Numerous studies have been conducted recently on the
characteristics of leader cells in vivo and in vitro,
demonstrating their important role in collective invasion (11-13).
Furthermore, follower cells, which comprise the majority of the
cell groups involved in collective invasion, have received
increasing attention. Optimized spatiotemporal genomic and cellular
analysis (SaGA) can specifically target, extract, separate and
amplify leader and follower cells from a 3D microenvironment
(11). The separated follower
cells proliferate rapidly and divide frequently but are not highly
invasive, whereas the separated leader cells are highly invasive,
and divide and proliferate slowly (12,13).
At the leading edge, specialized leader cells exert traction force
on the follower cells (14). In
addition to being pulled along by leader cells, follower cells are
actively involved in selecting a specific direction of travel by
extending protrusions underneath leader cells; these protrusions
are known as cryptic lamellipodia (c-lamellipodia) (15). Since they typically possess
c-lamellipodia, which are typically smaller and exhibit fewer
adhesions with the extracellular matrix (ECM), the follower cells
have less interactions with the ECM and exert less traction
(16). The biomechanics generated
by follower cells through c-lamellipodia facilitates collective
invasion, allowing follower cells to invade as a group. Death of
the leader cell causes the follower cells to cease migrating in the
same direction, leading to random, slow movement of cells and an
end to collective migration (17).
Additionally, follower cells help the leader cells initiate
appropriate polarization, strengthen leadership of the cells and
ensure that sufficient leader cells are available to lead the
invasion. This is achieved through the utilization of a multitude
of signalling molecules, including chemotactic factors and physical
contacts, whereby follower cells migrate in the wake of the leader
cells and constitute a significant portion of the multicellular
cluster. A variety of strategies are employed by follower cells to
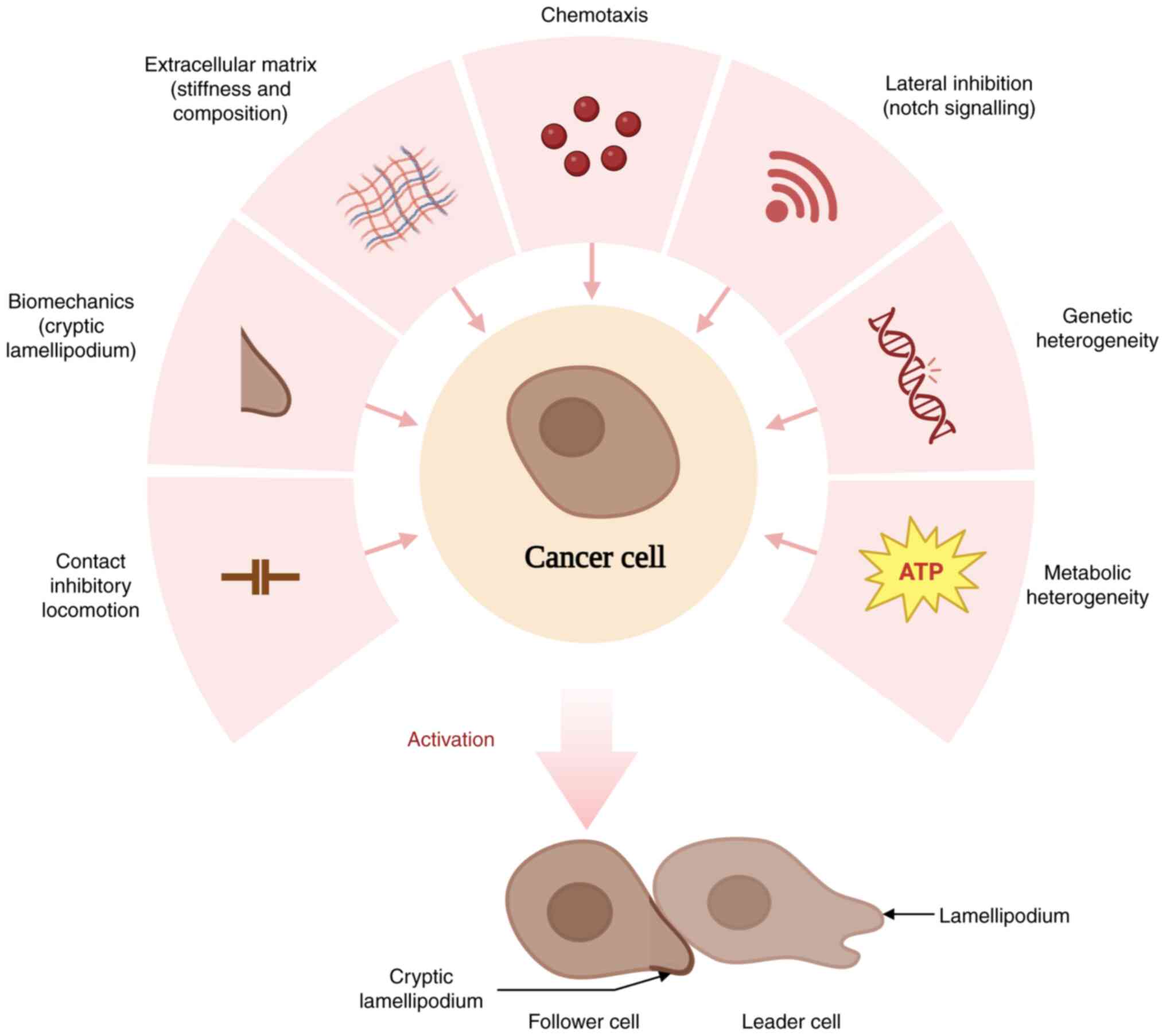
increase their invasive capacity, including contact inhibition
locomotion (CIL), biomechanics, matrix remodelling, chemotaxis,
lateral inhibition and exhibiting genetic and metabolic
heterogeneity (18-21) (Fig.
1). Elucidation of the role of follower cells in collective
invasion may identify new molecular targets for cancer treatment
and intervention.
CIL is a multifaceted procedure that changes cell
movement when one cell collides with another cell, and
inappropriate regulation of CIL may promote the spread of cancerous
cells. Normal cells exhibit strong CIL when adjacent cells come
into contact with each other, helping to maintain proper tissue
architecture and prevent overgrowth. This process is achieved
through the activation of proteins involved in cell adhesion,
cytoskeletal rearrangement and signalling pathways that suppress
cell proliferation and migration (22,23).
However, cancerous cells are defective in these pathways and
display weaker or disrupted CIL, which may be caused by mutations
or alterations in genes that regulate the aforementioned processes
(22,23). One of the key differences in CIL
mechanisms between normal cells and cancerous cells is the loss of
stable cell-cell adhesions in cancerous cells (24). Cancer cells decrease Ras homolog
family member A (RhoA) activation at the front of the cell cluster,
resulting in weaker intercellular adhesion and increased cell
migration (10). As a result,
cancerous cells can exhibit uncontrolled cell motility and tissue
invasion.
CIL between cells of the same type is known as
homotypic CIL, exhibited by both normal and tumour cells. By
contrast, CIL between cells of different types is known as
heterotypic CIL, which is often lost by tumour cells when they
encounter normal cells. Homotypic CIL is established between leader
and follower cells for collective migration. Initial contact
followed by varying degrees of protrusion inhibition at the contact
site enables the leader and follower cells to form protrusions
towards the basement membrane in the direction of the movement,
thus facilitating the directed migration of the cell cluster
(23) (Fig. 2A). Certain types of sarcoma, such
as the S180 and BAS56 cell lines as well as melanoma cells, with
inappropriately regulated CIL acquire the capacity for collective
motility in the cancer cell population, and they invade areas
occupied by other types of cells (25). These CIL properties can promote
tumour aggressiveness by preferentially directing cancer cells into
the stromal environment in the form of clusters. During this
process, leader-follower intercellular CIL is induced by
intracellular signalling and mechanical coupling.
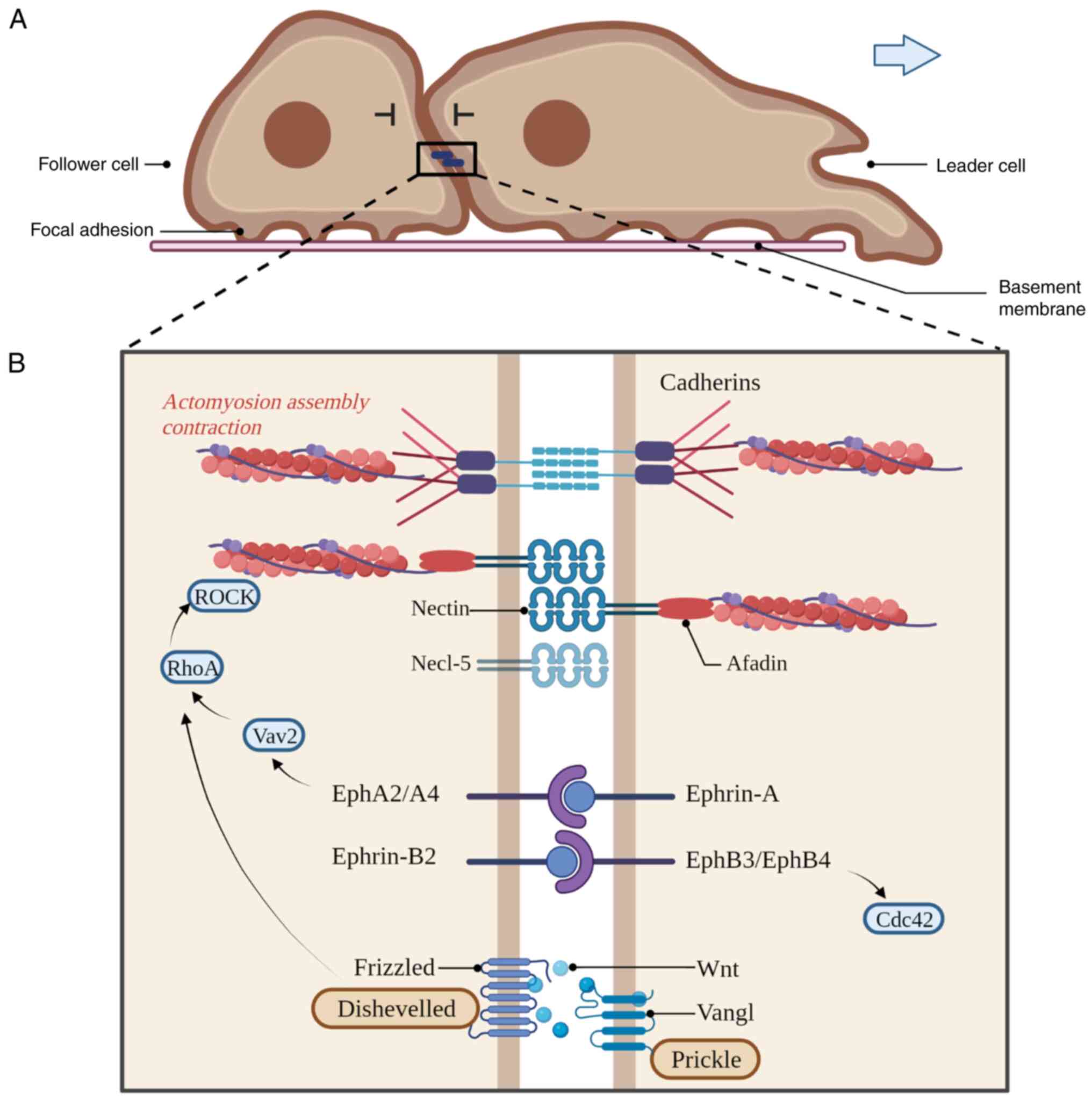
Contacts between leader and follower cells are
dynamic and continuous during collective invasion, which is the
foundation of CIL. The follower cells follow the leader cells very
precisely through cell-cell contact, exhibiting the characteristics
of CIL (26-28). A different distribution of adhesion
proteins has been observed between leader and follower cells when
exposed to different levels of extracellular signals (26-28).
The membrane proteins involved in the interaction between cells are
E-cadherin and N-cadherin-coordinated adherens junctions (AJs),
Ephrins/Eph receptors, Ig-superfamily proteins and planar cell
polarity (PCP) members (Fig. 2B)
(29). In addition to forming
mechanical bonds between cells, membrane proteins also function as
ligands/receptors that regulate intracellular signals, such as for
determining cell polarity and cytoskeletal dynamics. Moreover,
intercellular AJs combined with CIL prevent follower cell perimeter
integrins from contacting the ECM, resulting in common adhesion
structures and protrusions in any direction (30). As a result, this process
significantly increases the efficiency of collective invasion.
