suppression of KLB promoted HCC cell motility
under FGF21 overexpression. These findings suggest that FGF21-KLB
signaling may serve as a potential biomarker and a therapeutic
target for HCC.
EMT is a key process in tumor metastasis. Increasing
evidence has suggested that aberrant acetylation of EMT-related
genes is involved in tumorigenicity and HCC metastasis (39). HDAC-mediated histone acetylation
inhibits E-cadherin expression or induces mesenchymal protein
expression to facilitate HCC migration and invasion, thereby
promoting HCC metastasis (40).
Therefore, HDAC inhibitors serve as attractive targets for cancer
treatment. Panobinostat, a novel hydroxamic acid-derived HDAC
inhibitor, has demonstrated promising anticancer effects by
inhibiting HCC growth and metastasis (41). However, other studies found that
HDAC inhibitors promote the expression of Snail and induce EMT in
HCC cells, thus limiting the clinical outcome of HDAC
inhibitor-based therapies in HCC (42,43).
In the present study, it was revealed that HDAC3 is a potential
deacetylase for KLB. It was further demonstrated that HDAC3
inhibitor-mediated acetylation modification downregulated KLB
expression, causing a blockade of FGF21-KLB signaling, further
increasing the expression of EMT induction-related genes.
Consistently, a previous study reported that HDAC3 deficiency
promotes liver cancer through a defect in H3K9ac/H3K9me3 transition
(44). Since different target
genes of HDAC inhibitors may lead to distinct effects, further
identification of the key genes involved in HDAC-mediated
acetylation modifications may lead to the development of effective
therapeutic interventions for HCC.
There are certain limitations to the present study.
First, most of the in vitro experiments in the present study
were conducted on Huh7 cells; however, HCC cells are highly
heterogeneous and further studies on other HCC cell lines are
required to verify the results of the present study. Furthermore,
KLB serves as a co-receptor for both FGF21 and FGF19; thus, the
present findings may have been a result of KLB knockdown-mediated
blockade of FGF19 signaling. For instance, upregulation of the
FGF15 (FGF19 orthologue in mice)/FGFR4 signaling in a lipid
metabolism disorder mouse model promoted HCC development by
activating EMT and Wnt/β-catenin signaling, while activation of the
FGF21/KLB signaling induced the downregulation of β-catenin
signaling, which may explain why nuclear β-catenin expression is
not apparent after KLB knockdown (45,46).
In the present study, only FGF21-overexpressing cells and
sh-KLB-treated FGF21-overexpressing cells were used to observe the
effect of KLB knockdown on FGF21 signaling, and these results need
to be verified further in subsequent studies.
In summary, the present study revealed the role of
KLB in the regulation of EMT in HCC from an epigenetic perspective.
In addition, FGF21 was indicated to exert its anti-HCC metastatic
role in a KLB-dependent manner. Furthermore, HDAC3-mediated
suppression of KLB was found to accelerate HCC-cell migration and
invasion by blocking FGF21 signaling (Fig. 6), which may serve as a potential
therapeutic target for HCC treatment.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The graphical figure was generated by Figdraw
(www.figdraw.com).
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82270646), the Fundamental Research
Funds for the Central Universities (grant no. 0214-14380510), the
Nanjing Health Science and Technology Development Project for
Distinguished Young Scholars (grant no. JQX19002), Project of
Modern Hospital Management and Development Institute, Nanjing
University and Aid Project of Nanjing Drum Tower Hospital Health,
Education & Research Foundation (grant no. NDYG2022057),
fundings for Clinical Trials from the Affiliated Drum Tower
Hospital, Medical School of Nanjing University (grant no.
2022-LCYJ-PY-35) and the Chen Xiao-Ping Foundation for the
Development of Science and Technology of Hubei Province, China
(grant no. CXPJJH121001-2021073).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
JX and ZZ conducted the experiments and collected
the data; GW, YC, RA and SX analyzed the data; and HR and WG
conceived the study, checked and confirmed the authenticity of the
raw data, and prepared the manuscript. All the authors have read
and approved the final draft of the manuscript.
