Introduction
Prostate cancer (PCa), which usually occurs in
middle-aged and older men, is the top hereditary cancer type of all
major cancers and is also the most common non-skin cancer in men
worldwide. Despite new advances in diagnosis and treatment of PCa,
this disease remains a major medical problem in men due to the
overtreatment of inherently benign disease and the undertreatment
of metastatic PCa (1-3). Numerous patients with PCa are at an
advanced stage or have metastases at the time of diagnosis
(4). In patients with distant
metastases, the 5-year relative survival rate decreases from 80% in
patients with no metastases to 30% (5).
Early stages of PCa are known as androgen-dependent
PCa and almost all patients who receive androgen deprivation
therapy (ADT) will progress to castration-resistant PCa (CRPC)
(6,7). CRPC is prone to distant metastasis
and is more aggressive, making it very difficult to treat (7). PCa treated with ADT is also
susceptible to neuroendocrine transformation and even evolution to
neuroendocrine PCa (NEPC) (8). As
a very poor prognostic subtype of CRPC, the five-year survival rate
for NEPC patients is only 10% (9). Given the worse prognosis and limited
treatment options for CRPC and NEPC, the present study focused on
both.
The vascular endothelium controls the entry of
nutrients into tissues, maintains blood flow, and regulates the
trafficking of white blood cells. Evidence have revealed that
abnormalities in the tumor endothelium contribute to tumor growth
and metastasis (10). Given the
role of blood vessels in maintaining tumors, they have become an
increasingly attractive target for the development of anticancer
therapies. To improve understanding and manipulation of the
relevant pathological mechanisms, researchers must elucidate and
exploit the molecular mechanisms of angiogenesis (11,12).
For the aforementioned, the effects of CDK12/insulin
like growth factor binding protein 3 (IGFBP3) on cellular
biological behavior and angiogenesis in PCa were explored. CDK12 is
a serine/threonine protein kinase that regulates transcriptional
and post-transcriptional processes, thereby regulating various
cellular functions (13). This
molecule is critical for the development of cancer (14). After a previous study by the
authors identified the pro-oncogenic role of CDK12 in PCa and its
negative regulation of another important factor, IGFBP3, the
authors delved into the effect of IGFBP3 on PCa. There are no
studies on whether CDK12 plays a role in PCa by inhibiting IGFBP3,
thus it was first revealed that CDK12 promotes PCa by inhibiting
IGFBP3. IGFBP3 is a proapoptotic, anti-metastatic and
antiangiogenic protein that has previously been demonstrated to
induce apoptosis using insulin-like growth factor receptor
(IGF)-dependent and non-dependent mechanisms (15,16). There have been numerous cancer
studies on IGFBP3 as a proapoptotic molecule in vitro
(17). Growing evidence has
revealed that IGFBP regulates angiogenesis (18). After showing that IGFBP3 has a
strong inhibitory effect on angiogenesis in PCa (15), the authors subsequently linked
CDK12 to angiogenesis and designed experiments to verify the
proangiogenic effect of CDK12, which has not been reported by
others thus far.
Materials and methods
Cell culture
PCa cells (C4-2 and PC3) and human umbilical vein
endothelial cells (HUVECs) were provided by the Institute of
Urology, The First Affiliated Hospital of Anhui Medical University
(Hefei, China). The minimum number of passages of cells obtained
for the first time was 1-2 generations. C4-2 cells were originally
isolated from CRPC; PC3 cells were originally isolated from NEPC.
PCa cells and HUVECs were cultured in RPMI-1640 medium (cat. no.
SH30809.01; HyClone; Cytiva) with 10% fetal bovine serum (cat. no.
086-150; Wisent Biotechnology) and 1% penicillin/streptomycin (cat.
no. C0222; Beyotime Institute of Biotechnology). The environment of
the cell culture was 37°C, 5% CO2, 95% relative
humidity. Cells were digested with 0.25% trypsin digestion solution
(cat. no. C0201; Beyotime Institute of Biotechnology).