CIL involves the establishment of transient adhesion
sites between cells via cadherins. Leader cells exhibit
asymmetrical AJs, with integrin-based focal adhesions (FAs) at
their extending fronts and cadherin-based AJs at the intercellular
junctions on the trailing edges (17). However, follower cells possess
symmetrical cadherin-based AJs that inhibit protrusion formation
throughout their periphery (17,31).
During the CIL of different cell types, different types of
cadherins are found at the cell-cell contacts, generally at AJs,
such as E-cadherin, N-cadherin and cadherin11 (23,32).
The importance of E-cadherin for CIL in particular has been
demonstrated. A high level of E-cadherin expression within follower
cells is related to an epithelial phenotype and maintains
intercellular CIL (33).
E-cadherin is therefore generally considered an influential
molecule in maintaining epithelial differentiation and
counteracting cancer invasion (34). This finding may explain why the
loss of E-cadherin contributes to migration in vitro, and
its loss may adversely affect breast cancer metastasis in
vivo (35).
The differential cadherin expression in leader and
follower cells indicates that cadherin conversion might be
associated with CIL acquisition (36,37).
Through this cadherin modulation, cells can establish distinct
adhesion properties that enable the coordination of collective cell
migration (36). The ideal ratio
between E-cadherin and N-cadherin in follower and leader cell
populations remains unknown and this ratio may vary depending on
cell type and context. In general, E-cadherin is the cell-cell
adhesion molecule most abundant in adherent cells. As such, in
leader cells, which are highly mobile, the ratio of E-cadherin to
N-cadherin is low (36,37). Conversely, in a follower cell,
where adhesion activity is needed to maintain and guide its
migration, the ratio of E-cadherin to N-cadherin is higher
(38). Moreover, the ideal ratio
between follower and leader cells depends on the tumour
environment. For example, a higher ratio of N-cadherin has been
reported to enhance cell migration, a process that involves
expansive migration along tissues, environments and individual
cells (36,37). By contrast, in developing
epithelial layers, a higher ratio of E-cadherin to N-cadherin has
been found to assist strong cell-cell adhesion, allowing cells to
remain closely packed and form an epithelium (38).
When N-cadherin or E-cadherin expression is knocked
down, the number of multicellular invasion chains are greatly
decreased due to disrupted cell-cell junctions (38). Furthermore, in collective invasions
of cancer cells, e-cadherin is diminished as a result of partial
EMT, which results in the reprogramming that leads to the
destabilization of cell-cell junctions and leads to increased
number of invasive and metastatic cancer cells (24). As neural crest (NC) and cancer
cells undergo EMT, the switch from E-cadherin to N-cadherin
contributes to the invasiveness of CIL by promoting cell detachment
and enhancing migratory capacity (23,36,39).
CAFs and mesenchymal leader cells are capable of guiding follower
cells via heterotypic N- and E-cadherin interactions in
vitro and in vivo (40,41).
In addition, M2 macrophage leader cells isolated from an infection
and inflammation mouse model were found to express E-cadherin
(42). This observation suggests
that E-cadherin may be involved in homotypic (between M2
macrophages) and heterotypic (between different cell types)
interactions, possibly mediated by IL-4 and polyamine-induced
E-cadherin/catenin complexes (42). However, it is unclear whether
E-cadherin plays a role in collective tumour invasion dominated by
tumour-associated macrophages through CIL.
In addition to cadherin-based AJs, other receptor
families are involved in establishing initial cell contact during
CIL. Studies have found that nectin forms AJs before cadherin forms
intercellular adhesion between leader and follower cells in many
cultured cell lines, such as Madin-Darby Canine Kidney and MCF-7
cells (43,44). In addition, the removal of Necl-5
from the cell surface through endocytosis inhibits Ras-mediated
cell proliferation signalling and contributes to the induction of
CIL via the phosphorylation of sprouty2 by Src, as leader cells and
follower cells establish intercellular contact (45). Notably, a major function of
nectin-like molecule-5 (Necl-5) is to extend protrusions at the
leading edge and generate traction in the direction of collective
motility, which ultimately promotes cell invasion (46). Invasion and metastasis of cancer
cells may also be influenced by the upregulation of Necl-5
(47,48).
An Eph receptor is a tyrosine kinase receptor that
binds transmembrane ligands on adjacent cells, and bidirectional
signalling resulting from Eph-ephrin interactions can lead to
adhesion (49). The binding of
ephrin-A ligands to EphA2 and EphA4 receptors leads to RhoA
activation, contributing to homotypic CIL. As a result of RhoA
activation at cell contact, membrane protrusion collapses and cell
polarity is altered, resulting in directional migration (50). The failure of CIL depends on the
activation of EphB3 and EphB4 by ephrin-B2 at the contact site in
follower cells (50). However, the
cell migration response is regulated by the ratio of
ephrin-A/ephrin-B2 in follower cells, which determines whether
cancer cells exhibit CIL (Fig. 2B)
(50,51). Moreover, homotypic CIL is triggered
by EphA-Rho-Rho kinase (ROCK) signalling in prostate cancer cells
when two cells come into contact (50).
The contact between leader and follower cells
triggers the transmission of CIL to the migrating population
through Wnt/PCP signalling, which in turn activates RhoA (52-54).
The leader and follower cell populations of the Wnt/PCP core
component complex are asymmetrically localized, which regulates the
collective invasion of cancer, at least in part (55).
In addition to the leader-follower cell contact, the
actin cytoskeleton also plays a critical role in regulating CIL,
cell morphology and polarity. In CIL, nectin-based cell-cell
contact reorganizes the actin cytoskeleton and modulates cell
polarization (28). Protrusions
are typically inhibited through actin-mediated contraction at the
contact site. This sudden contraction also causes polarization of
cancer cells at the leading edge. In CIL, the initial contact of
cell surface proteins may regulate Rho GTPases. Specifically,
cadherin activates intracellular signals, such as RhoA and
Ena/vasodilator-stimulated phosphoprotein (VASP), which regulate
cell polarity and cytoskeleton dynamics (56). RhoA is located at the cell contact
site and induces stress fibre formation (57). In addition to inhibiting
protrusions at the site of contact, cadherin junctions also exert
local control over the expression and activity of Rac and
actin-related protein-2/3 (ARP2/3) (58,59).
Leader cells contain Rac, integrin β1 and PI3K proteins at their
leading edge, whereas the follower cells lack these proteins at
their leading edge, and blocking these proteins impairs the
movement of both types of cells (17). By binding and inhibiting RhoA, p120
regulates the cadherin-actin cytoskeleton through indirect
activation of Rac1 and Cdc42 via Vav2, promoting the formation of
protrusions and polarization (60-62).
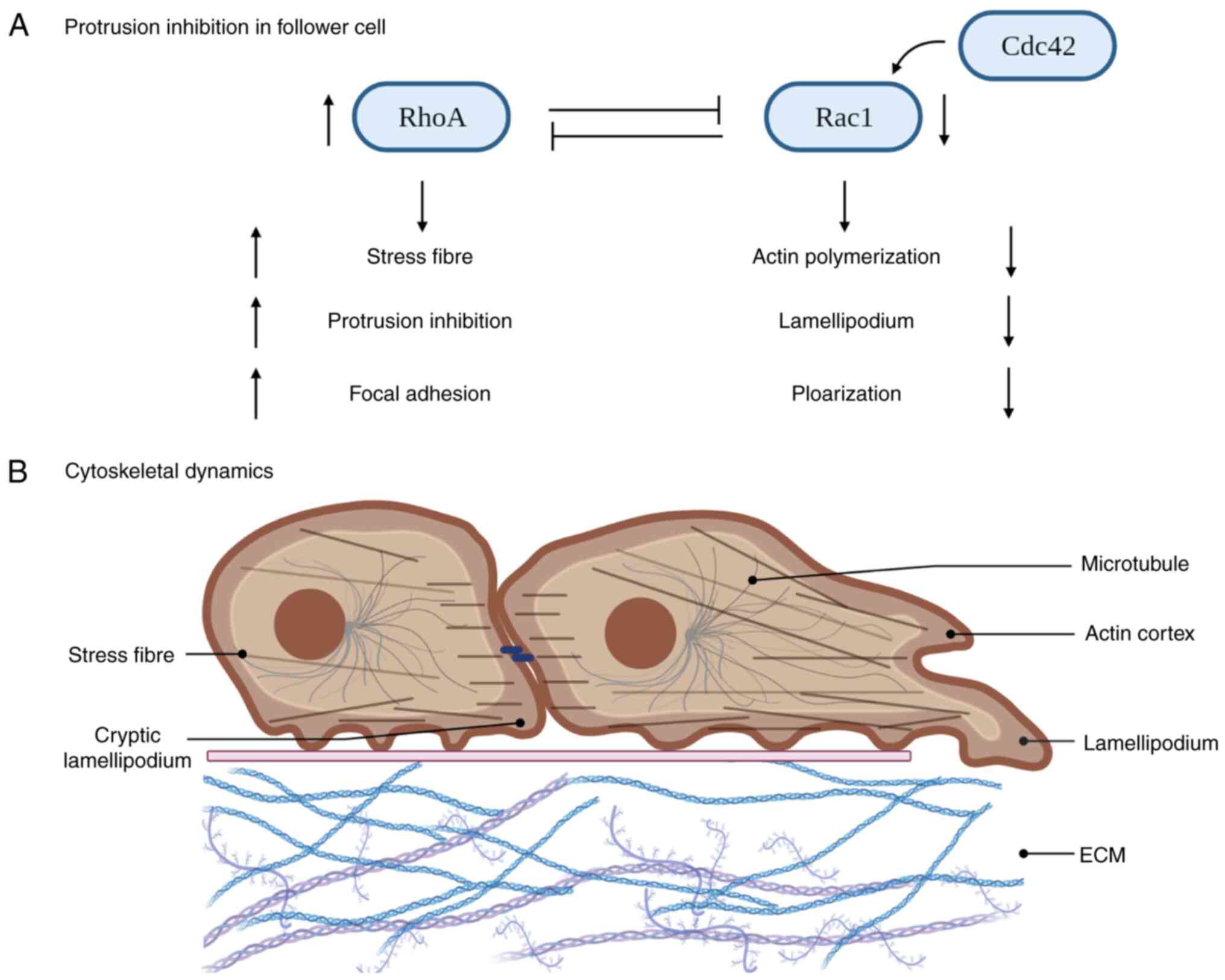
Rac1 and RhoA participate in the process of establishing front-rear
polarity through mutually antagonistic interactions (63). Activated RhoA suppresses Rac1
activity, preventing excessive protrusion at the leading edge and
ensuring proper cell adhesion. Cdc42 controls actomyosin
arrangement to generate the force necessary for follower cells to
follow the tracks of leader cells (63). Furthermore, Cdc42 is able to
activate Rac1, thereby contributing to the generation and
maintenance of cellular protrusions at the leading edge (63). The leading edge involves the
engagement of all three GTPases (Rac1, Rhoa and Cdc42), working
together to control and regulate key aspects of cell migration
(64). At the rear of leader cells
and among follower cells, Rho and related proteins control actin
contractility, resulting in the collapse of protrusions in response
to cell contact (Fig. 3A)
(65).
In addition, Rho GTPases regulate actomyosin
contractility through F-actin polymerization and myosin light chain
phosphorylation in both follower and leader cells (66). The carboxy terminus of discoidin
domain receptor 1 (DDR1) is suggested to regulate cell polarity by
recognizing the PDZ domains of Par3/Par6 (67). The DDR1/Par3/Par6 complex between
follower cells inhibits ROCK-mediated actomyosin contraction by
controlling RhoE recruitment to cell-cell interfaces. After
depletion of DDR1, Par3 or Par6, actomyosin contractability
increases, cohesion is lost and collective cell invasion is
defective (68). By contrast,
cancerous cells invade collectively due to a reduction in
actomyosin contractility that is controlled by DDR1 at the
intercellular junction between follower cells and leader cells
(54,68). After knockdown of either Cdc42 or
both Cdc42-binding protein kinases related to myotonic dystrophy
kinase isoforms (MRCKα and MRCKβ), the localisation and
phosphorylation of myosin light chains at the cortex is
significantly disrupted, affecting the invasion ability of follower
cells (10). As a result,
signalling events can be activated in a context-specific manner at
specific subcellular sites with precise kinetics (69).
