Introduction
Colorectal cancer (CRC) is the third most commonly
diagnosed cancer type and the second leading cause of
cancer-related deaths worldwide (1,2).
Patients with CRC develop metastatic disease in >50% of cases,
especially in the liver, which results in the death of >2/3 of
the patients (3–5). Advances in chemotherapy, radiotherapy
and immunotherapy have improved the survival of patients with
metastatic CRC (6,7); however, resistance to anticancer
drugs, radiation and immune therapies remains a major therapeutic
challenge (7–9).
Increasing evidence has demonstrated that the
combination of hyperthermia with the aforementioned therapies can
overcome tumor resistance to antitumor treatments and improve their
efficacy (10–12). In addition, heat shock (HS)
proteins (HSPs) are involved in enhancing the cytotoxic activity of
natural killer (NK) cells (11,13),
inducing maturation and antigen presentation of dendritic cells and
activating T-cells (11,14). By contrast, previous studies have
indicated that HSPs including HSP90 promote cancer cell
proliferation, invasion and metastasis, as well as tumor
angiogenesis, which can negatively affect the hyperthermia efficacy
(15,16). These major HSPs are highly
expressed in some malignant tumor types and are inversely
correlated with prognosis, leading to these HSPs being used as
therapeutic targets (15,16). Nonetheless, the overall involvement
of HSPs in hyperthermia remains unclear.
Heat shock-inducible tumor small protein (HITS) is
an 18 kDa protein that was originally identified as a molecule
upregulated upon HS treatment in Jurkat cells (17). The induction of HITS protein as
well as HSP90 protein has been confirmed in THP-1 cells in
vitro and transplanted rat walker 256 sarcoma cells in
vivo (17). HITS is highly
homologous to downregulated in renal cell carcinoma 1 (DRR1), a
putative tumor suppressor that plays important roles in actin and
microtubule cytoskeleton organization, and is downregulated in
renal cell carcinoma (17–19). Our previous studies indicate that
HITS expression can be observed in various cancer cells and is
downregulated during tumor progression in colon cancer as well as
cervix, thyroid and breast cancers compared with the corresponding
healthy tissues (17,20). Unlike the major HSPs,
overexpression of HITS shows tumor suppressive phenotype in the
mouse cervical cancer xenograft model (20).
The present study showed that HITS overexpression
increased the levels of glycogen synthase kinase-3β (GSK3β)
phosphorylated (p) at serine (S) 9 (pGSK3βS9), resulting
in its deactivation in CRC cells (21–23).
To the best of our knowledge, while GSK3β deactivation has been
reported to prevent β-catenin degeneration (23), no previous studies have shown a
direct association of cellular GSK3β deactivation with cancer
development or progression (22).
GSK3β does not participate in the canonical β-catenin destruction
complex in the majority of CRCs due to the mutations in either
adenomatous polyposis coli (APC; <90% of cases), catenin β-1
(CTNNB1; ~5% of cases) or axis inhibition protein 1 (AXIN1)
(22). By contrast, previous
studies have demonstrated that pharmacological inhibition of GSK3β
activity suppresses cancer progression by attenuating tumor cell
migration and invasion in several cancer types, including
colorectal, breast and pancreatic cancers, in addition to
glioblastoma (21–29). The present study showed that HITS
upregulation induced by HS exerted an anti-migratory effect via
GSK3β deactivation (S9 phosphorylation) in human CRCs, thereby
counteracting the pro-migratory effects of HSPs. This novel
anti-migratory mechanism may play an important role in reducing the
risk of cancer metastasis during hyperthermia.
Materials and methods
Cell culture
HCT 116, RKO and SW480 cells were purchased from
American Type Culture Collection and maintained in Dulbecco's
Modified Eagle's Medium-high glucose (cat. no. 043-30085; Fujifilm
Wako Pure Chemical Corporation) supplemented with 10% fetal bovine
serum (FBS) (cat. no. F7524; Sigma-Aldrich; Merck KGaA) in a
humidified incubator with 5% CO2 at 37°C. Cells were
tested for mycoplasma contamination.
Western blotting
Cells were lysed with 2X sample buffer containing 4%
SDS (cat. no. 08933-34; Nacalai Tesque, Inc.), 20% glycerol (cat.
no. 17018-25; Nacalai Tesque, Inc.), 0.001% bromophenol blue (cat.
no. 05808-61; Nacalai Tesque, Inc.), 0.125 M Tris HCl (cat. no.
T1503, Sigma-Aldrich; Merck KGaA) and 10% 2-mercaptoethanol (cat.
no. 21418-42; Nacalai Tesque, Inc.) and boiled for 5 min. The
protein concentration was measured using BCA Protein Assay Kit
(cat. no. 23225; Thermo Fisher Scientific, Inc.). Lysates (20
µg/lane) were separated by SDS-PAGE on an 8.5% gel and transferred
onto PVDF membrane (cat. no. IPVH00010; MilliporeSigma). To
calculate relative intensity, cell lysates to compare were loaded
in the same gel. Membranes were blocked with phosphate-buffered
saline [PBT; 0.01 M Na2HPO4; cat. no.
31801-05 and KH2PO4; cat. no. 28721-55; pH
7.4 with 0.15 M NaCl; cat. no. 31320-05; and 0.1% Tween-20 (cat.
no. 35624-02; Nacalai Tesque, Inc.)] containing 5% non-fat dry milk
or 5% bovine serum albumin (cat. no. 019-21272; Fujifilm Wako Pure
Chemical Corporation) for 1 h at room temperature. After washing
with PBT, the membranes were incubated with the primary antibodies
against GSK3β (1:20,000 or 40,000; cat. no. 9315; Cell Signaling
Technology, Inc.), pGSK3βS9 (1:1,000 or 2,000; cat. no.
9336; Cell Signaling Technology, Inc.) and β-tubulin (1:120,000 or
40,000; cat. no. 017-25031; Fujifilm Wako Pure Chemical
Corporation) overnight at 4°C. This was followed by incubation with
anti-rabbit (1:4,000 dilution; cat. no. 5220-0336; SeraCare Life
Sciences, Inc.) and anti-mouse (1:4,000 dilution; cat. no.
5220-0341; SeraCare Life Sciences, Inc.) horseradish
peroxidase-conjugated secondary antibodies for 1 h at room
temperature. The protein bands were developed using the ECL Prime
Western Blotting Detection Reagent (cat. no. RPN2232; Cytiva) and
the chemiluminescence was detected using a ChemiDoc Touch MP
Imaging System with Image Touch 2.4 software (Bio-Rad Laboratories,
Inc.) or C-Digit Blot scanner (LI-COR Biosciences).
RNA extraction and semi-quantitative
reverse transcription (RT)-PCR
Total RNA was extracted from cells using TRIzol™
(cat. no. 15596026; Thermo Fisher Scientific, Inc.) and
reverse-transcribed to complementary DNA (cDNA) using ReverTra Ace™
(cat. no. FSQ-201; Toyobo Life Science) with Random Primer (cat.
no. FSK-301; Toyobo Life Science) according to the manufacturer's
instructions. Semi-quantitative RT-PCR was performed using a
LifeECO thermal cycler (Yakukensha Co., Ltd.). The reaction mixture
comprised 0.25 µl Blend Taq plus (2.5 U/µl; cat. no. BTQ-201;
Toyobo Life Science), 2.5 µl 10X buffer, 2.5 µl dNTP (2 mM; cat.
no. NTP-501; Toyobo), 0.25 µl primers (10 µM; cat. no. FSK-301;
Toyobo) and 1–3 µl of synthesized cDNA. The following primer pairs
were used for semi-quantitative RT-PCR: HITS forward,
5′-CCACCTGAGGATATTGACCATAA-3′ and reverse,
5′-TTCTGTGCTTCTTCTTCCTTCTG-3′; matrix metalloproteinase-3 (MMP-3),
forward, 5′-CTCAGGAAGCTTGAACCTGAAT-3′ and reverse,
5′-CAGCTCGTACCTCATTTCCTCT-3′; MMP-13, forward,
5′-TTACCAGTCTCCGAGGAGAAAC-3′ and reverse,
5′-TTTTGGAAGACCCAGTTCAGAT-3′; and TATA-box binding protein (TBP)
forward, 5′-AGAAAGTGAACATCATGGATCAGA-3′ and reverse,
5′-GTTTACAACCAAGATTCACTGTGG-3′. TBP was amplified as an internal
control to normalize the expression of the target gene. The PCR
reaction was performed under the following conditions: Initial
denaturation at 94°C for 1 min; followed by cycling at 94°C for 20
sec, 55°C (HITS, MMP-13 and TBP) or 53°C (MMP-3) for 20 sec and
72°C for 20 sec; and final extension at 72°C for 1 min. The numbers
of cycles were 29 and 30 for HITS, TBP; 36 for MMP-3, 36 for MMP-13
and 33 for TBP. The DNA products were run on a 1.2% agarose gel
containing ethidium bromide (cat. no. 312-01193; Nippon Gene Co.,
Ltd.) and imaged using a ChemiDoc MP Imaging System (Bio-Rad
Laboratories, Inc.). The intensity of each band was quantified
using ImageJ software, ver. 1.50 (National Institutes of
Health).