Ethics approval and consent to
participate
Human HCC tissues were obtained in accordance with
the Declaration of Helsinki, and all of the patients concerned
provided written informed consent. Clinical experiments were
approved by the Medical Ethics Committee of the Affiliated Drum
Tower Hospital of Nanjing University Medical School (Nanjing,
China; no. 2019-257-02). Animal experiments were performed in
accordance with the international guidelines; they were approved by
the Animal Ethics Committee of the Affiliated Drum Tower Hospital
of Nanjing University Medical School University (Nanjing, China;
no. 2021AE01021).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
KLB
|
β-klotho
|
|
FGF21
|
fibroblast growth factor 21
|
|
FGFR
|
fibroblast growth factor receptor
|
|
HCC
|
hepatocellular carcinoma
|
|
GSVA
|
Gene Set Variation Analysis
|
|
EMT
|
epithelial-mesenchymal transition
|
|
HDAC3
|
histone deacetylase 3
|
|
IHC
|
immunohistochemistry
|
|
TCGA
|
The Cancer Genome Atlas
|
|
co-IP
|
co-immunoprecipitation
|
|
TSA
|
trichostatin A
|
|
RTK
|
receptor tyrosine kinase
|
|
NASH
|
nonalcoholic steatohepatitis
|
References
|
1
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J and
Finn RS: Hepatocellular carcinoma. Nat Rev Dis Primers. 7:62021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Biswas S and Rao CM: Epigenetics in
cancer: Fundamentals and beyond. Pharmacol Ther. 173:118–134. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kelley RK and Greten TF: Hepatocellular
carcinoma-origins and outcomes. N Engl J Med. 385:280–282. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giannelli G, Koudelkova P, Dituri F and
Mikulits W: Role of epithelial to mesenchymal transition in
hepatocellular carcinoma. J Hepatol. 65:798–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ito S, Kinoshita S, Shiraishi N, Nakagawa
S, Sekine S, Fujimori T and Nabeshima YI: Molecular cloning and
expression analyses of mouse betaklotho, which encodes a novel
Klotho family protein. Mech Dev. 98:115–119. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee S, Choi J, Mohanty J, Sousa LP, Tome
F, Pardon E, Steyaert J, Lemmon MA, Lax I and Schlessinger J:
Structures of β-klotho reveal a ‘zip code’-like mechanism for
endocrine FGF signalling. Nature. 553:501–505. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi SY, Lu YW, Richardson J, Min X,
Weiszmann J, Richards WG, Wang Z, Zhang Z, Zhang J and Li Y: A
systematic dissection of sequence elements determining β-Klotho and
FGF interaction and signaling. Sci Rep. 8:110452018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Triantis V, Saeland E, Bijl N,
Oude-Elferink RP and Jansen PL: Glycosylation of fibroblast growth
factor receptor 4 is a key regulator of fibroblast growth factor
19-mediated down-regulation of cytochrome P450 7A1. Hepatology.
52:656–666. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang Z, Xu A and Cheung BMY: The
potential role of fibroblast growth factor 21 in lipid metabolism
and hypertension. Curr Hypertens Rep. 19:282017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen J, Du F, Dang Y, Li X, Qian M, Feng
W, Qiao C, Fan D, Nie Y, Wu K and Xia L: Fibroblast growth factor
19-mediated up-regulation of SYR-related high-mobility group box 18
promotes hepatocellular carcinoma metastasis by transactivating
fibroblast growth factor receptor 4 and Fms-related tyrosine kinase
4. Hepatology. 71:1712–1731. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poh W, Wong W, Ong H, Aung MO, Lim SG,
Chua BT and Ho HK: Klotho-beta overexpression as a novel target for
suppressing proliferation and fibroblast growth factor receptor-4
signaling in hepatocellular carcinoma. Mol Cancer. 11:142012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anestopoulos I, Voulgaridou GP,
Georgakilas AG, Franco R, Pappa A and Panayiotidis MI: Epigenetic
therapy as a novel approach in hepatocellular carcinoma. Pharmacol
Ther. 145:103–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khalili-Tanha G and Moghbeli M: Long
non-coding RNAs as the critical regulators of doxorubicin
resistance in tumor cells. Cell Mol Biol Lett. 26:392021.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han TS, Ban HS, Hur K and Cho HS: The
epigenetic regulation of HCC metastasis. Int J Mol Sci.