Actin cytoskeletons between leader and follower
cells are connected by tight junctions, such as junctional adhesion
molecules-a (JAM-A) (70). The
downstream effects of Src are activated when JAM-A is deleted. This
activation leads to the activation of various proteins, including
extracellular signal-regulated kinase 1/2 (ERK1/2), Abi1, and
paxillin. Additionally, the activity of Rac1 is also increased at
the cell-cell contact site when JAM-A is deleted (71). As demonstrated in a study, a
follower cell lacking JAM-A also migrated more quickly and was
incapable of stopping when it collided with other cells (71). Consequently, CIL is severely
impaired in the absence of JAM-A.
Follower cells undergo morphological changes as well
as changes in their actin cytoskeleton (Fig. 3B) (72), which is maintained by the actin
cortex, located on the side and back of the plasma membrane. Myosin
II-dependent contractions and the formation of stress fibres
coordinate the CIL response of colliding cells (73). A tight collaboration between actin
and microtubule dynamics is observed in vivo during cell
polarity and CIL (73,74). Migration of follower and leader
cells may be initiated by retraction at the back of the cell
(22). A major motor of the actin
cytoskeleton is myosin II, which is activated within the cell body
and at the rear of the cell prior to a spatial bias in actin
polymerization at the leading edge of the cell (22). In contrast to normal cells,
malignant cells have a reduction in the absolute amount of F-actin
(75). Flowing actin networks act
as mechanotransmitters, providing tactile communication of CIL
between cells, and transient stress fibres are formed as a result
of the coupling of colliding actin networks (73). Transient stress fibres are also
formed when protrusion tension increases, which facilitates cell
migration. The tubulin of normal cells is extensively tyrosinated,
whereas the tubulin of cancer cells is often de-tyrosinated, as
observed in breast cancer tissues with poor prognosis (76). Microtubules can be linked to stress
fibres by an actin-microtubule crosslinker. These structures align
themselves between colliding cells during CIL based on the path of
least resistance within the actin network (73). Rac and Cdc42 can stabilize
microtubules, which inhibit the activity of stathmin (77). During CIL, Rac and Cdc42 activation
maintain the direction of polarization and migration by controlling
microtubule capture at the leading edge of the cell. Par3 can
prevent the activation of Rac1 at the contact between cells and
contribute to microtubule collapse (78). Migration may disrupt cell-cell
adhesion by releasing tension generated from engagement of the
actin-clutch, and tension may cause damage to microtubule bundles
or actin stress fibres (79,80).
Overall, the investigation of microtubule dynamics and their
relationship with actin stress fibres and signalling molecules
provides valuable insights into the mechanisms driving collective
invasion of tumor cells.
Follower cells migrate through a dynamic
cell-autonomous process by sending actin-dependent c-lamellipodia
underneath the cells in front of them (81). c-lamellipodia are formed by
adhesion proteins, such as wave and ARP2/3 complexes (82). These c-lamellipodia sporadically
grow around E-cadherin-based AJs in adenocarcinoma-derived
epithelial cells and tend to grow at junctions with mechanically
weak surfaces (82). AJs also
facilitate c-lamellipodia formation by recruiting actin regulators,
which allows follower cells to migrate in an orderly fashion.
C-lamellipodia growth can be uncontrolled when AJs are disrupted by
the removal of αE-catenin, which further results in myosin II
activation and contraction of actomyosin cables associated with AJs
(82). CIL and c-lamellipodia
formation at cell-cell contacts appear to contradict each other.
This contradiction arises due to the fact that c-lamellipodia
formation is commonly associated with cell migration, while CIL
involves the suppression of protrusion. A detailed examination of
follower cells, however, revealed that not all follower cells
strictly follow the CIL restrictions (82). In other words, follower cells
located away from the leading edge may form c-lamellipodia on their
basal surface, which is thought to exert small traction forces
(29). Overall, traction and
c-lamellipodia are less pronounced in follower cells. As WAVE and
ARP2/3 complexes are distributed along the AJs, inhibiting them has
the double effect of preventing the emergence of c-lamellipodia and
preventing follower cells from trailing leader cells (82).
When leader cells die, the c-lamellipodia of nearby
follower cells expand and exert substantial force, converting them
into new leader cells and continuing collective cell migration
(83). Breast cancer cell
invasion, endothelial cell budding and epithelial tracheal
branching are all instances in which leader cells appear
transiently and are replaced by follower cells (84-89).
To ensure that sufficiently qualified leader cells are present at
the front of the collective invasion, follower cells gradually
acquire the phenotype of leader cells when invading the complex
microenvironment. The emerging actin cable joins the follower cell
and two neighbouring leader cells together. After the follower cell
has advanced to the leader cell via the contractile force along the
actin cable, the original cable between them is interrupted and a
thin extension from the new leader cell stretches to a tear
(90). This distinct reconnection
of the actomyosin cable between the follower cell and the
neighbouring leader cell provides further insight into the
mechanism of collective invasion.
A previous study using monolayer stress microscopy
experiments demonstrated that mechanical interactions between
follower cells determine the emergence of leader cells (91). Generally, follower cells decide who
becomes leader cells, not vice versa (92). Before collective invasion, follower
cells generate local forces that can be transmitted to future
leader cells and can be used to pull on them. This traction occurs
before leader cells are formed. In a migrating follower cell, an
actin network assembles into a branch at the leading edge and
protrudes, extends and guides the cell, while an actin bundle near
the trailing edge provides the force that constricts the cell
posteriorly and propels it forwards (93). In response to this force, future
leader cells polarize and form protrusions. After the leader cell
forms, the leader cell pulls on the follower cell via actin
contraction, and this cell continues to migrate, causing the next
cell to be pulled and to transmit further directional signals
(93). In addition, leader cells
produce higher contractile capacity than follower cells (19,94).
Depending on the length to which forces can be transmitted, the
size of a cell group following leader cells may vary (91).
By interacting with its neighbours, a follower cell
experiences a more substantial cumulative force. As a result of
cell-cell adhesion proteins being distributed differently,
actomyosin cytoskeleton contraction is heterogeneous at cell-cell
junctions (27,95,96).
Cellular stresses equilibrate traction forces, which are
transmitted mainly by the cytoskeleton and intercellular junctions.
Stress builds up throughout the migrating tissues and becomes more
intense as the distance from the leading edge increases. It is also
produced spontaneously in possible follower cells by the formation
of c-lamellipodia (97). The
stress field of a cohesive cell monolayer can also be estimated
from traction force data. A substantial amount of tension builds
during collective cell migration in expanding monolayers, as
cell-cell junction stresses increase from edge to centre (98,99).
Two actin skeletons between cells are connected by
cadherin, which form an intercellular force chain. Changes in actin
contractility therefore affect cadherin tension (27). Mechanical forces may be converted
into biochemical responses through molecular effects (100). Actin-regulating proteins, such as
vinculin, formins, affadin, VASP, Zyxin and Testin, are recruited
to cadherin junctions when tension is increased (41,101-103). P-cadherin, which forms
associations with catenins, mediates force transmission between
cells (104). The magnitude of
intercellular tension is predicted by P-cadherin, whereas its
build-up rate is predicted by E-cadherin. There is a competitive
relationship between P-cadherin and E-cadherin during the response
to a mechanotransduction pathway involving vinculin. When the level
of E-cadherin is decreased, P-cadherin replaces its function to
regulate tension, preventing a decrease in intercellular tension
(104). P-cadherin-dependent
collective cell movement is induced by Cdc42 through the regulation
of intercellular stresses and traction force polarization (105). An acute stimulus by RhoA or an
exogenous pulling force stimulates rapid AJ growth (106). E-cadherin expression in follower
cells regulates intercellular mechanics through AJs, and these
junctions stiffen cell aggregates and facilitate the transmission
of traction forces (107-109). Via mechanical stimulation of
E-cadherin, EGFR activates PI3K, which contributes to integrin
adhesion and improves myosin II-dependent cell contractility in
breast carcinoma (110). In
conjunction with these force responses, cytoskeletal remodelling
and cortical stiffening are affected in junction-proximal regions,
which may influence membrane protrusions and their extent and
orientation (111). Increasing
tension on E-cadherin enhances phosphorylation of β-catenin,
lowering its stability at E-cadherin junctions and driving
transcription to promote mesenchymal and basal fates (112-114).
The waves of ERK activation propagate through the
leader and follower cells, linking them together and coordinating
their behaviours (115,116). The pulling forces from leader
cells can stretch the follower cells and activate the EGFR-ERK
pathway, which then generates contractile forces on the rear side
of the follower cells via ROCK signalling (115). ERK activation suppresses traction
forces while activating contractile forces through the accumulation
of F-actin in cell-cell contacts (116). It is possible that the impaired
traction caused by the activation of ERK is due to the breakdown of
actin stress fibres associated with FA, leading to the turnover of
FA (116). The cells, via FA
turnover, can contract efficiently upon contractile force
generation, and the traction force gradually decreases between the
follower cells. Consequently, collective invasion occurs through
sustained traction forces, cell polarity and ERK activation.
Additionally, the negative feedback loop of
Merlin-Rac interaction contributes to collective movement (117). Merlin, a tumour suppressor
protein located at cell junctions in static monolayers, can
translate the intercellular tension between leader and follower
cells into molecular signals (117). Merlin is mainly restricted to
cell junctions in follower cells by cell-cell tension but
accumulates in the cytoplasm of leader cells due to high traction
forces. Cell-cell tension induces Merlin to delocalize from cell
junctions to the cytoplasm, where it coordinates Rac1 polarization.
Rac1 polarization induces the growth of branching actin filaments
and the formation of c-lamellipodium, which facilitates the
migration of follower cells towards leader cells (117). The polarization of Rac1
activation in leader cells may stabilize merlin at the
intercellular junctions by reducing Rac1 activity at the rear of
leader cells and between follower cells (117). Aside from strengthening adhesion,
Merlin expression in follower cells also enables these cells to
pull and attract neighbours (118). Therefore, Merlin-Rac maintains
both leader and follower cell functions in collective cell
invasion.
The ECM, offering both a biochemical and
biomechanical context, plays a crucial role in cancer progression
by regulating the ability of cancer cells to cross its barrier. By
secreting diverse profibrotic growth factors and inflammatory
factors, follower cells are primarily responsible for recruiting
and activating stromal cells within the tumour microenvironment
(119). In response to
tumour-derived activation factors, stromal cells differentiate into
CAFs, and CAFs act as myofibroblasts to reconstruct the ECM to
facilitate tumour invasion (120-122). Follower cells themselves have
also been shown to express altered ECM components, such as collagen
I and III, and ECM-modifying enzymes (123-125). Lysyl oxidases are secreted by
leader and follower cells to crosslink collagen fibres (126). Crosslinked collagen stiffens the
ECM, leading to integrin-dependent invasive behaviour. Due to
increased ECM stiffness, force-loading rates at FAs change,
resulting in the stretching of talin1 and vinculin recruitment
(127). FA kinase (FAK), RhoA and
Src are activated as a result of this, increasing the contractility
of follower cells.
To regulate follower cell behaviour, integrins
transduce signals from the ECM by assembling FAs, thereby
stimulating follower cell cytoskeletal remodelling (128). Integrins act as major receptors
for ECM molecules during cancer metastasis (129). The activation of integrins and
downstream mechanotransduction adapters, such as p130CAS, occurs
when mechanical tension is increased (130). As a result, there is an increase
in FA and actin stress fibre formation in follower cells (130,131). In cancer, actin-rich protrusions
attach to ECM molecules bound to integrins and to contractile
structures within follower cells, causing the basement membrane to
breach without protein hydrolysis (132). Moreover, follower cells can exert
force on ECM networks, affect the ECM architecture reversibly or
permanently and enhance stiffness and ligand density locally
(132-134). A growing body of evidence
indicates that the ECM is significantly stiffer in this state,
compared with its normal state, indicating abnormalities in its
composition and structure (135,136). The ECM is undoubtedly an
important component of the tumour microenvironment, not only
promoting malignancy but also regulating tumour invasion (20,135,136).
Follower cells may exhibit lower levels of durotaxis
and matrix metalloproteinase (MMP) secretion than leader cells, but
still play a critical role in the collective invasive process
(137,138). Durotaxis is the directed
migration of cells towards regions of high mechanical resistance.
Moreover, cells migrate towards the 'optimal matrix stiffness'
region, where they can generate the greatest traction, and when in
a region above the optimal matrix stiffness, cells show an apparent
tendency to migrate towards softer regions (138,139). By undergoing durotaxis, the
follower cells are able to move towards regions of high mechanical
resistance and utilize secreted MMPs to degrade the ECM and
facilitate cell invasion. Follower cells are typically more
dependent on the behaviour of leader cells and the signals they
produce. Follower cells often follow the path carved out by the
leader cells and can contribute to the spreading of the tumour
(137,140-142). By traversing a larger space
created by the leader cells, follower cells encounter less
mechanical resistance and can spend less energy remodelling the ECM
(143). As a result of increased
crosslinking and force-mediated ECM remodelling, the
tumour-surrounding interstitial matrix becomes linearized. For
efficient cell migration, follower cells migrate along densely
aligned collagen fibres (126).