Plasmid construction and
overexpression
The amino acid sequence of the mouse HITS open
reading frame (ORF) is 98.47% homologous to the human HITS;
therefore, mouse HITS ORF was cloned for transfection (17). The HITS ORF fragment with EcoRV and
BamHI ends was amplified from HeLa-Tet-HITS (16) using PCR (initial denaturation at
94°C for 1 min; 25 cycles of 94°C for 20 sec, 55°C for 20 sec and
72°C for 20 sec; and a final extension at 72°C for 1 min) and the
following primers: forward, 5′-GATATCATGGCTGAGCCAGACTACATAGAAG-3′,
and reverse, 5′-GGATCCCTAGGACTCCTGGGCCTGAGCCACC-3′. The amplified
DNA was cloned into PCR2.1 vector using the TOPO™ TA™ Cloning Kit
(cat. no. K450002; Thermo Fisher Scientific, Inc.) and subcloned
into the EcoRV/BamHI sites of pCAG-IRES-EGFP, which was kindly
supplied by Dr T. Kawauchi (30).
The generated plasmid (0.5 µg/well), namely pCAG-HITS-IRES-EGFP,
and pEGFP (30) were mixed with
transfection reagents and kept for 10 min then transfected into HCT
116, RKO and SW480 cells, using Lipofectamine 3000™
(cat. no. L3000001; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. The cells were incubated for 36 h
and obtained cell lysates for western blotting, wound healing assay
or MTT assay as described later. Transfection efficiency was tested
using semi-quantitative RT-PCR as described in the previous section
(Fig. S1).
RNA interference (RNAi)
The sequences of small interfering RNAs (siRNAs)
targeting HITS were as follows: HITS-siRNA#1,
5′-GGAUAUUGACCAUAAGGACUCAUAU-3′; and HITS-siRNA#2,
5′-GCCUCAGAAACUGAUCAAUCCUGUA-3′ (Thermo Fisher Scientific, Inc.).
Lipofectamine™ RNAiMAX (cat. no. 13778030; Thermo Fisher
Scientific, Inc.) was used to transfect HITS siRNAs and control
siRNA (cat. no. 12935300; Thermo Fisher Scientific, Inc.). A total
of 10 pmol/well siRNAs were mixed with transfection reagents and
kept for 5 min according to the manufacturer's instructions. The
cells were incubated for 36 h and obtained cell lysates for western
blotting or wound healing assay as described later.
Wound healing assay
HCT 116 cells were seeded at a density of
5×105 cells/well in a 24-well plate and transfected with
HITS the following day. At 10 h after transfection, the cells were
trypsinized, re-seeded at a density of 8×105 cells/well,
and incubated for 18 h (the confluency of the cells reached
80–90%). The cells were then scratched with a 200-µl pipette tip
and the medium was replaced to medium containing 1% FBS to prevent
the loose cells from settling back down. Images of the cells were
captured at 0 and 24 h under a phase-contrast and fluorescence
microscope (BZ-X700; Keyence Corp.). Multiple images were captured
with the microscope to cover an entire scratched area and merged
into one image to compare the same wound gap positions between 0
and 24 h. The wound healing rate was calculated as follows: Wound
healing rate (%)=(wound area at 0 h-wound area at 24 h)/(initial
wound at 0 h) ×100. For the assay performed on HS-induced cells (HS
at 42°C for 1 h), 8.5×105 cells/well were plated. For
RNAi followed by HS and inhibitors treatment, the CytoSelect
24-well Wound Healing Assay Kit (Cell Biolabs, Inc.) was used.
Plastic inserts provided with the kit were placed onto the wells to
create wound gaps where cells were plated at a density of
8.5×105 cells/well. Some groups of cells were treated
with the HSP90 inhibitor 17-AAG (cat. no. CS-0161; Funakoshi Co.,
Ltd.) or the GSK3β inhibitor AR-A014418 (cat. no. A3230;
Sigma-Aldrich; Merck KGaA), both of which were dissolved in
dimethyl sulfoxide (DMSO) to prepare 10 mM stock solutions. DMSO
was added to control and 17-AAG samples to adjust the DMSO
concentration to 0.2%. The concentration of 17-AAG was optimized by
testing different concentrations, which showed suppression of cell
migration but no effect against cell survival (data not shown). The
concentration of AR-A014418 was determined based on our previous
studies (29).
MTT assay
HCT 116 cells were seeded at a density of
1×104 cells/well in a 96-well plate. The expression
vectors pCAG-HITS-IRES-EGFP and pEGFP were transfected into the
cells the following day, as previously described. The MTT assay
(cat. no. 11465007001; Sigma-Aldrich; Merck KGaA) was performed 2
days after transfection, according to the manufacturer's
instructions. Methanol was used to dissolve formazan. Colorimetric
detection was done at a wavelength of 550 nm and reference
wavelength was 690 nm.
Statistical analysis
Data are expressed as the mean ± standard error.
Statistical analysis was performed using the unpaired Student's
t-test to compare two groups of data and one-way analysis of
variance (ANOVA) followed by Dunnett's test or two-way ANOVA
followed by Tukey's test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
HITS promotes the phosphorylation of
GSK3β S9 in CRC cells
The effects of HITS on GSK3β, which plays important
roles in tumorigenesis (21,22),
were investigated to study the role of HITS in cancer progression.
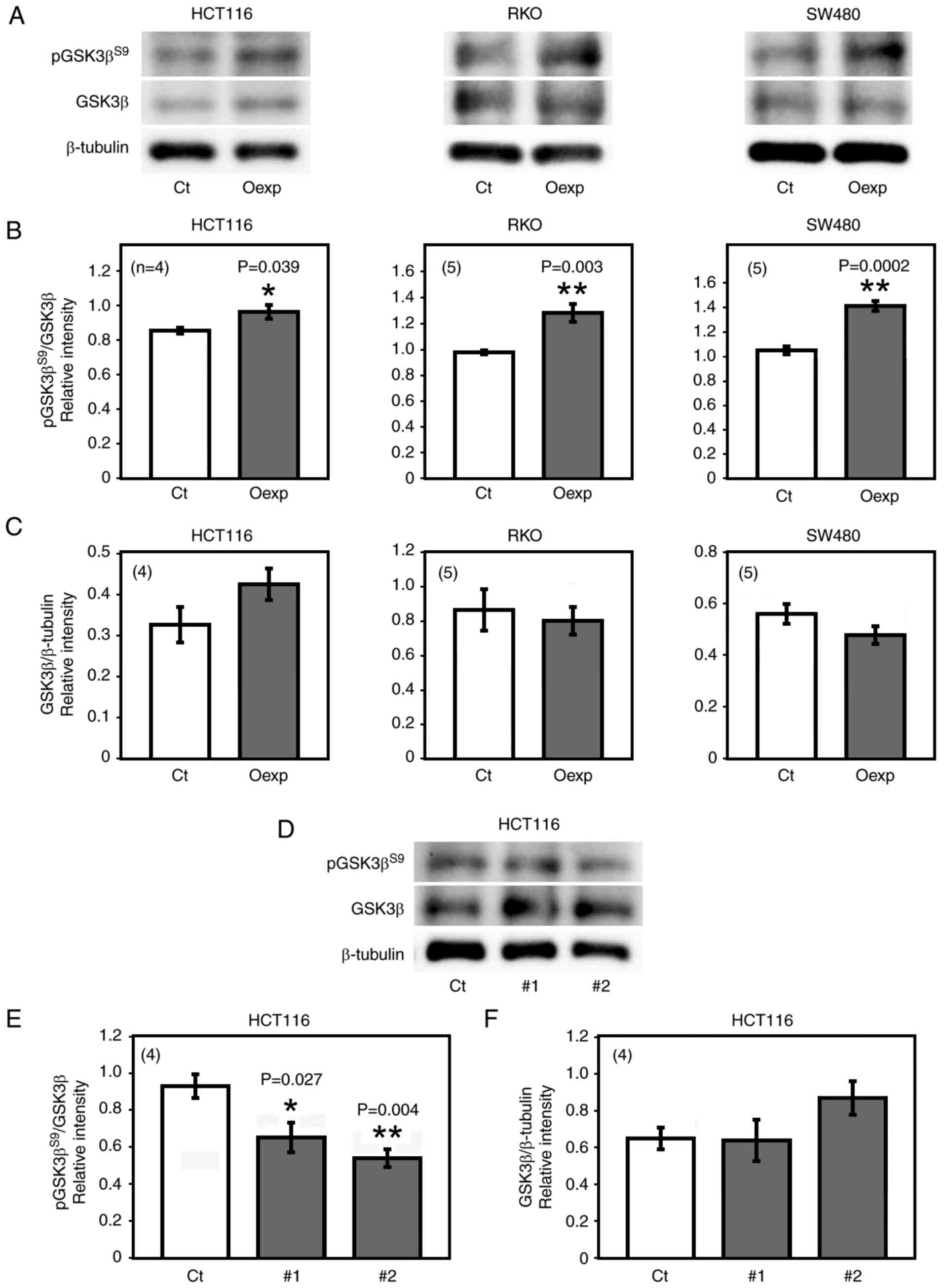
Overexpression of HITS in HCT 116, RKO and SW480 cells were
followed by a significant increase in pGSK3βS9 level,
while no significant changes were observed in the expression of
GSK3β (Fig. 1A-C) nor in the
levels of GSK3β phosphorylation at tyrosine (Y)216
(pGSK3βY216; active form; data not shown).