19:39782018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen Y, Wei W and Zhou DX: Histone
acetylation enzymes coordinate metabolism and gene expression.
Trends Plant Sci. 20:614–621. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Falkenberg KJ and Johnstone RW: Histone
deacetylases and their inhibitors in cancer, neurological diseases
and immune disorders. Nat Rev Drug Discov. 13:673–691. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsilimigras DI, Ntanasis-Stathopoulos I,
Moris D, Spartalis E and Pawlik TM: Histone deacetylase inhibitors
in hepatocellular carcinoma: A therapeutic perspective. Surg Oncol.
27:611–618. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
You H, Li Q, Kong D, Liu X, Kong F, Zheng
K and Tang R: The interaction of canonical Wnt/β-catenin signaling
with protein lysine acetylation. Cell Mol Biol Lett. 27:72022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu ZY, Tang N, Wang MF, Zhou JC, Wang JL,
Ren HZ and Shi XL: Comprehensive pan-cancer genomic analysis
reveals PHF19 as a carcinogenic indicator related to immune
infiltration and prognosis of hepatocellular carcinoma. Front
Immunol. 12:7810872022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shang L, Ren H, Wang S, Liu H, Hu A, Gou
P, Lin Y, Zhou J, Zhu W and Shi X: SS-31 protects liver from
ischemia-reperfusion injury via modulating macrophage polarization.
Oxid Med Cell Longev. 2021:66621562021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ren HZ, Xia SZ, Qin XQ, Hu AY and Wang JL:
FOXO1 alleviates liver ischemia-reperfusion injury by regulating
the Th17/Treg ratio through the AKT/Stat3/FOXO1 pathway. J Clin
Transl Hepatol. 10:1138–1147. 2022.PubMed/NCBI
|
|
22
|
Wang C, Yang Z, Xu E, Shen X, Wang X, Li
Z, Yu H, Chen K, Hu Q, Xia X, et al: Apolipoprotein C-II induces
EMT to promote gastric cancer peritoneal metastasis via
PI3K/AKT/mTOR pathway. Clin Transl Med. 11:e5222021.PubMed/NCBI
|
|
23
|
Zhang L, Ren CF, Yang Z, Gong LB, Wang C,
Feng M and Guan WX: Forkhead Box S1 mediates epithelial-mesenchymal
transition through the Wnt/β-catenin signaling pathway to regulate
colorectal cancer progression. J Transl Med. 20:3272022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Zhang P, Martin RC, Cui G, Wang G,
Tan Y, Cai L, Lv G and Li Y: Lack of fibroblast growth factor 21
accelerates metabolic liver injury characterized by
steatohepatities in mice. Am J Cancer Res. 6:1011–1125.
2016.PubMed/NCBI
|
|
25
|
Suh Y, Yoon CH, Kim RK, Lim EJ, Oh YS,
Hwang SG, An S, Yoon G, Gye MC, Yi JM, et al: Claudin-1 induces
epithelial-mesenchymal transition through activation of the
c-Abl-ERK signaling pathway in human liver cells. Oncogene.
32:4873–4882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen F, Gao Q, Wei A, Chen X, Shi Y, Wang
H and Cao W: Histone deacetylase 3 aberration inhibits Klotho
transcription and promotes renal fibrosis. Cell Death Differ.
28:1001–1012. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun W, Fu S, Wu S and Tu R: Growing
evidence of exosomal MicroRNA-related metastasis of hepatocellular
carcinoma. Biomed Res Int. 2020:45014542020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B,
Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al: The mechanisms of
sorafenib resistance in hepatocellular carcinoma: Theoretical basis
and therapeutic aspects. Signal Transduct Target Ther. 5:872020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang W, Sun HC, Wang WQ, Zhang QB, Zhuang
PY, Xiong YQ, Zhu XD, Xu HX, Kong LQ, Wu WZ, et al: Sorafenib
down-regulates expression of HTATIP2 to promote invasiveness and
metastasis of orthotopic hepatocellular carcinoma tumors in mice.