Membrane-type-1-MMP (MT1-MMP) is typically localized to invasive
actin-rich cell structures (144). The delivery of MT1-MMP and other
proteinases by exosomes outside the cell can also facilitate
invasive lamellipodia maturation and degradation of the ECM
(145). By inhibiting MMP
activity in CAFs before adding SCC follower cells, the invasion of
follower cells was effectively halted (10). MMP function is not required by
follower cells once the matrix has been remodelled by CAFs
(10). In addition, as follower
cells tend to undergo glycolysis, increased lactate generation and
excretion favour protease-mediated matrix remodelling and thus
enhance the invasion of cancer cells (146-148) (Fig.
4A). Lactate-induced acidity has also been found to enhance the
activity of certain proteases, namely MMP-2, MMP-9, cathepsin B and
cathepsin L (149).
In glioblastoma, follower cells remodel the
extracellular hyaluronic acid (HA) through a combination of
synthesis, by hyaluronan synthases (HASs), and degradation, by
hyaluronidases and MMPs (150,151). In vitro models have
indicated that incorporating HA into gelatin matrices enhances the
invasiveness of the follower cells (150). The production of HA by HASs
(particularly HAS2) is significantly activated during invasion into
the HA-rich ECM (151). The CD44
gene, which is a representative HA receptor, shares a close
relationship with HAS, suggesting that there may be crosstalk
between these two genes that stimulates signal cascades for
glioblastoma follower cell invasion (152). Therefore, the invasion of
glioblastoma follower cells is primarily influenced by the HA-rich
ECM environments. This is mainly due to the high involvement of
CD44 receptors and HASs (151,153,154).
According to extensive data, chemotactic cell
migration guided by soluble signalling molecules facilitates
invasion and metastasis (155,156). Chemoattractants are soluble
proteins secreted into the extracellular space. These molecules are
believed to be retained in this space by binding to
glycosaminoglycans in the ECM, thereby establishing immobilized
concentration gradients. Chemotactic gradients tend to be local and
transient in nature. Furthermore, cells can navigate through
complex topologies with self-generated chemotaxis (156). In self-generated gradients,
follower cells produce an outwards-facing gradient by breaking down
an attractive chemical, which serves as a guiding cue for leader
cells. Leader cells need chemical gradients generated by follower
cells in order to respond to external signals (157). As leader cells interact with
their surroundings, their local gradient moves with them, resulting
in directed movement that is exceptionally robust and capable of
operating over long distances. However, if the attractant level is
too high, then the follower cells cannot migrate but instead break
down the molecule until the concentration is low enough for a
gradient to be observed (157).
Additionally, if the cell leading wave does not have enough leader
cells, the attractant is left behind, attracting more follower
cells. In summary, cells show chemotaxis over long distances by
moving in waves, with saturating chemoattractants in front and low
levels behind (158). Without
physically visiting their environment, follower cells acquire
information about their surroundings (156,159) (Fig.
4B). Chemotaxis is a crucial concept for the understanding of
the physiology of cells since it helps elucidate matrix
composition.
It is believed that chemotaxis promotes the invasion
and metastasis of follower cells. When a follower cell migrates, it
senses a gradient of external chemoattractants, of which
chemokines, chemotactic growth factors and lysophosphatidic acid
(LPA) are major families (118,156,160). For example, as zebrafish
posterior lateral line primordium collectively migrate, follower
cells sense the attractant, CXCL12a, to migrate efficiently
(118). The CXCL12/CXCR4 pathway
also influences breast cancer cell metastasis to the lungs
(161). Follower cells are
CXCR4+ and invade along CXCL12 gradients into organs
expressing CXCL12 (for example, the lymph nodes and lungs).
Moreover, when malignant lymphocytes are exposed to CCL19
gradients, their surface is stripped of CCR7, resulting in the
retraction of protrusions and loss of polarity, which indicates
follower cell behaviour. On the basis of intermediate gradients of
CCL19, follower cells migrate towards the invasion front in cell
migration (162). In addition,
chemical gradients formed by EGF uptake can guide the movement of
malignant epithelial cells within confined spaces (156). However, Muinonen-Martin et
al (160) noted that rather
than acting as chemoattractants that could guide melanoma follower
cells, growth factors acted as accessory factors that increased
cell speed and chemotaxis efficiency, regulating melanoma cell
behaviour. As such, the chemoattractant relay is rearranged to
generate positional information using positive feedback.
Furthermore, soluble chemicals are either degraded by enzymes or
scavenged by decoy receptors (via endocytic internalization). In
addition, follower cells are strongly induced to invade outwards by
the LPA gradient (160). It has
been demonstrated that follower cells can degrade LPA during
melanoma metastasis, resulting in a gradient that the cells then
respond to by migrating (160).
Furthermore, expression of the LPA receptor (LPAR) in follower
cells promotes metastasis and cell growth in metastatic breast
cancer xenografts (163,164). An instrumental factor in the
metastasis of pancreatic ductal adenocarcinoma cells from primary
tumours is the neural Wiskott-Aldrich syndrome protein, which
controls the recognition of LPA gradients by controlling LPAR1
(165). LPAR signalling mediates
actin-myosin contractility and control of cell orientation, as well
as matrix remodelling.
According to a mathematical analysis of the tracks
of cells, follower cells are chemotactic and are attracted
directionally by attractants (166). As follower cells respond to
chemotactic gradients, they can alter them, resulting in robust
chemotactic gradients. Furthermore, follower cells can reduce
attractant concentrations when the attractant concentrations are
too high. Depending on their needs follower cells alter themselves
accordingly by inducing enzymes or cell division (166). However, the role of follower
cells in generating chemokine gradients and their potential
regulation by endocytosis have not yet, to the best of our
knowledge, been studied.
Among neighbouring cells, Notch signalling can
coordinate divergent cell fate, which is termed lateral inhibition
(167). Typically, follower cells
upregulate Notch1 and Jagged1 (Jag1) expression, while leader cells
upregulate Delta-like 4 (Dll4) expression to promote collective
invasion (13,92,168). Notch also coordinates the
adoption of similar follower cell fates by modulating lateral
induction, a process through which it promotes the acquisition of
comparable cell fates in adjacent cells (11,169). Through lateral inhibition,
Notch-Delta signalling suppresses leader cell behaviour in follower
cells. A ligand can bind to the Notch receptor in one of two ways:
Delta-like or Jagged-like. In response to ligand-receptor binding
and forces originating from endocytosis, the Notch receptor
undergoes a specific conformational change, releasing the Notch
intracellular domain (NICD) into the cytoplasm, leading to lateral
inhibition and induction, which regulate collective invasion
(170). NICD translocates to the
nucleus and forms an active transcriptional complex with the
DNA-binding protein, Rbpj, and the coactivator, Mastermind-like
(Maml). Moreover, the NICD-Rbpj-Maml complex regulates the
transcription of the Notch receptor and its ligands, thereby
promoting the transcription of Hey/Hes1, an inhibitor of Delta but
an activator of Jagged and Notch target genes (171,172). Therefore, lateral inhibition is
observed between follower and leader cells when Notch-Delta
signalling is dominant compared with Notch-Jagged signalling, while
lateral induction takes place amongst follower cells when
Notch-Jagged signalling is dominant (173). Moreover, Notch1 exhibits a higher
affinity to Dll4 than to Jag1 following Fringe-mediated
glycosylation of Notch1 (174).
Based on the results of a study using mathematical modelling of
Notch-Delta-Jagged signalling, Fringe proteins glycosylate the
Notch receptor, resulting in a conformational change in the
extracellular domain (175).
Fringe can stabilize leader and follower cells by inhibiting
Notch-Jagged binding, whereas its absence may shift the balance
towards Notch-Jagged signalling (176).
VEGF stimulates leader cells by binding VEGFR-2 or
VEGFR-3 in the microenvironment (177). Leader cells express Dll4
following VEGFR signalling, which activates intercellular Notch
signalling to inhibit adjacent follower cells from turning into
leader cells by targeting Notch1 (178-180). In follower cells, Notch
signalling inhibits VEGFR function. Conversely, leader cells
increase VEGFA secretion, which induces follower cell motility and
invasion (89). In
Drosophila oogenesis, upregulating Rac expression or
PDGF/VEGF receptor expression can make follower cells switch
positions from posterior to anterior and maintain their position as
precursor cells, controlling migration throughout clusters
(89). A landscape perspective on
how Notch signalling affects leader and follower cell stability and
transition may be a useful future direction of study.
Compared with follower cells, leader cells display
a variety of mitotic defects, the most prominent of which is
cytokinetic instability (11).
Follower cells are proliferative and may be able to rescue
defective leader cells during collective movement. A study of NSCLC
cells isolated using SaGA technology found that gene expression in
follower and leader cells was different (11). In follower cells, lysine
demethylase 5B (KDM5B) mutations lead to phenotypic heterogeneity,
and expression of ARP3 K240R promotes invasion, even in less
invasive follower cells, and confers leader cell behaviour
(12).
In a previous study, through a SaGA-based capture
and amplification procedure, wild-type KDM5B was selectively
enriched in leader cells, while mutant KDM5B L685W was selectively
enriched in follower cells (12).
KDM5B is a lysine demethylase enzyme responsible for catalysing the
elimination of di- and trimethylation from histone H3 molecules
that have been methylated at K4 (known as H3K4me2 and H3K4me3,
respectively). KDM5B, functioning as a transcriptional repressor,
promotes the leader cell phenotype by restricting follower cell
behaviour (12). The mutation of
KDM5B directly impacts invasive behaviour, serving as a unique
epigenetic regulator that affects multiple pathways (181,182). Follower cells contain mutations
in L685W near the zinc finger structural domain of KDM5B, which is
essential for demethylase activity (183). Overexpression of KDM5B L685W,
however, may enhance collective migration behaviour by enhancing
heterogeneity and resulting in the emergence of cells with follower
characteristics (184). Luminal
breast cancer cells with upregulated KDM5B expression possess
enhanced phenotypic stability, while cells with KDM5B depletion or
repression exhibit enhanced transcriptional plasticity and can
overcome therapeutic resistance (12).
The K240R mutation in ARP3, an essential component
of the ARP2/3 complex, may interfere with its ubiquitylation at
K240, thus resulting in either reduced ARP3 turnover or augmented
activity (185). This mutation
has been associated with enhanced leader cell behaviour, since it
amplifies directional cellular protrusion events and accelerates
the speed of cell motility, allowing the cell to detect signals
that direct migration more easily. Further experiments are
necessary to investigate the conjecture that ARP3 K240R is
resistant to ubiquitylation, and if so, ascertain its effect on
leader cell behaviour. Genetic heterogeneity can be identified in
an invasion model using multiple cell lines and cancer types.
Collective invasion may be influenced by a combination of multiple
genetic and epigenetic changes rather than by isolated changes
alone. Finally, identifying these key changes may support clinical
judgement by monitoring relevant predictive biomarkers.
Glycolysis is sustained by glucose transporter 1
(GLUT1) expression in follower cells, while mitochondrial
respiration is sustained by active pyruvate dehydrogenase (PDH)
expression in leader cells (184). Specifically, follower cells
exhibit a higher level of GLUT1 expression and glucose uptake than
leader cells during collective NSCLC invasion in vitro
(190). GLUT1 is ubiquitous in
all tumour types that have poor patient prognosis (191) and maintains glucose uptake by
cancer cells (192-194). Follower cells may divert glucose
uptake to the pentose phosphate pathway (PPP), which supports
ribulobiogenesis and proliferation without altering citric acid
cycle flux. The PPP begins with glucose-6-phosphate dehydrogenase,
which is also more highly expressed in follower cells (190). In response to a decrease in
glycolytic intermediates entering glycolysis and the PPP, follower
cells switch from proliferation to invasion (190). In leader cells, GLUT1 expression
is reduced by regulators, such as tumour suppressor p53 and
hypoxia-inducible factor-1 (195,196). By contrast, GLUT1 upregulation in
leader cells hinders their collective invasive ability (195-198). In follower cells, mitochondria
are primarily found around the nucleus, whereas in leader cells,
they are more common at the edge of the cytoplasm (190). The presence of mitochondria at
the edge of the cytoplasm suggests a higher energy demand in the
leading edge, where cellular protrusions and migratory activity
occur. As PDH activity increases, mitochondrial distribution to the
periphery of the cell increases (190). Since dichloroacetate (DCA)
inhibits PDH kinases, DCA-treated follower cells have lower levels
of phosphorylated PDH at position S293 and increase invasion,
particularly in chains, similar to leader cells (190).