HCT 116 is one of the human CRC cell lines that were
established to extrapolate clinical CRC (31,32).
Our previous studies showed the distinct and common tumor-promoting
roles of active GSK3β (lower and higher phosphorylation of its S9
and Y216 residues, respectively) in multiple human CRC cell lines
including HCT 116 cells (29,33–35).
In addition, no biological association between the pathological
(tumor-promoting) property of deregulated GSK3β and the pathways
mediated by activated β-catenin and phosphoinositide 3-kinase
(PI3K)/Akt in human CRC cell lines including HCT 116 cells as well
as in clinical CRC tumors (29).
Therefore, the present study used HCT 116 cells for the subsequent
analyses.
To confirm the effects of HITS on the expression and
phosphorylation of GSK3β, HITS RNAi was performed on CRC cells.
Semi-quantitative RT-PCR indicated that transfection with either
HITS siRNA#1 or HITS siRNA#2 significantly reduced HITS mRNA
expression by >90% compared with the that in cells transfected
with control siRNA (Fig. S2).
Transfection of these HITS siRNAs in HCT 116 cells significantly
decreased the level of pGSK3βS9 compared with that in
cells transfected with control siRNA, but did not affect the GSK3β
expression (Fig. 1D-F). The
present data demonstrated that HITS promoted the phosphorylation of
GSK3β at S9 in CRC cells.
HS induces GSK3β S9 phosphorylation
via HITS upregulation
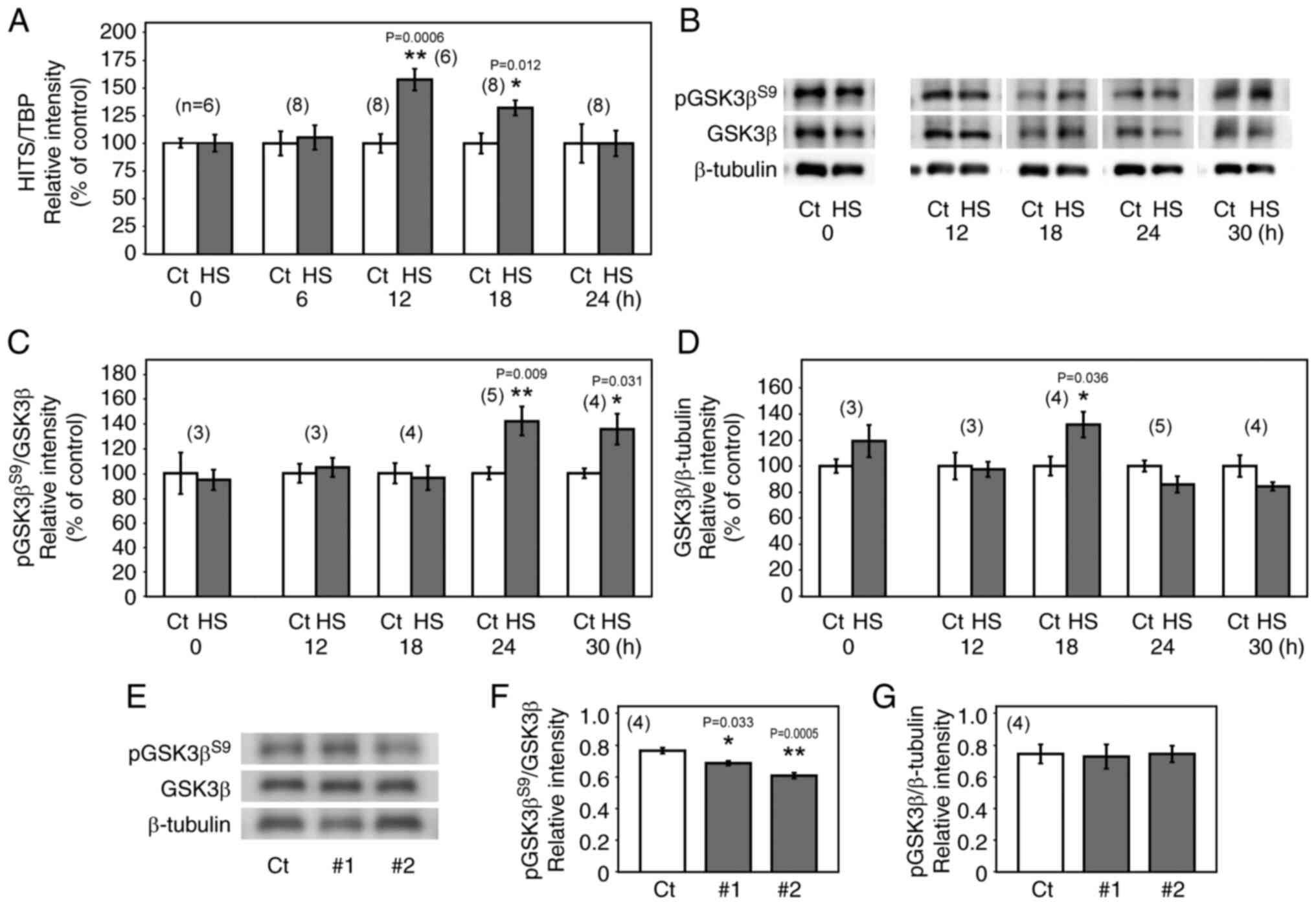
Our previous study showed that HS induced HITS
protein expression both in vitro and in vivo
(17). To study the
transcriptional response to HS in detail, the levels of HITS mRNA
were monitored in HCT 116 cells in response to HS (42°C, 1 h).
Semi-quantitative RT-PCR analysis showed that the level of HITS
mRNA increased relatively slowly following HS, with its expression
peaking at 12 h after HS (Fig.
2A). The level of pGSK3βS9 in the cells, measured
using western blotting, increased significantly at 24 and 30 h
after HS compared with their respective controls (Fig. 2B and C). This was likely caused by
HITS upregulation in response to HS, since HITS knockdown in the
heat-shocked cells significantly decreased the level of
pGSK3βS9 at 24 h after HS compared with the control
group (Fig. 2E and F).
Significantly increased expression of GSK3β was observed 18 h after
HS compared with the control (Fig. 2B
and D); however, HITS knockdown induced no significant change
in the GSK3β expression (Fig. 2E and
G).