Gastroenterology. 143:1641–1649.e5. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim E, Lisby A, Ma C, Lo N, Ehmer U, Hayer
KE, Furth EE and Viatour P: Promotion of growth factor signaling as
a critical function of β-catenin during HCC progression. Nat
Commun. 10:19092019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Potthoff MJ: FGF21 and metabolic disease
in 2016: A new frontier in FGF21 biology. Nat Rev Endocrinol.
13:74–76. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lewis JE, Ebling FJP, Samms RJ and
Tsintzas K: Going back to the biology of FGF21: New insights.
Trends Endocrinol Metab. 30:491–504. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Geng L, Lam KSL and Xu A: The therapeutic
potential of FGF21 in metabolic diseases: From bench to clinic. Nat
Rev Endocrinol. 16:654–667. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu W, Li X and Luo Y: FGF21 in obesity and
cancer: New insights. Cancer Lett. 499:5–13. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang X, Yu C, Jin C, Yang C, Xie R, Cao
D, Wang F and McKeehan WL: Forced expression of hepatocyte-specific
fibroblast growth factor 21 delays initiation of chemically induced
hepatocarcinogenesis. Mol Carcinog. 45:934–942. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng Q, Martin RC, Shi X, Pandit H, Yu Y,
Liu X, Guo W, Tan M, Bai O, Meng X and Li Y: Lack of FGF21 promotes
NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling.
Theranostics. 10:9923–9936. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu ZY, Luo Y, Fang AP, Wusiman M, He TT,
Liu XZ, Yishake D, Chen S, Lu XT, Zhang YJ and Zhu HL: High serum
fibroblast growth factor 21 is associated with inferior
hepatocellular carcinoma survival: A prospective cohort study.
Liver Int. 42:663–673. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin YT and Wu KJ: Epigenetic regulation of
epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β
signaling. J Biomed Sci. 27:392020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu Y, Zheng Y, Dai M, Wang X, Wu J, Yu B,
Zhang H, Cui Y, Kong W, Wu H and Yu X: G9a and histone deacetylases
are crucial for Snail2-mediated E-cadherin repression and
metastasis in hepatocellular carcinoma. Cancer Sci. 110:3442–3452.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song X, Wang J, Zheng T, Song R, Liang Y,
Bhatta N, Yin D, Pan S, Liu J, Jiang H and Liu L: LBH589 inhibits
proliferation and metastasis of hepatocellular carcinoma via
inhibition of gankyrin/STAT3/Akt pathway. Mol Cancer. 12:1142013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiao Q, Liu H, Wang HS, Cao MT, Meng XJ,
Xiang YL, Zhang YQ, Shu F, Zhang QG, Shan H and Jiang GM: Histone
deacetylase inhibitors promote epithelial-mesenchymal transition in
hepatocellular carcinoma via AMPK-FOXO1-ULK1 signaling
axis-mediated autophagy. Theranostics. 10:10245–10261. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu W, Liu H, Liu ZG, Wang HS, Zhang F,
Wang H, Zhang J, Chen JJ, Huang HJ, Tan Y, et al: Histone
deacetylase inhibitors upregulate Snail via Smad2/3 phosphorylation
and stabilization of Snail to promote metastasis of hepatoma cells.
Cancer Lett. 420:1–13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ji H, Zhou Y, Zhuang X, Zhu Y, Wu Z, Lu Y,
Li S, Zeng Y, Lu QR, Huo Y, et al: HDAC3 deficiency promotes liver
cancer through a defect in H3K9ac/H3K9me3 transition. Cancer Res.
79:3676–3688. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cui G, Martin RC, Jin H, Liu X, Pandit H,
Zhao H, Cai L, Zhang P, Li W and Li Y: Up-regulation of FGF15/19
signaling promotes hepatocellular carcinoma in the background of
fatty liver. J Exp Clin Cancer Res. 37:1362018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim GY, Kwon JH, Cho JH, Zhang L,
Mansfield BC and Chou JY: Downregulation of pathways implicated in
liver inflammation and tumorigenesis of glycogen storage disease
type Ia mice receiving gene therapy. Hum Mol Genet. 26:1890–1899.
2017. View Article : Google Scholar : PubMed/NCBI
|