In summary, CIL, biomechanics, remodelling of the
ECM, chemotaxis, lateral inhibition and the genetic and metabolic
heterogeneity associated with follower cells were discussed in the
present review. These mechanisms determine movement polarity by
identifying leader and follower cells and guiding cancer cells to
acquire follower-like or leader-like morphology, function and
behaviour during collective invasion. The follower cells receive
signals from the leader cells and the microenvironment due to
intracellular and intercellular signalling cascades as well as
mechanotransduction. The signals are then transmitted to the entire
mass of cells. When collective movements occur, follower cells
maintain their phenotype and consolidate the status of the leader
cell. Additionally, cell behaviour and fate can be changed by
biomechanics and the microenvironment, resulting in follower cells
adopting a leadership phenotype, which ultimately leads to genetic
changes. In addition, follower cells may take over the positions of
leader cells when their energy level drops below a certain level.
For cells to move collectively, follower and leader cells must
coordinate their movements to be controlled by physical
(mechanical) and chemical (signalling) interactions.
However, the factors that drive follower cell
formation and the mechanisms that regulate follower cell migration
remain unknown. Currently, the impact of the molecular mechanism of
cadherin mechanotransduction on the behaviour of leader-follower
cells remains mostly unclear. To improve understanding of the
interaction between cadherin junctions and cell mechanics,
researchers should focus on the interaction between cadherin
junctions and Rho-GTPases. In vivo, mechanocoupling is
spatially controlled in a number of ways, such as by cell polarity
and ECM remodelling. The more complex and physiologically relevant
collective migration within 3D matrices is being replicated in a
growing number of in silico models. The use of computational
models can provide insights into the migration of follower cells
and overcome limitations associated with experimental research
(200,201). Furthermore, it is unclear how
bioenergetic status affects the emergence of follower cells when
taking over from failing leader cells. The metabolic, morphological
and migration functions of cells are closely intertwined, so
targeting cellular metabolism may provide a novel strategy for
treating cancer by inhibiting the production of cellular energy. It
is necessary to conduct additional experimental research to
investigate the function of follower cells in collective movement
and to analyse the mechanism of coordinated invasion.
Not applicable.
XW conceived the outline of the manuscript by
searching the literature, wrote a draft of the manuscript and
created the figures. XL and YT reviewed and edited the manuscript.
Data authentication is not applicable. All authors read and
approved the final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This work was supported by The National Natural Science
Foundation of China grants (grant nos. 82173326, 81972542 and
82073000), The National Science Foundation of Sichuan Province
(grant no. 2022YFS0289) and The Exploration and Research Projects
of West China College of Stomatology, Sichuan University (grant no.
RD-03-202004).
|
1
|
Place AE, Jin Huh S and Polyak K: The
microenvironment in breast cancer progression: Biology and
implications for treatment. Breast Cancer Res. 13:2272011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Almendro V, Marusyk A and Polyak K:
Cellular heterogeneity and molecular evolution in cancer. Annu Rev
Pathol. 8:277–302. 2013. View Article : Google Scholar
|
|
3
|
Friedl P, Locker J, Sahai E and Segall JE:
Classifying collective cancer cell invasion. Nat Cell Biol.
14:777–783. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haeger A, Wolf K, Zegers MM and Friedl P:
Collective cell migration: Guidance principles and hierarchies.
Trends Cell Biol. 25:556–566. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Westcott JM, Prechtl AM, Maine EA, Dang
TT, Esparza MA, Sun H, Zhou Y, Xie Y and Pearson GW: An
epigenetically distinct breast cancer cell subpopulation promotes
collective invasion. J Clin Invest. 125:1927–1943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pandya P, Orgaz JL and Sanz-Moreno V:
Actomyosin contractility and collective migration: May the force be
with you. Curr Opin Cell Biol. 48:87–96. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Poujade M Grasland-Mongrain E, Hertzog A,
Jouanneau J, Chavrier P, Ladoux B, Buguin A and Silberzan P:
Collective migration of an epithelial monolayer in response to a
model wound. Proc Natl Acad Sci USA. 104:15988–15993. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park J and Chronopolous A: Abstract B029:
Flip-flopping of fusion-positive rhabdomyosarcoma regulating
intratumoral heterogeneity for metastasis. Clin Cancer Res. 28(18
Suppl): B0292022. View Article : Google Scholar
|
|
9
|
Yamamoto E, Kohama G, Sunakawa H, Iwai M
and Hiratsuka H: Mode of invasion, bleomycin sensitivity, and
clinical course in squamous cell carcinoma of the oral cavity.
Cancer. 51:2175–2180. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gaggioli C, Hooper S, Hidalgo-Carcedo C,
Grosse R, Marshall JF, Harrington K and Sahai E: Fibroblast-led
collective invasion of carcinoma cells with differing roles for
RhoGTPases in leading and following cells. Nat Cell Biol.
9:1392–1400. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Konen J, Summerbell E, Dwivedi B, Galior
K, Hou Y, Rusnak L, Chen A, Saltz J, Zhou W, Boise LH, et al:
Image-guided genomics of phenotypically heterogeneous populations
reveals vascular signalling during symbiotic collective cancer
invasion. Nat Commun. 8:150782017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zoeller EL, Pedro B, Konen J, Dwivedi B,
Rupji M, Sundararaman N, Wang L, Horton JR, Zhong C, Barwick BG, et
al: Genetic heterogeneity within collective invasion packs drives
leader and follower cell phenotypes. J Cell Sci. 132:jcs2315142019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Riahi R, Sun J, Wang S, Long M, Zhang DD
and Wong PK: Notch1-Dll4 signalling and mechanical force regulate
leader cell formation during collective cell migration. Nat Commun.
6:65562015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tse JM, Cheng G, Tyrrell JA,
Wilcox-Adelman SA, Boucher Y, Jain RK and Munn LL: Mechanical
compression drives cancer cells toward invasive phenotype. Proc
Natl Acad Sci USA. 109:911–916. 2012. View Article : Google Scholar :
|
|
15
|
Farooqui R and Fenteany G: Multiple rows
of cells behind an epithelial wound edge extend cryptic
lamellipodia to collectively drive cell-sheet movement. J Cell Sci.
118:51–63. 2005. View Article : Google Scholar
|
|
16
|
Reffay M, Parrini MC, Cochet-Escartin O,
Ladoux B, Buguin A, Coscoy S, Amblard F, Camonis J and Silberzan P:
Interplay of RhoA and mechanical forces in collective cell
migration driven by leader cells. Nat Cell Biol. 16:217–223. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamaguchi N, Mizutani T, Kawabata K and
Haga H: Leader cells regulate collective cell migration via Rac
activation in the downstream signaling of integrin β1 and PI3K. Sci
Rep. 5:76562015. View Article : Google Scholar
|
|
18
|
Mayor R and Etienne-Manneville S: The
front and rear of collective cell migration. Nat Rev Mol Cell Biol.
17:97–109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Goliwas KF, Wang W, Taufalele PV,
Bordeleau F and Reinhart-King CA: Energetic regulation of
coordinated leader-follower dynamics during collective invasion of
breast cancer cells. Proc Natl Acad Sci USA. 116:7867–7872. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wolf K, Wu YI, Liu Y, Geiger J, Tam E,
Overall C, Stack MS and Friedl P: Multi-step pericellular
proteolysis controls the transition from individual to collective
cancer cell invasion. Nat Cell Biol. 9:893–904. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jolly MK, Somarelli JA, Sheth M, Biddle A,
Tripathi SC, Armstrong AJ, Hanash SM, Bapat SA, Rangarajan A and
Levine H: Hybrid epithelial/mesenchymal phenotypes promote
metastasis and therapy resistance across carcinomas. Pharmacol
Ther. 194:161–184. 2019. View Article : Google Scholar
|
|
22
|
Mayor R and Carmona-Fontaine C: Keeping in
touch with contact inhibition of locomotion. Trends Cell Biol.
20:319–328. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stramer B and Mayor R: Mechanisms and in
vivo functions of contact inhibition of locomotion. Nat Rev Mol
Cell Biol. 18:43–55. 2017. View Article : Google Scholar
|
|
24
|
Wendt MK, Taylor MA, Schiemann BJ and
Schiemann WP: Down-regulation of epithelial cadherin is required to
initiate metastatic outgrowth of breast cancer. Mol Biol Cell.
22:2423–2435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abercrombie M: Contact inhibition and
malignancy. Nature. 281:259–262. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rørth P: Fellow travellers: Emergent
properties of collective cell migration. EMBO Rep. 13:984–991.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Khalil AA and de Rooij J: Cadherin
mechanotransduction in leader-follower cell specification during
collective migration. Exp Cell Res. 376:86–91. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rorth P: Collective cell migration. Annu
Rev Cell Dev Biol. 25:407–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qin L, Yang D, Yi W, Cao H and Xiao G:
Roles of leader and follower cells in collective cell migration.
Mol Biol Cell. 32:1267–1272. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Camand E, Peglion F, Osmani N, Sanson M
and Etienne-Manneville S: N-cadherin expression level modulates
integrin-mediated polarity and strongly impacts on the speed and
directionality of glial cell migration. J Cell Sci. 125:844–857.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ladoux B, Mège RM and Trepat X: Front-rear
polarization by mechanical cues: From single cells to tissues.
Trends Cell Biol. 26:420–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abbruzzese G, Becker SF, Kashef J and
Alfandari D: ADAM13 cleavage of cadherin-11 promotes CNC migration
independently of the homophilic binding site. Dev Biol.
415:383–390. 2016. View Article : Google Scholar :
|
|
33
|
Quan Q, Wang X, Lu C, Ma W, Wang Y, Xia G,
Wang C and Yang G: Cancer stem-like cells with hybrid
epithelial/mesenchymal phenotype leading the collective invasion.
Cancer Sci. 111:467–476. 2020. View Article : Google Scholar :
|
|
34
|
Ye X and Weinberg RA:
Epithelial-mesenchymal plasticity: A central regulator of cancer
progression. Trends Cell Biol. 25:675–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Padmanaban V, Krol I, Suhail Y, Szczerba
BM, Aceto N, Bader JS and Ewald AJ: E-cadherin is required for
metastasis in multiple models of breast cancer. Nature.
573:439–444. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Scarpa E, Szabó A, Bibonne A, Theveneau E,
Parsons M and Mayor R: Cadherin switch during EMT in neural crest
cells leads to contact inhibition of locomotion via repolarization
of forces. Dev Cell. 34:421–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Moriwaki K, Wada M, Kuwabara H, Ayani Y,
Terada T, Higashino M, Kawata R and Asahi M: BDNF/TRKB axis
provokes EMT progression to induce cell aggressiveness via
crosstalk with cancer-associated fibroblasts in human parotid gland
cancer. Sci Rep. 12:175532022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shih W and Yamada S: N-cadherin-mediated
cell-cell adhesion promotes cell migration in a three-dimensional
matrix. J Cell Sci. 125:3661–3670. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saénz-de-Santa-María I, Celada L and
Chiara MD: The leader position of mesenchymal cells expressing
N-cadherin in the collective migration of epithelial cancer. Cells.
9:7312020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Labernadie A, Kato T, Brugués A,
Serra-Picamal X, Derzsi S, Arwert E, Weston A, González-Tarragó V,
Elosegui-Artola A, Albertazzi L, et al: A mechanically active
heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to
drive cancer cell invasion. Nat Cell Biol. 19:224–237. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Van den Bossche J, Bogaert P, van Hengel
J, Guérin CJ, Berx G, Movahedi K, Van den Bergh R,
Pereira-Fernandes A, Geuns JM, Pircher H, et al: Alternatively
activated macrophages engage in homotypic and heterotypic
interactions through IL-4 and polyamine-induced E-cadherin/catenin
complexes. Blood. 114:4664–4674. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takai Y, Irie K, Shimizu K, Sakisaka T and
Ikeda W: Nectins and nectin-like molecules: Roles in cell adhesion,
migration, and polarization. Cancer Sci. 94:655–667. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Izumi G, Sakisaka T, Baba T, Tanaka S,
Morimoto K and Takai Y: Endocytosis of E-cadherin regulated by Rac
and Cdc42 small G proteins through IQGAP1 and actin filaments. J
Cell Biol. 166:237–248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takai Y, Miyoshi J, Ikeda W and Ogita H:
Nectins and nectin-like molecules: Roles in contact inhibition of
cell movement and proliferation. Nat Rev Mol Cell Biol. 9:603–615.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ikeda W, Kakunaga S, Takekuni K, Shingai
T, Satoh K, Morimoto K, Takeuchi M, Imai T and Takai Y: Nectin-like
molecule-5/Tage4 enhances cell migration in an integrin-dependent,
Nectin-3-independent manner. J Biol Chem. 279:18015–18025. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bevelacqua V, Bevelacqua Y, Candido S,
Skarmoutsou E, Amoroso A, Guarneri C, Strazzanti A, Gangemi P,
Mazzarino MC, D'Amico F, et al: Nectin like-5 overexpression
correlates with the malignant phenotype in cutaneous melanoma.