HITS suppresses cell migration but not
proliferation
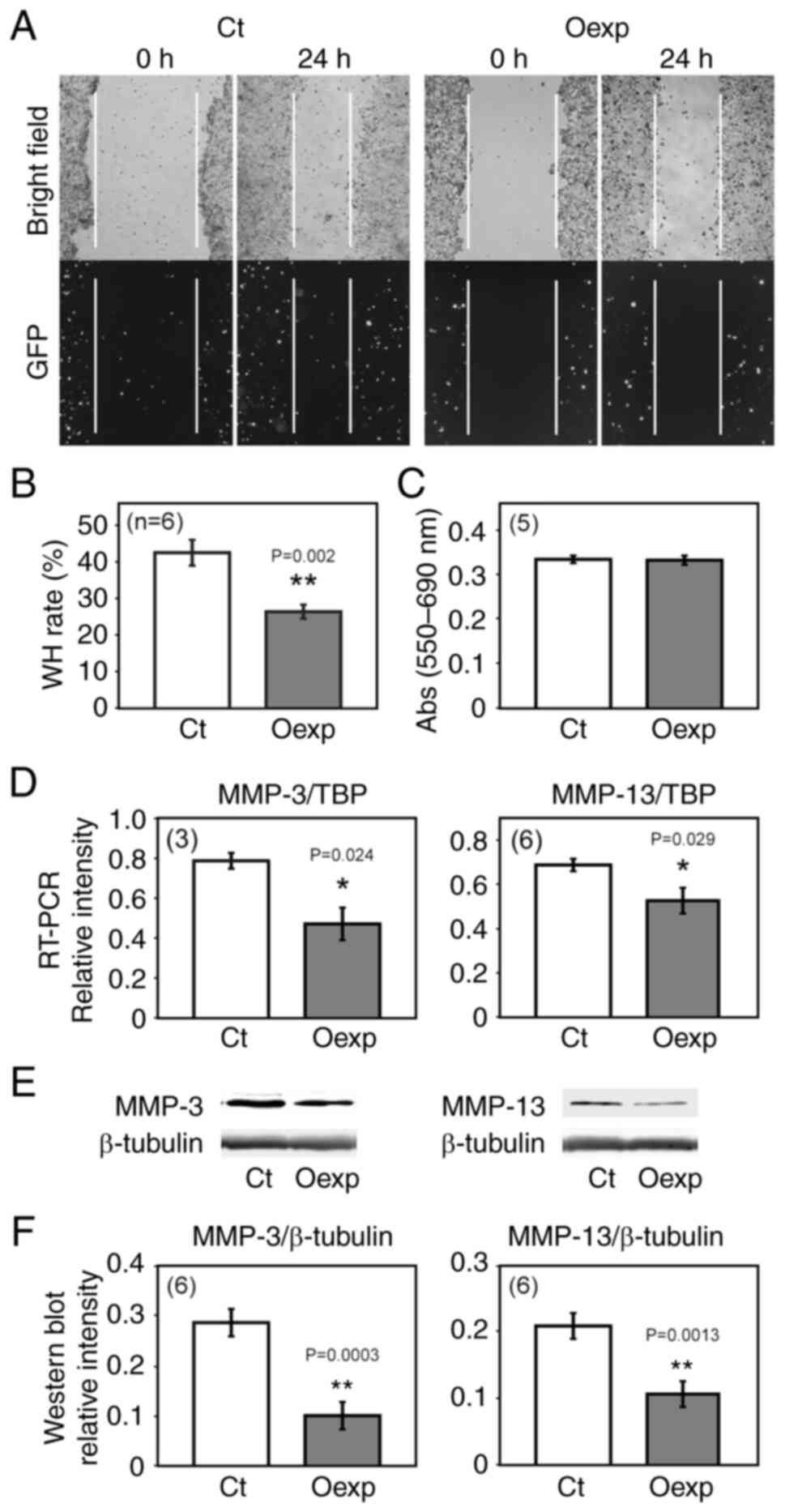
It is hypothesized that HITS is involved in
suppressing tumor progression via phosphorylation-dependent
deactivation of GSK3β. To investigate the effects of HITS on the
migration of CRC cells, a wound healing assay was performed on HCT
116 cells transfected with a pEGFP control vector expressing GFP or
pCAG-HITS-IRES-EGFP vector expressing both HITS and GFP. Compared
with that in the control cells, cells overexpressing HITS showed a
significant suppression of cell migration (Fig. 3A and B). This was not due to a cell
proliferation suppression caused by HITS because no significant
changes were observed in cell proliferation upon HITS
overexpression (Fig. 3C).
MMPs are key proteases involved in cancer cell
migration, invasion and metastasis by degrading the extracellular
matrix (ECM) (36). MMP-3 and
MMP-13 have been reported to be regulated by GSK3β (37); therefore, the present study
investigated if HITS caused any changes in the expression of MMPs.
Semi-quantitative RT-PCR and western blotting analysis revealed
significant downregulation of MMP-3 and MMP-13 mRNA and protein in
the HITS overexpressing cells (Fig.
3D-F), indicating that HITS suppressed cell motility, at least
in part, by downregulating MMP-3 and MMP-13.
Anti-migratory effect of the
HITS-GSK3β pathway offsets the pro-migratory effects of HSPs after
HS
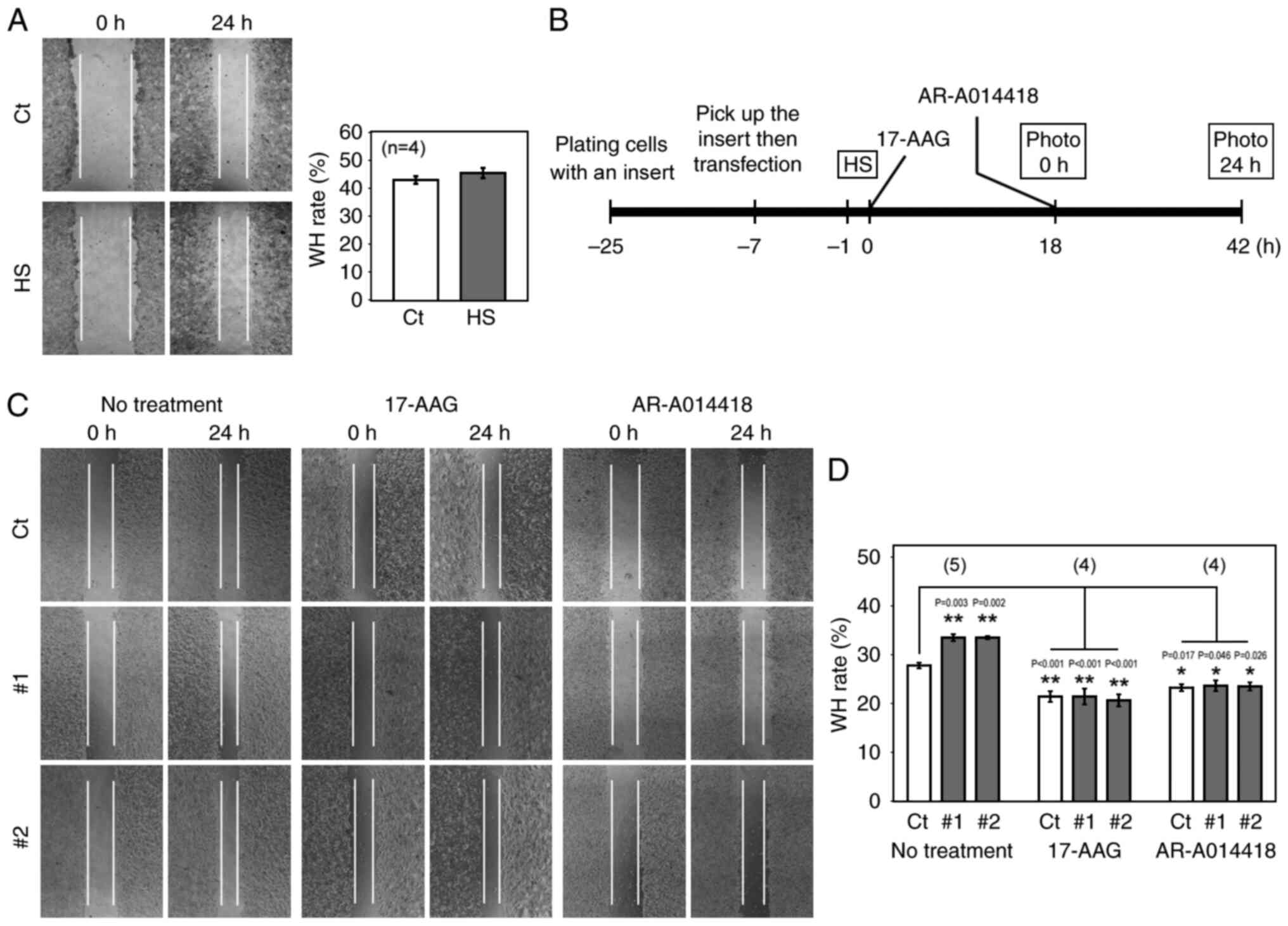
Previous studies have indicated that, while HS
increases HITS expression, HS also induces expression of HSPs,
including HSP90 (17,38,39),
which in turn promotes the migration of cancer cells (15,16,40–42).
Therefore, it remains unclear whether the overall effects of HS on
CRC cell migration are promotive or suppressive. Thus, a wound
healing assay was performed to compare the migration of cells with
and without HS. To cover all the time points where the S9
phosphorylation or the protein expression of GSK3β was upregulated
by HS, HCT 116 cells were scratched and observed at 14 and 38 h (0
and 24 h in the wound healing timeframe, respectively) after HS. No
significant change was observed in the migration of these cells
(Fig. 4A), suggesting that the
anti-migratory effect of the HITS-GSK3β pathway offset the
pro-migratory activities of HSPs after HS. To understand the
HITS-GSK3β effect, HITS siRNAs were transfected into HCT 116 cells
under the same HS conditions, and a wound healing assay was
performed following the time schedule shown in Fig. 4B. The wound healing rate was
significantly increased in cells transfected with HITS siRNAs
compared with that in the control, demonstrating that HITS
decreased the migration of the cells induced with HS (Fig. 4C and D). Subsequently, the
pro-migratory effect of HSP90 was further demonstrated using 17-AAG
which is metabolized by NAD(P)H:quinone oxidoreductase 1 (NQO1) and
selectively inhibits HSP90 (43).
17-AAG decreased the wound healing rate of HS-treated cells
(Fig. 4C and D). Moreover, the
migratory effect of the HITS knockdown was not observed in cells
treated with 17-AAG, which suggested that the pro-migratory pathway
mediated by HSP90 was the target of HITS. The function of HITS was
mediated by GSK3β because the treatment with the specific GSK3β
inhibitor, AR-A014418 (44)
counteracted the effects of the HITS knockdown on cell migration
(Fig. 4C and D). Taken together,
the present findings indicated that HS not only induced the
pro-migratory pathways mediated by HSPs, but also induced the
anti-migratory pathway involving HITS and GSK3β, and that
HITS-mediated GSK3β deactivation sufficiently offset the
pro-migratory activity in CRC cells.
Discussion
The present study explored the effects of HS on CRC
cell migration by focusing on HITS and its putative downstream
target GSK3β. The level of pGSK3βS9 (GSK3β inactive
form) was significantly increased in CRC cells overexpressing HITS,
whereas its knockdown showed an opposite effect. These data
demonstrated that HITS expression suppressed GSK3β activity by
increasing the levels of pGSK3βS9. Our previous studies
have indicated that the deactivation of GSK3β by S9 phosphorylation
suppresses tumor progression in various CRC cells such as RKO,
SW480, SW48, SW620 and HT29, as well as HCT 116 in vitro
(29) and xenograft in vivo
(35). The wound healing assay
showed that overexpression of HITS in the CRC cells significantly
suppressed cell migration compared with that of the control cells.
This was most likely mediated by GSK3β deactivation because the
positive effect of HITS knockdown on cell migration was abolished
by treatment with the GSK3β inhibitor AR-A014418.
The molecular mechanisms underlying the
anti-migratory and anti-invasive effects of GSK3β inhibition are of
interest. Previous studies have indicated that the inhibition of
GSK3β suppresses F-actin assembly and decreases the formation of
lamellipodia and invadopodia via suppression of the
phosphorylation-dependent activity of focal adhesion kinase (FAK)
(27,28,45),
c-Jun N-terminal kinase (JNK) (27) and adenylyl cyclase-associated
protein 1 (CAP1) (46,47), the deactivation of guanine
nucleotide-exchange factors (GEF) and Ras-related C3 botulinum
toxin substrate 1 (RAC1) (27,28,48)
and the degradation of nuclear factor of activated T cells (NFAT)
(49). Potential involvement of
these functional molecules in suppression of CRC cells migration
upon the S9 phosphorylation-mediated deactivation of GSK3β is an
aim of our future studies to investigate mechanisms by which HITS
regulates tumor cell migration. The deactivation of GEF/RAC1 and
JNK pathways reduces the expression of MMP-2 and membrane type 1
(MT1)-MMP, resulting in the suppression of invadopodia formation in
glioblastoma and pancreatic cancer cells (27,28).
The present study showed that HITS decreased the expression of
MMP-3 and MMP-13 in CRC cells in association with attenuated cell
migration (50,51). Therefore, the anti-migratory effect
of HITS in cancer cells might be mediated by suppressing these
MMPs.
A previous study reported that DRR1, which shares a
highly homologous region with HITS but is not induced by HS
(18,19,52),
can directly bind actin (53).
This study elucidated the two actin-binding regions of DRR1, of
which the N-terminal region is highly homologous to HITS (70% amino
acid identity) and is necessary for suppressing F-actin elongation
in HeLa cells. It is possible that HITS also directly binds to
actin to suppress F-actin assembly. Guo et al (54) indicated that the downregulation of
HITS by S100A4, a pro-metastatic protein, increases the migration
of gastric cancer MGC803 cells. Considering that S100A4 has been
reported to bind F-actin and myosin II heavy chain to promote
cytoskeletal formation (55,56),
the direct binding of HITS to actin may disturb the interaction
between S100A4 and the actin/myosin II heavy chain, resulting in
the suppression of cell migration. In conjunction with these
previous studies, the present findings suggested that HITS was
involved in suppressing actin cytoskeleton formation directly
and/or via S9 phosphorylation-mediated deactivation of GSK3β, which
suppresses the formation of lamellipodia and invadopodia to prevent
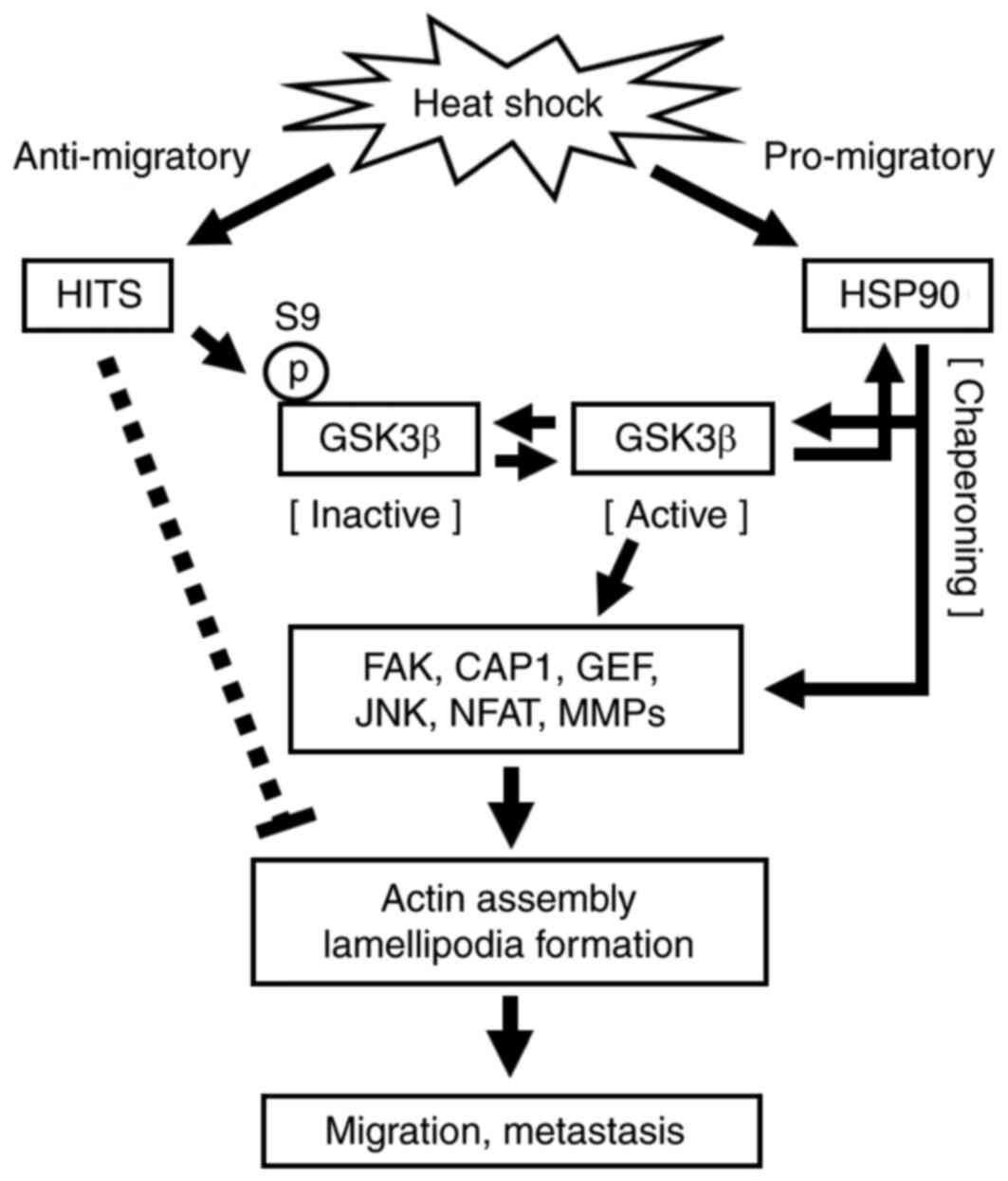
the CRC cell migration that is necessary for invasion (Fig. 5).
A number of previous studies have indicated that
HSPs promote the migration, invasion and metastasis of cancer cells
(15,16,57–59).
Among the HSPs, HSP90 was shown to act as a molecular chaperone
able to stabilize and maintain oncoproteins involved in CRC
progression such as mutated TP53 (gain-of-function), HER2 and BRAF,
especially in stress conditions caused by chemotherapy,
radiotherapy and hyperthermia (60,61).
GSK3β has also been shown to be one of the client proteins of HSP90
(62–64), which in turn phosphorylates HSP90
and HSP70 to facilitate cancer progression (65). The upregulation of HSP90 in HCT 116
cells was indicated in a previous report (66). Consistently, the increased
expression of GSK3β observed after HS might be due to the
chaperoning function of HSP90 and other HSPs. It has been indicated
that HSP90 chaperoning function is required for maintaining the
expression and/or function of FAK, NFAT, JNK and MMPs (67–71)
that promote the cell migration under the control of GSK3β, as
discussed above. The present study indicated that GSK3β was
involved in the major pro-migratory pathways mediated by HSP90
after HS, and that the inhibition of GSK3β caused by HITS played a
critical role in attenuating the pro-migratory effect of GSK3β
(Fig. 5). Indeed, HS did not
induce significant changes in the migration of HCT 116 cells,
although HSP90 was activated and showed pro-migratory activity.
This was most likely due to the deactivation of GSK3β caused by
HITS because HITS knockdown, under the same HS condition, showed an
increase in wound healing rate, which in turn was decreased by
treatment with AR-A014418. Treatment with 17-AAG counteracted the
effect of the HITS knockdown, confirming the HSP90 chaperoning
effect on GSK3β and its downstream pathways (Fig. 5). Other studies have also reported
the anti-migratory and anti-metastatic effects of hyperthermia
in vitro or in animal models, although the underlying
mechanisms remain unclear (72–74).