Oncotarget. 3:882–892. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nakai R, Maniwa Y, Tanaka Y, Nishio W,
Yoshimura M, Okita Y, Ohbayashi C, Satoh N, Ogita H, Takai Y and
Hayashi Y: Overexpression of Necl-5 correlates with unfavorable
prognosis in patients with lung adenocarcinoma. Cancer Sci.
101:1326–1330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kania A and Klein R: Mechanisms of
ephrin-Eph signalling in development, physiology and disease. Nat
Rev Mol Cell Biol. 17:240–256. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Astin JW, Batson J, Kadir S, Charlet J,
Persad RA, Gillatt D, Oxley JD and Nobes CD: Competition amongst
Eph receptors regulates contact inhibition of locomotion and
invasiveness in prostate cancer cells. Nat Cell Biol. 12:1194–1204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Batson J, Astin JW and Nobes CD:
Regulation of contact inhibition of locomotion by Eph-ephrin
signalling. J Microsc. 251:232–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matthews HK, Marchant L, Carmona-Fontaine
C, Kuriyama S, Larraín J, Holt MR, Parsons M and Mayor R:
Directional migration of neural crest cells in vivo is regulated by
Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development.
135:1771–1780. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kurayoshi M, Oue N, Yamamoto H, Kishida M,
Inoue A, Asahara T, Yasui W and Kikuchi A: Expression of Wnt-5a is
correlated with aggressiveness of gastric cancer by stimulating
cell migration and invasion. Cancer Res. 66:10439–10448. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
VanderVorst K, Dreyer CA, Konopelski SE,
Lee H, Ho HH and Carraway KL III: Wnt/PCP signaling contribution to
carcinoma collective cell migration and metastasis. Cancer Res.
79:1719–1729. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Luga V, Zhang L, Viloria-Petit AM,
Ogunjimi AA, Inanlou MR, Chiu E, Buchanan M, Hosein AN, Basik M and
Wrana JL: Exosomes mediate stromal mobilization of autocrine
Wnt-PCP signaling in breast cancer cell migration. Cell.
151:1542–1556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Halbleib JM and Nelson WJ: Cadherins in
development: Cell adhesion, sorting, and tissue morphogenesis.
Genes Dev. 20:3199–3214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wheeler AP and Ridley AJ: Why three Rho
proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res.
301:43–49. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Theveneau E, Marchant L, Kuriyama S, Gull
M, Moepps B, Parsons M and Mayor R: Collective chemotaxis requires
contact-dependent cell polarity. Dev Cell. 19:39–53. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Drees F, Pokutta S, Yamada S, Nelson WJ
and Weis WI: Alpha-catenin is a molecula r switch that binds
E-cadherin-beta-catenin and regulates actin-filament assembly.
Cell. 123:903–915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Anastasiadis PZ and Reynolds AB:
Regulation of Rho GTPases by p120-catenin. Curr Opin Cell Biol.
13:604–610. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Macpherson IR, Hooper S, Serrels A,
McGarry L, Ozanne BW, Harrington K, Frame MC, Sahai E and Brunton
VG: p120-catenin is required for the collective invasion of
squamous cell carcinoma cells via a phosphorylation-independent
mechanism. Oncogene. 26:5214–5228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Noren NK, Liu BP, Burridge K and Kreft B:
p120 catenin regulates the actin cytoskeleton via Rho family
GTPases. J Cell Biol. 150:567–580. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nobes CD and Hall A: Rho, rac, and cdc42
GTPases regulate the assembly of multimolecular focal complexes
associated with actin stress fibers, lamellipodia, and filopodia.
Cell. 81:53–62. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kurokawa K and Matsuda M: Localized RhoA
activation as a requirement for the induction of membrane ruffling.
Mol Biol Cell. 16:4294–4303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Krause M and Gautreau A: Steering cell
migration: Lamellipodium dynamics and the regulation of directional
persistence. Nat Rev Mol Cell Biol. 15:577–590. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Haga RB and Ridley AJ: Rho GTPases:
Regulation and roles in cancer cell biology. Small GTPases.
7:207–221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kim SY, Lee S, Lee E, Lim H, Shin JY, Jung
J, Kim SG and Moon A: Sex-biased differences in the correlation
between epithelial-to-mesenchymal transition-associated genes in
cancer cell lines. Oncol Lett. 18:6852–6868. 2019.PubMed/NCBI
|
|
68
|
Hidalgo-Carcedo C, Hooper S, Chaudhry SI,
Williamson P, Harrington K, Leitinger B and Sahai E: Collective
cell migration requires suppression of actomyosin at cell-cell
contacts mediated by DDR1 and the cell polarity regulators Par3 and
Par6. Nat Cell Biol. 13:49–58. 2011. View Article : Google Scholar :
|
|
69
|
Zaritsky A, Tseng YY, Rabadán MA, Krishna
S, Overholtzer M, Danuser G and Hall A: Diverse roles of guanine
nucleotide exchange factors in regulating collective cell
migration. J Cell Biol. 216:1543–1556. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Runkle EA and Mu D: Tight junction
proteins: From barrier to tumorigenesis. Cancer Lett. 337:41–48.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kummer D, Steinbacher T, Thölmann S,
Schwietzer MF, Hartmann C, Horenkamp S, Demuth S, Peddibhotla SSD,
Brinkmann F, Kemper B, et al: A JAM-A-tetraspanin-αvβ5 integrin
complex regulates contact inhibition of locomotion. J Cell Biol.
221:e2021051472022. View Article : Google Scholar
|
|
72
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Davis JR, Luchici A, Mosis F, Thackery J,
Salazar JA, Mao Y, Dunn GA, Betz T, Miodownik M and Stramer BM:
Inter-cellular forces orchestrate contact inhibition of locomotion.
Cell. 161:361–373. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Davis JR, Huang CY, Zanet J, Harrison S,
Rosten E, Cox S, Soong DY, Dunn GA and Stramer BM: Emergence of
embryonic pattern through contact inhibition of locomotion.
Development. 139:4555–4560. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Guck J, Schinkinger S, Lincoln B, Wottawah
F, Ebert S, Romeyke M, Lenz D, Erickson HM, Ananthakrishnan R,
Mitchell D, et al: Optical deformability as an inherent cell marker
for testing malignant transformation and metastatic competence.
Biophys J. 88:3689–3698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mialhe A, Lafanechère L, Treilleux I,
Peloux N, Dumontet C, Brémond A, Panh MH, Payan R, Wehland J,
Margolis RL and Job D: Tubulin detyrosination is a frequent
occurrence in breast cancers of poor prognosis. Cancer Res.
61:5024–5027. 2001.PubMed/NCBI
|
|
77
|
Daub H, Gevaert K, Vandekerckhove J, Sobel
A and Hall A: Rac/Cdc42 and p65PAK regulate the
microtubule-destabilizing protein stathmin through phosphorylation
at serine 16. J Biol Chem. 276:1677–1680. 2001. View Article : Google Scholar
|
|
78
|
Moore R, Theveneau E, Pozzi S, Alexandre
P, Richardson J, Merks A, Parsons M, Kashef J, Linker C and Mayor
R: Par3 controls neural crest migration by promoting microtubule
catastrophe during contact inhibition of locomotion. Development.
140:4763–4775. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cramer LP: Forming the cell rear first:
Breaking cell symmetry to trigger directed cell migration. Nat Cell
Biol. 12:628–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yam PT, Wilson CA, Ji L, Hebert B,
Barnhart EL, Dye NA, Wiseman PW, Danuser G and Theriot JA:
Actin-myosin network reorganization breaks symmetry at the cell
rear to spontaneously initiate polarized cell motility. J Cell
Biol. 178:1207–1221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Olson HM and Nechiporuk AV: Using
zebrafish to study collective cell migration in development and
disease. Front Cell Dev Biol. 6:832018. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ozawa M, Hiver S, Yamamoto T, Shibata T,
Upadhyayula S, Mimori-Kiyosue Y and Takeichi M: Adherens junction
regulates cryptic lamellipodia formation for epithelial cell
migration. J Cell Biol. 219:e2020061962020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yokoyama S, Matsui TS and Deguchi S: New
wrinkling substrate assay reveals traction force fields of leader
and follower cells undergoing collective migration. Biochem Biophys
Res Commun. 482:975–979. 2017. View Article : Google Scholar
|
|
84
|
Jakobsson L, Franco CA, Bentley K, Collins
RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G,
Medvinsky A, et al: Endothelial cells dynamically compete for the
tip cell position during angiogenic sprouting. Nat Cell Biol.
12:943–953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ghabrial AS and Krasnow MA: Social
interactions among epithelial cells during tracheal branching
morphogenesis. Nature. 441:746–749. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Nguyen-Ngoc KV, Cheung KJ, Brenot A,
Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z and Ewald AJ: ECM
microenvironment regulates collective migration and local
dissemination in normal and malignant mammary epithelium. Proc Natl
Acad Sci USA. 109:E2595–E2604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cheung KJ, Gabrielson E, Werb Z and Ewald
AJ: Collective invasion in breast cancer requires a conserved basal
epithelial program. Cell. 155:1639–1651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cai D, Chen SC, Prasad M, He L, Wang X,
Choesmel-Cadamuro V, Sawyer JK, Danuser G and Montell DJ:
Mechanical feedback through E-cadherin promotes direction sensing
during collective cell migration. Cell. 157:1146–1159. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Inaki M, Vishnu S, Cliffe A and Rørth P:
Effective guidance of collective migration based on differences in
cell states. Proc Natl Acad Sci USA. 109:2027–2032. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Okimura C, Iwanaga M, Sakurai T, Ueno T,
Urano Y and Iwadate Y: Leading-edge elongation by follower cell
interruption in advancing epithelial cell sheets. Proc Natl Acad
Sci USA. 119:e21199031192022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Vishwakarma M, Di Russo J, Probst D,
Schwarz US, Das T and Spatz JP: Mechanical interactions among
followers determine the emergence of leaders in migrating
epithelial cell collectives. Nat Commun. 9:34692018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bocci F, Onuchic JN and Jolly MK:
Understanding the principles of pattern formation driven by notch
signaling by integrating experiments and theoretical models. Front
Physiol. 11:9292020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
DeMali KA and Burridge K: Coupling
membrane protrusion and cell adhesion. J Cell Sci. 116:2389–2397.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
DeCamp SJ, Tsuda VMK, Ferruzzi J, Koehler
SA, Giblin JT, Roblyer D, Zaman MH, Weiss ST, Kılıç A, De Marzio M,
et al: Epithelial layer unjamming shifts energy metabolism toward
glycolysis. Sci Rep. 10:183022020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Weber GF, Bjerke MA and DeSimone DW: A
mechanoresponsive cadherin-keratin complex directs polarized
protrusive behavior and collective cell migration. Dev Cell.
22:104–115. 2012. View Article : Google Scholar :
|
|
96
|
Chen T, Saw TB, Mège RM and Ladoux B:
Mechanical forces in cell monolayers. J Cell Sci.
131:jcs2181562018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tambe DT, Hardin CC, Angelini TE,
Rajendran K, Park CY, Serra-Picamal X, Zhou EH, Zaman MH, Butler
JP, Weitz DA, et al: Collective cell guidance by cooperative
intercellular forces. Nat Mater. 10:469–475. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Trepat X, Wasserman MR, Angelini TE,
Millet E, Weitz DA, Butler JP and Fredberg JJ: Physical forces
during collective cell migration. Nat Phys. 5:426–430. 2009.
View Article : Google Scholar
|
|
99
|
Gayrard C, Bernaudin C, Déjardin T, Seiler
C and Borghi N: Src- and confinement-dependent FAK activation
causes E-cadherin relaxation and β-catenin activity. J Cell Biol.
217:1063–1077. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Desai RA, Gopal SB, Chen S and Chen CS:
Contact inhibition of locomotion probabilities drive solitary
versus collective cell migration. J R Soc Interface.