In these reports, the HITS-GSK3β pathway might have played a
critical role in suppressing cell migration in these reports.
The single or combined use of HSPs inhibitors with
other therapies were proposed as attractive strategies for cancer
therapy (75,76). Several studies have proposed to use
hyperthermia in combination with HSP inhibitors, such as HSP90
inhibitors, which provides successful in vitro results
(77–79). However, HSPs are involved in the
activation of the immune response, including the cytotoxicity of NK
cells, maturation and antigen presentation of dendritic cells, and
activation of T cells (11,13,14).
Previous studies have shown that CRC is a heterogeneous disease
consisting of the right-side colon cancer and the left-side
colorectal cancer with different molecular, biological and
clinicopathological properties (80–82).
Consistently, the antitumor immunity activated by HSPs would be
beneficial to prevent peritoneal metastasis of the right-sided
(proximal) colon cancer with highly immunogenic and intensive
lymphocytic infiltration (81,82).
Therefore, the combined use of HSPs inhibitors with hyperthermia
may reduce the clinical benefits of hyperthermia. The present
findings showed that the HITS-GSK3β pathway prevented
hyperthermia-induced cancer cell migration caused by HSP90. One
concern of the present study is that, in certain cancer types, HS
may not induce HITS considering that the endogenous HITS expression
has been reported to be suppressed in various cancer types
(17,20,83,84)
possibly due to epigenetic modification (84–86).
The present data showed that treatment with a GSK3β inhibitor
reinforced the anti-migratory effect via HITS, while other studies
also showed the benefit of GSK3β inhibitors to activate NK cells
and dendritic cells and suppress the immune checkpoint protein PD-1
to enhance CD8+ T cells activity (87–90).
Grassilli et al (91) also
indicated another benefit of GSK3β inhibitors to overcome drug
resistance of p53 null colon carcinoma by inducing necroptosis.
Accordingly, as well as an investigation of the putative roles of
the pathways mediated by HITS and GSK3β in CRC biology as discussed
in the former paragraph, the combination use of GSK3β inhibitors
with hyperthermia would be an aim of our future studies.
Hyperthermia in cancer treatment shows clinical
benefits because of its antitumor effects exerted through several
processes, such as direct heat-induced cell death, induction of
oxidative stress and molecular damage, antitumor immunity boosting,
radio- and chemo-sensitivity enhancement, tumor microenvironment
modifications, and inhibition of epithelial-to-mesenchymal
transition (10–14). While the HSF1-HSPs system, major
cellular responses to HS, has long been considered to have a
promigratory function that leads to cancer progression, the present
study clarified that HITS could counterbalance the migratory
effects caused by major HSPs (15,16),
such as HSP90 in CRC cells. Further investigation is required to
fully understand the molecular mechanisms underlying the antitumor
effects of hyperthermia.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank Dr Keiko Nakao (Saitama Medical
University, Moroyama, Japan) for providing DNA vectors and
technical support.
Funding
This study was supported in part by the Extramural Collaborative
Research Grant of Cancer Research Institute, Kanazawa University.
This study was also supported by a fund from Ageo Central General
Hospital.
Availability of data and materials
The datasets used and/or analyzed during of current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KK conceptualized the study, designed the
experiments, acquired data and wrote the primary manuscript. TD
acquired data and resources, designed the methodology and
critically revised and proofread the manuscript. TM and KS acquired
resources, designed methodology and critically revised and
proofread the manuscript. HN conceptualized the study, acquired
resources, designed methodology and critically revised and
proofread the manuscript. All authors read and approved the final
version of the manuscript. KK and HN confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leporrier J, Maurel J, Chiche L, Bara S,
Segol P and Launoy G: A population-based study of the incidence,
management and prognosis of hepatic metastases from colorectal
cancer. Br J Surg. 93:465–474. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zarour LR, Anand S, Billingsley KG, Bisson
WH, Cercek A, Clarke MF, Coussens LM, Gast CE, Geltzeiler CB,
Hansen L, et al: Colorectal cancer liver metastasis: Evolving
paradigms and future directions. Cell Mol Gastroenterol Hepatol.
3:163–173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dillekås H, Rogers MS and Straume O: Are
90% of deaths from cancer caused by metastases? Cancer Med.
8:5574–5576. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chow FC and Chok KS: Colorectal liver
metastases: An update on multidisciplinary approach. World J
Hepatol. 11:150–172. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ganesh K, Stadler ZK, Cercek A, Mendelsohn
RB, Shia J, Segal NH and Diaz LA Jr: Immunotherapy in colorectal
cancer: rationale, challenges and potential. Nat Rev Gastroenterol
Hepatol. 16:361–375. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Van der Jeught K, Xu HC, Li YJ, Lu XB and
Ji G: Drug resistance and new therapies in colorectal cancer. World
J Gastroenterol. 24:3834–3848. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
George TJ, Franke AJ, Chakravarthy AB, Das
P, Dasari A, El-Rayes BF, Hong TS, Kinsella TJ, Landry JC, Lee JJ,
et al: National Cancer Institute (NCI) state of the science:
Targeted radiosensitizers in colorectal cancer. Cancer.
125:2732–2746. 2019.PubMed/NCBI
|
|
10
|
Hettinga JV, Konings AW and Kampinga HH:
Reduction of cellular cisplatin resistance by hyperthermia-a
review. Int J Hyperthermia. 13:439–457. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yagawa Y, Tanigawa K, Kobayashi Y and
Yamamoto M: Cancer immunity and therapy using hyperthermia with
immunotherapy, radiotherapy, chemotherapy, and surgery. J Cancer
Metastasis Treat. 3:218–230. 2017. View Article : Google Scholar
|
|
12
|
Vassos N and Piso P: Metastatic colorectal
cancer to the peritoneum: Current treatment options. Curr Treat
Options Oncol. 19:492018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dayanc BE, Beachy SH, Ostberg JR and
Repasky EA: Dissecting the role of hyperthermia in natural killer
cell mediated anti-tumor responses. Int J Hyperthermia. 24:41–56.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsan MF and Gao B: Heat shock proteins and
immune system. J Leukoc Biol. 85:905–910. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ciocca DR and Calderwood SK: Heat shock
proteins in cancer: Diagnostic, prognostic, predictive, and
treatment implications. Cell Stress Chaperones. 10:86–103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boroumand N, Saghi H, Avan A, Bahreyni A,
Ryzhikov M, Khazaei M and Hassanian SM: Therapeutic potency of
heat-shock protein-90 pharmacological inhibitors in the treatment
of gastrointestinal cancer, current status and perspectives. J
Pharm Pharmacol. 70:151–158. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakajima H, Ishigaki Y, Xia QS, Ikeda T,
Yoshitake Y, Yonekura H, Nojima T, Tanaka T, Umehara H, Tomosugi N,
et al: Induction of HITS, a newly identified family with sequence
similarity 107 protein (FAM107B), in cancer cells by heat shock
stimulation. Int J Oncol. 37:583–593. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Q, Zhao XY, Bai RZ, Liang SF, Nie CL,
Yuan Z, Wang CT, Wu Y, Chen LJ and Wei YQ: Induction of tumor
inhibition and apoptosis by a candidate tumor suppressor gene DRR1
on 3p21.1. Oncol Rep. 22:1069–1075. 2009.PubMed/NCBI
|
|
19
|
Schmidt MV, Schülke JP, Liebl C, Stiess M,
Avrabos C, Bock J, Wochnik GM, Davies HA, Zimmermann N, Scharf SH,
et al: Tumor suppressor down-regulated in renal cell carcinoma 1
(DRR1) is a stress-induced actin bundling factor that modulates
synaptic efficacy and cognition. Proc Natl Acad Sci USA.
108:17213–17218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakajima H, Koizumi K, Tanaka T, Ishigaki
Y, Yoshitake Y, Yonekura H, Sakuma T, Fukushima T, Umehara H, Ueno
S, et al: Loss of HITS (FAM107B) expression in cancers of multiple
organs: Tissue microarray analysis. Int J Oncol. 41:1347–1357.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Domoto T, Pyko IV, Furuta T, Miyashita K,
Uehara M, Shimasaki T, Nakada M and Minamoto T: Glycogen synthase
kinase-3β is a pivotal mediator of cancer invasion and resistance
to therapy. Cancer Sci. 107:1363–1372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Domoto T, Uehara M, Bolidong D and
Minamoto T: Glycogen synthase kinase 3β in cancer biology and
treatment. Cells. 9:13882020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Beurel E, Grieco SF and Jope RS: Glycogen
synthase kinase-3 (GSK3): Regulation, actions, and diseases.
Pharmacol Ther. 148:114–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Turano M, Costabile V, Cerasuolo A,
Duraturo F, Liccardo R, Delrio P, Pace U, Rega D, Dodaro CA, Milone
M, et al: Characterisation of mesenchymal colon tumour-derived
cells in tumourspheres as a model for colorectal cancer
progression. Int J Oncol. 53:2379–2396. 2018.PubMed/NCBI
|
|
25
|
Kazi A, Xiang S, Yang H, Delitto D,
Trevino J, Jiang RHY, Ayaz M, Lawrence HR, Kennedy P and Sebti SM:
GSK3 suppression upregulates β-catenin and c-Myc to abrogate
KRas-dependent tumors. Nat Commun. 9:51542018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshino Y, Suzuki M, Takahashi H and
Ishioka C: Inhibition of invasion by glycogen synthase kinase-3
beta inhibitors through dysregulation of actin re-organisation via
down-regulation of WAVE2. Biochem Biophys Res Commun. 464:275–280.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chikano Y, Domoto T, Furuta T, Sabit H,
Kitano-Tamura A, Pyko IV, Takino T, Sai Y, Hayashi Y, Sato H, et
al: Glycogen synthase kinase 3β sustains invasion of glioblastoma
via the focal adhesion kinase, Rac1, and c-Jun N-terminal
kinase-mediated pathway. Mol Cancer Ther. 14:564–574. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kitano A, Shimasaki T, Chikano Y, Nakada
M, Hirose M, Higashi T, Ishigaki Y, Endo Y, Takino T, Sato H, et
al: Aberrant glycogen synthase kinase 3β is involved in pancreatic
cancer cell invasion and resistance to therapy. PLoS One.
8:e552892013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shakoori A, Ougolkov A, Yu ZW, Zhang B,
Modarressi MH, Billadeau DD, Mai M, Takahashi Y and Minamoto T:
Deregulated GSK3β activity in colorectal cancer: Its association
with tumor cell survival and proliferation. Biochem Biophys Res
Commun. 334:1365–1373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kawauchi T, Chihama K, Nabeshima Y and
Hoshino M: The in vivo roles of STEF/Tiam1, Rac1 and JNK in
cortical neuronal migration. EMBO J. 22:4190–4201. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gayet J, Zhou XP, Duval A, Rolland S,
Hoang JM, Cottu P and Hamelin R: Extensive characterization of
genetic alterations in a series of human colorectal cancer cell
lines. Oncogene. 20:5025–5032. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahmed D, Eide PW, Eilertsen IA, Danielsen
SA, Eknæs M, Hektoen M, Lind GE and Lothe RA: Epigenetic and
genetic features of 24 colon cancer cell lines. Oncogenesis.
2:e712013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mai W, Miyashita K, Shakoori A, Zhang B,
Yu ZW, Takahashi Y, Motoo Y, Kawakami K and Minamoto T: Detection
of active fraction of GSK3β in cancer cells by nonradioisotopic in
vitro kinase assay. Oncology. 71:297–305. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shakoori A, Mai W, Miyashita K, Yasumoto
K, Takahashi Y, Ooi A, Kawakami K and Minamoto T: Inhibition of
GSK-3β activity attenuates proliferation of human colon cancer
cells in rodents. Cancer Sci. 98:1388–1393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mai W, Kawakami K, Shakoori A, Kyo S,
Miyashita K, Yokoi K, Jin MJ, Shimasaki T, Motoo Y and Minamoto T:
Deregulated glycogen synthase kinase 3β sustains gastrointestinal
cancer cells survival by modulating human telomerase reverse
transcriptase and telomerase. Clin Cancer Res. 15:6810–6819. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Turunen SP, Tatti-Bugaeva O and Lehti K:
Membrane-type matrix metalloproteases as diverse effectors of
cancer progression. Biochim Biophys Acta Mol Cell Res.
1864:1974–1988. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ning Q, Gan YH, Shi RR and Meng JH:
Effects of HDAC4 on IL-1β-induced matrix metalloproteinase
expression regulated partially through the WNT3A/β-catenin pathway.
Chin Med J (Engl). 134:963–970. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tu Y, Tian Y, Wu Y and Cui S: Clinical
significance of heat shock proteins in gastric cancer following
hyperthermia stress: Indications for hyperthermic intraperitoneal
chemoperfusion therapy. Oncol Lett. 15:9385–9391. 2018.PubMed/NCBI
|
|
39
|
Grimmig T, Moll EM, Kloos K, Thumm R,
Moench R, Callies S, Kreckel J, Vetterlein M, Pelz J, Polat B, et
al: Upregulated heat shock proteins after hyperthermic chemotherapy
point to induced cell survival mechanisms in affected tumor cells
from peritoneal carcinomatosis. Cancer Growth Metastasis.
10:11790644177305592017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen JS, Hsu YM, Chen CC, Chen LL, Lee CC
and Huang TS: Secreted heat shock protein 90α induces colorectal
cancer cell invasion through CD91/LRP-1 and NF-κB-mediated integrin
αV expression. J Biol Chem. 285:25458–25466. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song D, Guo M, Xu S, Song X, Bai B, Li Z,
Chen J, An Y, Nie Y, Wu K, et al: HSP90-dependent PUS7
overexpression facilitates the metastasis of colorectal cancer
cells by regulating LASP1 abundance. J Exp Clin Cancer Res.
40:1702021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sims JD, McCready J and Jay DG:
Extracellular heat shock protein (HSP)70 and HSP90α assist in
matrix metalloproteinase-2 activation and breast cancer cell
migration and invasion. PLoS One. 6:e188482011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Guo W, Reigan P, Siegel D, Zirrolli J,
Gustafson D and Ross D: Formation of
17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by
NAD(P)H:quinone oxidoreductase 1: Role of 17-AAG hydroquinone in
heat shock protein 90 inhibition. Cancer Res. 65:10006–10015. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bhat R, Xue Y, Berg S, Hellberg S, Ormö M,
Nilsson Y, Radesäter AC, Jerning E, Markgren PO, Borgegård T, et
al: Structural insights and biological effects of glycogen synthase
kinase 3-specific inhibitor AR-A014418. J Biol Chem.
278:45937–45945. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
John JK, Paraiso KH, Rebecca VW, Cantini
LP, Abel EV, Pagano N, Meggers E, Mathew R, Krepler C, Izumi V, et
al: GSK3β inhibition blocks melanoma cell/host interactions by
downregulating N-cadherin expression and decreasing FAK
phosphorylation. J Invest Dermatol. 132:2818–2827. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu H, Hasan R, Zhang H, Gray J, Williams
D, Miller M, Allen F, Lee V, Kelly T and Zhou GL: Phosphorylation
regulates CAP1 (cyclase-associated protein 1) functions in the
motility and invasion of pancreatic cancer cells. Sci Rep.
9:49252019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhou GL, Zhang H, Wu H, Ghai P and Field
J: Phosphorylation of the cytoskeletal protein CAP1 controls its
association with cofilin and actin. J Cell Sci. 127:5052–5065.
2014.PubMed/NCBI
|
|
48
|
Rom S, Fan S, Reichenbach N, Dykstra H,
Ramirez SH and Persidsky Y: Glycogen synthase kinase 3β inhibition
prevents monocyte migration across brain endothelial cells via
Rac1-GTPase suppression and down-regulation of active integrin
conformation. Am J Pathol. 181:1414–1425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yoeli-Lerner M, Chin YR, Hansen CK and
Toker A: Akt/protein kinase B and glycogen synthase kinase-3β
signaling pathway regulates cell migration through the NFAT1
transcription factor. Mol Cancer Res. 7:425–432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao J, Xu J, Zhao J and Zhang R: EFEMP2
promotes colon cancer cell invasion and growth through the ERK1/2
signaling pathway. Int J Clin Exp Pathol. 12:851–856.
2019.PubMed/NCBI
|
|
51
|
Rath T, Stöckle J, Roderfeld M,
Tschuschner A, Graf J and Roeb E: Matrix metalloproteinase-13 is
regulated by toll-like receptor-9 in colorectal cancer cells and
mediates cellular migration. Oncol Lett. 2:483–488. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nakajima H and Koizumi K: Family with
sequence similarity 107: A family of stress responsive small
proteins with diverse functions in cancer and the nervous system
(Review). Biomed Rep. 2:321–325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kretzschmar A, Schülke JP, Masana M, Dürre
K, Müller MB, Bausch AR and Rein T: The stress-inducible protein
DRR1 exerts distinct effects on actin dynamics. Int J Mol Sci.
19:39932018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo J, Bian Y, Wang Y, Chen L, Yu A and
Sun X: FAM107B is regulated by S100A4 and mediates the effect of
S100A4 on the proliferation and migration of MGC803 gastric cancer
cells. Cell Biol Int. 41:1103–1109. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Allgöwer C, Kretz AL, von Karstedt S,
Wittau M, Henne-Bruns D and Lemke J: Friend or foe: S100 proteins
in cancer. Cancers (Basel). 12:20372020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen M, Bresnick AR and O'Connor KL:
Coupling S100A4 to rhotekin alters Rho signaling output in breast
cancer cells. Oncogene. 32:3754–3764. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huang CY, Wei PL, Chen WY, Chang WC and
Chang YJ: Silencing heat shock protein 27 inhibits the progression
and metastasis of colorectal cancer (CRC) by maintaining the
stability of stromal interaction molecule 1 (STIM1) proteins.