10:201307172013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Thomas WA, Boscher C, Chu YS, Cuvelier D,
Martinez-Rico C, Seddiki R, Heysch J, Ladoux B, Thiery JP, Mege RM
and Dufour S: α-Catenin and vinculin cooperate to promote high
E-cadherin-based adhesion strength. J Biol Chem. 288:4957–4969.
2013. View Article : Google Scholar
|
|
102
|
Seddiki R, Narayana GHNS, Strale PO,
Balcioglu HE, Peyret G, Yao M, Le AP, Teck Lim C, Yan J, Ladoux B
and Mège RM: Force-dependent binding of vinculin to α-catenin
regulates cell-cell contact stability and collective cell behavior.
Mol Biol Cell. 29:380–388. 2018. View Article : Google Scholar :
|
|
103
|
Matsuzawa K, Himoto T, Mochizuki Y and
Ikenouchi J: α-Catenin controls the anisotropy of force
distribution at cell-cell junctions during collective cell
migration. Cell Rep. 23:3447–3456. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bazellières E, Conte V, Elosegui-Artola A,
Serra-Picamal X, Bintanel-Morcillo M, Roca-Cusachs P, Muñoz JJ,
Sales-Pardo M, Guimerà R and Trepat X: Control of cell-cell forces
and collective cell dynamics by the intercellular adhesome. Nat
Cell Biol. 17:409–420. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Plutoni C, Bazellieres E, Le Borgne-Rochet
M, Comunale F, Brugues A, Séveno M, Planchon D, Thuault S, Morin N,
Bodin S, et al: P-cadherin promotes collective cell migration via a
Cdc42-mediated increase in mechanical forces. J Cell Biol.
212:199–217. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Liu Z, Tan JL, Cohen DM, Yang MT,
Sniadecki NJ, Ruiz SA, Nelson CM and Chen CS: Mechanical tugging
force regulates the size of cell-cell junctions. Proc Natl Acad Sci
USA. 107:9944–9949. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Crawford AJ, Gomez-Cruz C, Russo GC, Huang
W, Bhorkar I, uñoz-Barrutia A, Wirtz D and Garcia-Gonzalez D: Tumor
proliferation and invasion are coupled through cell-extracellular
matrix friction. bioRxiv. 2022.2011:2015.5165482022.
|
|
108
|
Lee MH, Russo G, Rahmanto YS, Du W,
Crawford AJ, Wu PH, Gilkes D, Kiemen A, Miyamoto T, Yu Y, et al:
Multi-compartment tumor organoids. Mater Today. 61:104–116. 2022.
View Article : Google Scholar
|
|
109
|
Russo GC, Crawford AJ, Clark D, Cui J,
Carney R, Karl MN, Su B, Starich B, Lih T, Kamat P, et al:
E-cadherin interacts with EGFR resulting in hyper-activation of ERK
in multiple models of breast cancer. bioRxiv. 2020.
|
|
110
|
Muhamed I, Wu J, Sehgal P, Kong X, Tajik
A, Wang N and Leckband DE: E-cadherin-mediated force transduction
signals regulate global cell mechanics. J Cell Sci. 129:1843–1854.
2016.PubMed/NCBI
|
|
111
|
Barry AK, Tabdili H, Muhamed I, Wu J,
Shashikanth N, Gomez GA, Yap AS, Gottardi CJ, de Rooij J, Wang N
and Leckband DE: α-catenin cytomechanics-role in cadherin-dependent
adhesion and mechanotransduction. J Cell Sci. 127:1779–1791. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Benham-Pyle BW, Pruitt BL and Nelson WJ:
Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1
and β-catenin activation to drive cell cycle entry. Science.
348:1024–1027. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Röper JC, Mitrossilis D, Stirnemann G,
Waharte F, Brito I, Fernandez-Sanchez ME, Baaden M, Salamero J and
Farge E: The major β-catenin/E-cadherin junctional binding site is
a primary molecular mechano-transductor of differentiation in vivo.
Elife. 7:e333812018. View Article : Google Scholar
|
|
114
|
Fernández-Sánchez ME, Barbier S, Whitehead
J, Béalle G, Michel A, Latorre-Ossa H, Rey C, Fouassier L, Claperon
A, Brullé L, et al: Mechanical induction of the tumorigenic
β-catenin pathway by tumour growth pressure. Nature. 523:92–95.
2015. View Article : Google Scholar
|
|
115
|
Hino N, Rossetti L, Marín-Llauradó A, Aoki
K, Trepat X, Matsuda M and Hirashima T: ERK-mediated
mechanochemical waves direct collective cell polarization. Dev
Cell. 53:646–660.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Coló GP, Hernández-Varas P, Lock J,
Bartolomé RA, Arellano-Sánchez N, Strömblad S and Teixidó J: Focal
adhesion disassembly is regulated by a RIAM to MEK-1 pathway. J
Cell Sci. 125:5338–5352. 2012.PubMed/NCBI
|
|
117
|
Das T, Safferling K, Rausch S, Grabe N,
Boehm H and Spatz JP: A molecular mechanotransduction pathway
regulates collective migration of epithelial cells. Nat Cell Biol.
17:276–287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Colak-Champollion T, Lan L, Jadhav AR,
Yamaguchi N, Venkiteswaran G, Patel H, Cammer M,
Meier-Schellersheim M and Knaut H: Cadherin-mediated cell coupling
coordinates chemokine sensing across collectively migrating cells.
Curr Biol. 29:2570–2579.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Heneberg P: Paracrine tumor signaling
induces transdifferentiation of surrounding fibroblasts. Crit Rev
Oncol Hematol. 97:303–311. 2016. View Article : Google Scholar
|
|
120
|
Barbazán J and Matic Vignjevic D: Cancer
associated fibroblasts: Is the force the path to the dark side?
Curr Opin Cell Biol. 56:71–79. 2019. View Article : Google Scholar
|
|
121
|
Ishii G, Ochiai A and Neri S: Phenotypic
and functional heterogeneity of cancer-associated fibroblast within
the tumor microenvironment. Adv Drug Deliv Rev. 99:186–196. 2016.
View Article : Google Scholar
|
|
122
|
Attaran S, Skoko JJ, Hopkins BL, Wright
MK, Wood LE, Asan A, Woo HA, Feinberg A and Neumann CA:
Peroxiredoxin-1 Tyr194 phosphorylation regulates LOX-dependent
extracellular matrix remodelling in breast cancer. Br J Cancer.
125:1146–1157. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ
and Werb Z: Concepts of extracellular matrix remodelling in tumour
progression and metastasis. Nat Commun. 11:51202020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Kai F, Drain AP and Weaver VM: The
extracellular matrix modulates the metastatic journey. Dev Cell.
49:332–346. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Poltavets V, Kochetkova M, Pitson SM and
Samuel MS: The role of the extracellular matrix and its molecular
and cellular regulators in cancer cell plasticity. Front Oncol.
8:4312018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Levental KR, Yu H, Kass L, Lakins JN,
Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et
al: Matrix crosslinking forces tumor progression by enhancing
integrin signaling. Cell. 139:891–906. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Elosegui-Artola A, Oria R, Chen Y,
Kosmalska A, Pérez-González C, Castro N, Zhu C, Trepat X and
Roca-Cusachs P: Mechanical regulation of a molecular clutch defines
force transmission and transduction in response to matrix rigidity.
Nat Cell Biol. 18:540–548. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Miranti CK and Brugge JS: Sensing the
environment: A historical perspective on integrin signal
transduction. Nat Cell Biol. 4:E83–E90. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Maritzen T, Schachtner H and Legler DF: On
the move: Endocytic trafficking in cell migration. Cell Mol Life
Sci. 72:2119–2134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Sawada Y, Tamada M, Dubin-Thaler BJ,
Cherniavskaya O, Sakai R, Tanaka S and Sheetz MP: Force sensing by
mechanical extension of the Src family kinase substrate p130Cas.
Cell. 127:1015–1026. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Tamada M, Sheetz MP and Sawada Y:
Activation of a signaling cascade by cytoskeleton stretch. Dev
Cell. 7:709–718. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Van Helvert S, Storm C and Friedl P:
Mechanoreciprocity in cell migration. Nat Cell Biol. 20:8–20. 2018.
View Article : Google Scholar :
|
|
133
|
Keating M, Kurup A, Alvarez-Elizondo M,
Levine AJ and Botvinick E: Spatial distributions of pericellular
stiffness in natural extracellular matrices are dependent on
cell-mediated proteolysis and contractility. Acta Biomater.
57:304–312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Bi D, Lopez JH, Schwarz JM and Manning ML:
A density-independent rigidity transition in biological tissues.
Nat Phys. 11:1074–1079. 2015. View Article : Google Scholar
|
|
135
|
Wolf K, Te Lindert M, Krause M, Alexander
S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ and Friedl
P: Physical limits of cell migration: Control by ECM space and
nuclear deformation and tuning by proteolysis and traction force. J
Cell Biol. 201:1069–1084. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Han YL, Ronceray P, Xu G, Malandrino A,
Kamm RD, Lenz M, Broedersz CP and Guo M: Cell contraction induces
long-ranged stress stiffening in the extracellular matrix. Proc
Natl Acad Sci USA. 115:4075–4080. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Roomi MW, Monterrey JC, Kalinovsky T, Rath
M and Niedzwiecki A: Patterns of MMP-2 and MMP-9 expression in
human cancer cell lines. Oncol Rep. 21:1323–1333. 2009.PubMed/NCBI
|
|
138
|
Isomursu A, Park KY, Hou J, Cheng B,
Mathieu M, Shamsan GA, Fuller B, Kasim J, Mahmoodi MM, Lu TJ, et
al: Directed cell migration towards softer environments. Nat Mater.
21:1081–1090. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Sunyer R, Conte V, Escribano J,
Elosegui-Artola A, Labernadie A, Valon L, Navajas D, García-Aznar
JM, Muñoz JJ, Roca-Cusachs P and Trepat X: Collective cell
durotaxis emerges from long-range intercellular force transmission.
Science. 353:1157–1161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D
and Semenza GL: Hypoxia-inducible factor 1 (HIF-1) promotes
extracellular matrix remodeling under hypoxic conditions by
inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol
Chem. 288:10819–10829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Chandler EM, Saunders MP, Yoon CJ, Gourdon
D and Fischbach C: Adipose progenitor cells increase fibronectin
matrix strain and unfolding in breast tumors. Phys Biol.
8:0150082011. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Guiet R, Van Goethem E, Cougoule C, Balor
S, Valette A, Al Saati T, Lowell CA, Le Cabec V and
Maridonneau-Parini I: The process of macrophage migration promotes
matrix metalloproteinase-independent invasion by tumor cells. J
Immunol. 187:3806–3814. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Vilchez Mercedes SA, Bocci F, Levine H,
Onuchic JN, Jolly MK and Wong PK: Decoding leader cells in
collective cancer invasion. Nat Rev Cancer. 21:592–604. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Artym VV, Zhang Y, Seillier-Moiseiwitsch
F, Yamada KM and Mueller SC: Dynamic interactions of cortactin and
membrane type 1 matrix metalloproteinase at invadopodia: Defining
the stages of invadopodia formation and function. Cancer Res.
66:3034–3043. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Hoshino D, Kirkbride KC, Costello K, Clark
ES, Sinha S, Grega-Larson N, Tyska MJ and Weaver AM: Exosome
secretion is enhanced by invadopodia and drives invasive behavior.
Cell Rep. 5:1159–1168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Han T, Kang D, Ji D, Wang X, Zhan W, Fu M,
Xin HB and Wang JB: How does cancer cell metabolism affect tumor
migration and invasion? Cell Adh Migr. 7:395–403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kim KS, Sengupta S, Berk M, Kwak YG,
Escobar PF, Belinson J, Mok SC and Xu Y: Hypoxia enhances
lysophosphatidic acid responsiveness in ovarian cancer cells and
lysophosphatidic acid induces ovarian tumor metastasis in vivo.
Cancer Res. 66:7983–7990. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Egeblad M, Rasch MG and Weaver VM: Dynamic
interplay between the collagen scaffold and tumor evolution. Curr
Opin Cell Biol. 22:697–706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Rofstad EK, Mathiesen B, Kindem K and
Galappathi K: Acidic extracellular pH promotes experimental
metastasis of human melanoma cells in athymic nude mice. Cancer
Res. 66:6699–6707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Pedron S, Becka E and Harley BA:
Regulation of glioma cell phenotype in 3D matrices by hyaluronic
acid. Biomaterials. 34:7408–7417. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Cha J, Kang SG and Kim P: Strategies of
mesenchymal invasion of patient-derived brain tumors:
Microenvironmental adaptation. Sci Rep. 6:249122016. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Kim Y and Kumar S: CD44-mediated adhesion
to hyaluronic acid contributes to mechanosensing and invasive
motility. Mol Cancer Res. 12:1416–1429. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Belousov A, Titov S, Shved N, Garbuz M,
Malykin G, Gulaia V, Kagansky A and Kumeiko V: The extracellular
matrix and biocompatible materials in glioblastoma treatment. Front
Bioeng Biotechnol. 7:3412019. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Serres E, Debarbieux F, Stanchi F,
Maggiorella L, Grall D, Turchi L, Burel-Vandenbos F,
Figarella-Branger D, Virolle T, Rougon G and Van
Obberghen-Schilling E: Fibronectin expression in glioblastomas
promotes cell cohesion, collective invasion of basement membrane in
vitro and orthotopic tumor growth in mice. Oncogene. 33:3451–3462.
2014. View Article : Google Scholar
|
|
155
|
Condeelis J, Singer RH and Segall JE: The
great escape: When cancer cells hijack the genes for chemotaxis and
motility. Annu Rev Cell Dev Biol. 21:695–718. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Tweedy L, Thomason PA, Paschke PI, Martin
K, Machesky LM, Zagnoni M and Insall RH: Seeing around corners:
Cells solve mazes and respond at a distance using attractant
breakdown. Science. 369:eaay97922020. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Susanto O, Koh YWH, Morrice N, Tumanov S,
Thomason PA, Nielson M, Tweedy L, Muinonen-Martin AJ, Kamphorst JJ,
Mackay GM and Insall RH: LPP3 mediates self-generation of
chemotactic LPA gradients by melanoma cells. J Cell Sci.
130:3455–3466. 2017.PubMed/NCBI
|
|
158
|
Tweedy L, Knecht DA, Mackay GM and Insall
RH: Self-generated chemoattractant gradients: Attractant depletion
extends the range and robustness of chemotaxis. PLoS Biol.
14:e10024042016. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Scherber C, Aranyosi AJ, Kulemann B,
Thayer SP, Toner M, Iliopoulos O and Irimia D: Epithelial cell
guidance by self-generated EGF gradients. Integr Biol (Camb).
4:259–269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Muinonen-Martin AJ, Susanto O, Zhang Q,
Smethurst E, Faller WJ, Veltman DM, Kalna G, Lindsay C, Bennett DC,
Sansom OJ, et al: Melanoma cells break down LPA to establish local
gradients that drive chemotactic dispersal. PLoS Biol.
12:e10019662014. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Sobolik T, Su YJ, Wells S, Ayers GD, Cook
RS and Richmond A: CXCR4 drives the metastatic phenotype in breast
cancer through induction of CXCR2 and activation of MEK and PI3K
pathways. Mol Biol Cell. 25:566–582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Malet-Engra G, Yu W, Oldani A, Rey-Barroso
J, Gov NS, Scita G and Dupré L: Collective cell motility promotes
chemotactic prowess and resistance to chemorepulsion. Curr Biol.
25:242–250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Boucharaba A, Serre CM, Grès S,
Saulnier-Blache JS, Bordet JC, Guglielmi J, Clézardin P and
Peyruchaud O: Platelet-derived lysophosphatidic acid supports the
progression of osteolytic bone metastases in breast cancer. J Clin
Invest. 114:1714–1725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Zhao C, Sardella A, Chun J, Poubelle PE,
Fernandes MJ and Bourgoin SG: TNF-alpha promotes LPA1- and
LPA3-mediated recruitment of leukocytes in vivo through CXCR2
ligand chemokines. J Lipid Res. 52:1307–1318. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Juin A, Spence HJ, Martin KJ, McGhee E,
Neilson M, Cutiongco MFA, Gadegaard N, Mackay G, Fort L, Lilla S,
et al: N-WASP control of LPAR1 trafficking establishes response to
self-Generated LPA gradients to promote pancreatic cancer cell
metastasis. Dev Cell. 51:431–445.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Tweedy L and Insall RH: Self-generated
gradients yield exceptionally robust steering cues. Front Cell Dev
Biol. 8:1332020. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Bray SJ: Notch signalling in context. Nat
Rev Mol Cell Biol. 17:722–735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Bocci F, Gearhart-Serna L, Boareto M,
Ribeiro M, Ben-Jacob E, Devi GR, Levine H, Onuchic JN and Jolly MK:
Toward understanding cancer stem cell heterogeneity in the tumor
microenvironment. Proc Natl Acad Sci USA. 116:148–157. 2019.
View Article : Google Scholar :
|
|
169
|
Lewis J: Notch signalling and the control
of cell fate choices in vertebrates. Semin Cell Dev Biol.
9:583–589. 1998. View Article : Google Scholar
|
|
170
|
Tashima Y and Okajima T: Congenital
diseases caused by defective O-glycosylation of notch receptors.
Nagoya J Med Sci. 80:299–307. 2018.PubMed/NCBI
|
|
171
|
Shimojo H, Ohtsuka T and Kageyama R:
Dynamic expression of notch signaling genes in neural
stem/progenitor cells. Front Neurosci. 5:782011. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Manderfield LJ, High FA, Engleka KA, Liu
F, Li L, Rentschler S and Epstein JA: Notch activation of Jagged1
contributes to the assembly of the arterial wall. Circulation.
125:314–323. 2012. View Article : Google Scholar :
|
|
173
|
Boareto M, Jolly MK, Lu M, Onuchic JN,
Clementi C and Ben-Jacob E: Jagged-Delta asymmetry in notch
signaling can give rise to a sender/receiver hybrid phenotype. Proc
Natl Acad Sci USA. 112:E402–E409. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Luca VC, Jude KM, Pierce NW, Nachury MV,
Fischer S and Garcia KC: Structural biology. Structural basis for
Notch1 engagement of Delta-like 4. Science. 347:847–853. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Rana NA and Haltiwanger RS: Fringe
benefits: Functional and structural impacts of O-glycosylation on
the extracellular domain of notch receptors. Curr Opin Struct Biol.
21:583–589. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Jolly MK, Boareto M, Lu M, Onuchic JN,
Clementi C and Ben-Jacob E: Operating principles of
Notch-Delta-Jagged module of cell-cell communication. New J Phys.
17:0550212015. View Article : Google Scholar
|
|
177
|
Ruhrberg C, Gerhardt H, Golding M, Watson
R, Ioannidou S, Fujisawa H, Betsholtz C and Shima DT: Spatially
restricted patterning cues provided by heparin-binding VEGF-A
control blood vessel branching morphogenesis. Genes Dev.
16:2684–2698. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Lobov IB, Renard RA, Papadopoulos N, Gale
NW, Thurston G, Yancopoulos GD and Wiegand SJ: Delta-like ligand 4
(Dll4) is induced by VEGF as a negative regulator of angiogenic
sprouting. Proc Natl Acad Sci USA. 104:3219–3224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Benedito R, Rocha SF, Woeste M, Zamykal M,
Radtke F, Casanovas O, Duarte A, Pytowski B and Adams RH:
Notch-dependent VEGFR3 upregulation allows angiogenesis without
VEGF-VEGFR2 signalling. Nature. 484:110–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Noguera-Troise I, Daly C, Papadopoulos NJ,
Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD and Thurston
G: Blockade of Dll4 inhibits tumour growth by promoting
non-productive angiogenesis. Nature. 444:1032–1037. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Han M, Xu W, Cheng P, Jin H and Wang X:
Histone demethylase lysine demethylase 5B in development and
cancer. Oncotarget. 8:8980–8991. 2017. View Article : Google Scholar :
|
|
182
|
Zheng YC, Chang J, Wang LC, Ren HM, Pang
JR and Liu HM: Lysine demethylase 5B (KDM5B): A potential
anti-cancer drug target. Eur J Med Chem. 161:131–140. 2019.
View Article : Google Scholar
|
|
183
|
Horton JR, Engstrom A, Zoeller EL, Liu X,
Shanks JR, Zhang X, Johns MA, Vertino PM, Fu H and Cheng X:
Characterization of a linked jumonji domain of the KDM5/JARID1
family of histone H3 lysine 4 demethylases. J Biol Chem.
291:2631–2646. 2016. View Article : Google Scholar :
|
|
184
|
Hinohara K, Wu HJ, Vigneau S, McDonald TO,
Igarashi KJ, Yamamoto KN, Madsen T, Fassl A, Egri SB, Papanastasiou
M, et al: KDM5 histone demethylase activity links cellular
transcriptomic heterogeneity to therapeutic resistance. Cancer
Cell. 34:939–953.e9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Goley ED and Welch MD: The ARP2/3 complex:
An actin nucleator comes of age. Nat Rev Mol Cell Biol. 7:713–726.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Faubert B, Solmonson A and DeBerardinis
RJ: Metabolic reprogramming and cancer progression. Science.
368:eaaw54732020. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Warhurg O, Posener K and Negelein E: Über
den Stoffwechsel der Carcinomzelle. Naturwissenschaften.
12:1131–1137. 1924. View Article : Google Scholar
|
|
189
|
Zanotelli MR, Rahman-Zaman A, VanderBurgh
JA, Taufalele PV, Jain A, Erickson D, Bordeleau F and Reinhart-King
CA: Energetic costs regulated by cell mechanics and confinement are
predictive of migration path during decision-making. Nat Commun.
10:41852019. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Commander R, Wei C, Sharma A, Mouw JK,
Burton LJ, Summerbell E, Mahboubi D, Peterson RJ, Konen J, Zhou W,
et al: Subpopulation targeting of pyruvate dehydrogenase and GLUT1
decouples metabolic heterogeneity during collective cancer cell
invasion. Nat Commun. 11:15332020. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Tan Z, Yang C, Zhang X, Zheng P and Shen
W: Expression of glucose transporter 1 and prognosis in non-small
cell lung cancer: A pooled analysis of 1665 patients. Oncotarget.
8:609542017. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Yu M, Yongzhi H, Chen S, Luo X, Lin Y,
Zhou Y, Jin H, Hou B, Deng Y, Tu L and Jian Z: The prognostic value
of GLUT1 in cancers: A systematic review and meta-analysis.
Oncotarget. 8:43356–43367. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Carvalho KC, Cunha IW, Rocha RM, Ayala FR,
Cajaíba MM, Begnami MD, Vilela RS, Paiva GR, Andrade RG and Soares
FA: GLUT1 expression in malignant tumors and its use as an
immunodiagnostic marker. Clinics (Sao Paulo). 66:965–972. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Bos R, van Der Hoeven JJ, van Der Wall E,
van Der Groep P, van Diest PJ, Comans EF, Joshi U, Semenza GL,
Hoekstra OS, Lammertsma AA and Molthoff CF: Biologic correlates of
(18) fluorodeoxyglucose uptake in human breast cancer measured by
positron emission tomography. J Clin Oncol. 20:379–387. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Schwartzenberg-Bar-Yoseph F, Armoni M and
Karnieli E: The tumor suppressor p53 down-regulates glucose
transporters GLUT1 and GLUT4 gene expression. Cancer Res.
64:2627–2633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Chen C, Pore N, Behrooz A, Ismail-Beigi F
and Maity A: Regulation of glut1 mRNA by hypoxia-inducible
factor-1. Interaction between H-ras and hypoxia. J Biol Chem.
276:9519–9525. 2001. View Article : Google Scholar
|
|
197
|
Tanegashima K, Sato-Miyata Y, Funakoshi M,
Nishito Y, Aigaki T and Hara T: Epigenetic regulation of the
glucose transporter gene Slc2a1 by β-hydroxybutyrate underlies
preferential glucose supply to the brain of fasted mice. Genes
Cells. 22:71–83. 2017. View Article : Google Scholar
|
|
198
|
Barthel A, Okino ST, Liao J, Nakatani K,
Li J, Whitlock JP Jr and Roth RA: Regulation of GLUT1 gene
transcription by the serine/threonine kinase Akt1. J Biol Chem.
274:20281–20286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Cunniff B, McKenzie AJ, Heintz NH and Howe
AK: AMPK activity regulates trafficking of mitochondria to the
leading edge during cell migration and matrix invasion. Mol Biol
Cell. 27:2662–2674. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Alert R and Trepat X: Physical models of
collective cell migration. Annu Rev Condens Matter Phys. 11:77–101.
2020. View Article : Google Scholar
|
|
201
|
Merino-Casallo F, Gomez-Benito MJ,
Hervas-Raluy S and Garcia-Aznar JM: Unravelling cell migration:
Defining movement from the cell surface. Cell Adh Migr. 16:25–64.
2022. View Article : Google Scholar : PubMed/NCBI
|