Cells. 7:2622018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lin Y, Peng N, Zhuang H, Zhang D, Wang Y
and Hua ZC: Heat shock proteins HSP70 and MRJ cooperatively
regulate cell adhesion and migration through urokinase receptor.
BMC Cancer. 14:6392014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Moser C, Lang SA, Kainz S, Gaumann A,
Fichtner-Feigl S, Koehl GE, Schlitt HJ, Geissler EK and Stoeltzing
O: Blocking heat shock protein-90 inhibits the invasive properties
and hepatic growth of human colon cancer cells and improves the
efficacy of oxaliplatin in p53-deficient colon cancer tumors in
vivo. Mol Cancer Ther. 6:2868–2878. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hagn F, Lagleder S, Retzlaff M, Rohrberg
J, Demmer O, Richter K, Buchner J and Kessler H: Structural
analysis of the interaction between Hsp90 and the tumor suppressor
protein p53. Nat Struct Mol Biol. 18:1086–1093. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lacey T and Lacey H: Linking hsp90′s role
as an evolutionary capacitator to the development of cancer. Cancer
Treat Res Commun. 28:1004002021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dou F, Chang X and Ma D: Hsp90 maintains
the stability and function of the Tau phosphorylating kinase GSK3β.
Int J Mol Sci. 8:51–60. 2007. View Article : Google Scholar
|
|
63
|
Banz VM, Medová M, Keogh A, Furer C,
Zimmer Y, Candinas D and Stroka D: Hsp90 transcriptionally and
post-translationally regulates the expression of NDRG1 and
maintains the stability of its modifying kinase GSK3β. Biochim
Biophys Acta. 1793:1597–1603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tang W, Wu Y, Qi X, Yu R, Lu Z, Chen A,
Fan X and Li J: PGK1-coupled HSP90 stabilizes GSK3β expression to
regulate the stemness of breast cancer stem cells. Cancer Biol Med.
19:486–503. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Muller P, Ruckova E, Halada P, Coates PJ,
Hrstka R, Lane DP and Vojtesek B: C-terminal phosphorylation of
Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP
and HOP to determine cellular protein folding/degradation balances.
Oncogene. 32:3101–3110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang C, Li S and Zhao Z: β-Elemene
promotes apoptosis induced by hyperthermia via inhibiting HSP70.
Dis Markers. 2022:73130262022.PubMed/NCBI
|
|
67
|
Schwock J, Dhani N, Cao MP, Zheng J,
Clarkson R, Radulovich N, Navab R, Horn LC and Hedley DW: Targeting
focal adhesion kinase with dominant-negative FRNK or Hsp90
inhibitor 17-DMAG suppresses tumor growth and metastasis of SiHa
cervical xenografts. Cancer Res. 69:4750–4759. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Taiyab A and Rao ChM: HSP90 modulates
actin dynamics: Inhibition of HSP90 leads to decreased cell
motility and impairs invasion. Biochim Biophys Acta. 1813:213–221.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu Z, Li H, He L, Xiang Y, Tian C, Li C,
Tan P, Jing J, Tian Y, Du L, et al: Discovery of small-molecule
inhibitors of the HSP90-calcineurin-NFAT pathway against
glioblastoma. Cell Chem Biol. 26:352–365.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lu C, Chen D, Zhang Z, Fang F, Wu Y, Luo L
and Yin Z: Heat shock protein 90 regulates the stability of c-Jun
in HEK293 Cells. Mol Cells. 24:210–214. 2007.PubMed/NCBI
|
|
71
|
Stellas D, El Hamidieh A and Patsavoudi E:
Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by
disrupting their interaction with extracellular HSP90 and inhibits
formation of metastatic breast cancer cell deposits. BMC Cell Biol.
11:512010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jia D, Rao W, Wang C, Jin C, Wang S, Chen
D, Zhang M, Guo J, Chang Z and Liu J: Inhibition of B16 murine
melanoma metastasis and enhancement of immunity by fever-range
whole body hyperthermia. Int J Hyperthermia. 27:275–285. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Byun YH, Gwak HS, Kwon JW, Song MK, Shin
SH, Jo YH, Yoo H and Lee SH: Local recurrence of brain metastasis
reduced by intra-operative hyperthermia treatment. Int J
Hyperthermia. 35:168–175. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhao J, Lv Y, Cai Y, Wei W, Yin C, Wang X,
Hao Z, Shen C and Wang H: Hyperthermic carbon dioxide
pneumoperitoneum reinforces the inhibition of 5-FU on the
proliferation and invasion of colon cancer. Oncol Rep. 37:492–500.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kumar S, Stokes J III, Singh UP, Scissum
Gunn K, Acharya A, Manne U and Mishra M: Targeting HSP70: A
possible therapy for cancer. Cancer Lett. 374:156–166. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kryeziu K, Bruun J, Guren TK, Sveen A and
Lothe RA: Combination therapies with HSP90 inhibitors against
colorectal cancer. Biochim Biophys Acta Rev Cancer. 1871:240–247.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhou L, Zhang M, Fu Q, Li J and Sun H:
Targeted near infrared hyperthermia combined with immune
stimulation for optimized therapeutic efficacy in thyroid cancer
treatment. Oncotarget. 7:6878–6890. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Vriend LEM, van den Tempel N, Oei AL,
L'Acosta M, Pieterson FJ, Franken NAP, Kanaar R and Krawczyk PM:
Boosting the effects of hyperthermia-based anticancer treatments by
HSP90 inhibition. Oncotarget. 8:97490–97503. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Daunys S, Matulis D and Petrikaitė V:
Synergistic activity of HSP90 inhibitors and anticancer agents in
pancreatic cancer cell cultures. Sci Rep. 9:161772019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Baran B, Mert Ozupek N, Yerli Tetik N,
Acar E, Bekcioglu O and Baskin Y: Difference between left-sided and
right-sided colorectal cancer: A focused review of literature.
Gastroenterology Res. 11:264–273. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
de Vries NL, Swets M, Vahrmeijer AL,
Hokland M and Kuppen PJ: The immunogenicity of colorectal cancer in
relation to tumor development and treatment. Int J Mol Sci.
17:10302016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Picard E, Verschoor CP, Ma GW and Pawelec
G: Relationships between immune landscapes, genetic subtypes and
responses to immunotherapy in colorectal cancer. Front Immunol.
11:3692020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang H, Du Y, Wang Z, Lou R, Wu J and
Feng J: Integrated analysis of oncogenic networks in colorectal
cancer identifies GUCA2A as a molecular marker. Biochem Res Int.
2019:64694202019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mah WC, Thurnherr T, Chow PK, Chung AY,
Ooi LL, Toh HC, Teh BT, Saunthararajah Y and Lee CG: Methylation
profiles reveal distinct subgroup of hepatocellular carcinoma
patients with poor prognosis. PLoS One. 9:e1041582014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Song MA, Tiirikainen M, Kwee S, Okimoto G,
Yu H and Wong LL: Elucidating the landscape of aberrant DNA
methylation in hepatocellular carcinoma. PLoS One. 8:e557612013.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kan S, Chai S, Chen W and Yu B: DNA
methylation profiling identifies potentially significant
epigenetically-regulated genes in glioblastoma multiforme. Oncol
Lett. 18:1679–1688. 2019.PubMed/NCBI
|
|
87
|
Parameswaran R, Ramakrishnan P, Moreton
SA, Xia Z, Hou Y, Lee DA, Gupta K, deLima M, Beck RC and Wald DN:
Repression of GSK3 restores NK cell cytotoxicity in AML patients.
Nat Commun. 7:111542016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cichocki F, Valamehr B, Bjordahl R, Zhang
B, Rezner B, Rogers P, Gaidarova S, Moreno S, Tuininga K, Dougherty
P, et al: GSK3 inhibition drives maturation of NK cells and
enhances their antitumor activity. Cancer Res. 77:5664–5675. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Noh KT, Son KH, Jung ID, Kang TH, Choi CH
and Park YM: Glycogen synthase kinase-3β (GSK-3β) inhibition
enhances dendritic cell-based cancer vaccine potency via
suppression of interferon-γ-induced indoleamine 2,3-dioxygenase
expression. J Biol Chem. 290:12394–12402. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Taylor A, Harker JA, Chanthong K,
Stevenson PG, Zuniga EI and Rudd CE: Glycogen synthase kinase 3
inactivation drives T-bet-mediated downregulation of co-receptor
PD-1 to enhance CD8(+) cytolytic T cell responses. Immunity.
44:274–286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Grassilli E, Narloch R, Federzoni E,
Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE,
Bonin S, et al: Inhibition of GSK3B bypass drug resistance of
p53-null colon carcinomas by enabling necroptosis in response to
chemotherapy. Clin Cancer Res. 19:3820–3831. 2013. View Article : Google Scholar : PubMed/NCBI
|