RNA sequencing (RNA-Seq) analysis
The mRNA of C4-2 cells with and without CDK12
knockdown was extracted following the RNeasy Pure mRNA Bead Kit
(cat. no. 180244; Qiagen China Co., Ltd.) and the mRNA levels were
detected using Duke University's Illumina sequencing platform. The
type of sequencing performed was 150 bp for length and 'paired end'
for sequencing direction. The loading concentration of the final
library was measured using NanoDrop detection at 10 nM. R studio
3.0.1 was used to analyze the data (https://cran-archive.r-project.org/bin/windows/base/old/3.0.1/).
The C4-2 cell aliases used in RNA-Seq are LNCaP-C4-2; LNCaP subline
C4-2; C4-2; C42; Sp 2817 (cat. no. CRL-3314; American Type Culture
Collection). Knockdown of CDK12 in C4-2 cells was performed using
CRISPR technology, the oligonucleotides were as follows: forward,
5′-CAC CGC GGC GAC GTC AGC GAC AAA G-3′; and reverse, 5′-AAA CCT
TTG TCG CTG ACG TCG CCG C-3′. Control cells were transfected with
the empty vector plasmid Lenti-CRISPR-V2. The oligo sequences were
designed using Benchling (https://www.benchling.com). The RNA sequencing data
was uploaded to the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) with the GEO
number GSE246983.
CRISPR/CAS9 dual vector lentiviral
infection of cells
The products were provided by Shanghai Genechem Co.,
Ltd. and included Lenti-single guide (sg)RNA-EGFP virus, expressing
the target gene sgRNA sequence and control sgRN sequence and
Lenti-CAS9-puro virus, expressing the CAS9 protein with puromycin
resistance. The CRISPR/CAS9 dual-vector lentivirus enables the
knockout of the target gene by introducing the CAS9 protein and the
sgRNA sequence expression frame into the cell via two lentiviruses,
the CAS9 protein vector with puromycin labeling and sgRNA with a
green fluorescent label. C4-2 and PC3 cells were digested and the
cell suspension was then seeded in 6-well plates and cultured until
the cell confluence reached ~30%. According to the cellular MOI
value of 50, an appropriate amount of virus was added. The cell
status was observed after 12 h; if there was no obvious
cytotoxicity, the medium was replaced after another 24 h; if there
was any significant cytotoxicity, the medium was replaced
immediately. Three days after infection, an appropriate
concentration of puromycin was added to screen for 3 days, after
which a low concentration of puromycin was used for continued
culture and continued infection with sgRNA lentivirus. The three
sgRNA sequences and the control insertion sequences were as
follows: PCA09470, 5′-GGG GGA GAC AGA TCT CCA CC-3′; PCA09471,
5′-AGA TAC AGG GAA AGT AAA GT-3′; PCA09472, 5′-TAT GAG ATC GTG AGG
GAC TA-3′; and CON246, 5′-CGC TTC CGC GGC CCG TTC AA-3′. The oligo
sequences were designed using the CRISPOR tool version 5.01
(http://crispor.tefor.net/). Cells
infected successfully with sgRNA were sorted by green fluorescence.
Lenti-CAS9-puro viruses were infected at 37°C for a duration of at
least 3 days and then screened for 3 days with the addition of
Puromycin. After that, Lenti-sgRNA-EGFP viruses were infected at
37°C for a duration of at least 3 days. The duration of cellular
infection by both lentiviruses was at least 3 days each. After the
Lenti-sgRNA-EGFP viruses were also infected, fluorescence
photography revealed the presence of ~80% cells infected.
Subsequent experiments were then started. The antibiotic
concentration used for selection was 2.00 μg/ml, after which
this low concentration was maintained until the Lenti-sgRNA-EGFP
viruses had also completed infection. The antibiotic concentration
was not maintained after the completion of this experiment.
Transfection of siR NA
RNA primers (20 μM) provided by Shanghai
GenePharma Co., Ltd., included: IGFBP3-Homo-915 forward, 5′-GCU GGU
GUG UGG AUA AGU ATT-3′; and reverse, 5′-UAC UUA UCC ACA CAC CAG
CTT-3′; and negative control FAM forward, 5′-UUC UCC GAA CGU GUC
ACG UTT-3′ and reverse, 5′-ACG UGA CAC GUU CGG AGA ATT-3′.
Transfection with siRNA was started when the cell density reached
40 to 70%. Subsequently, 200 μl of Opti-MEM™ I Reduced Serum
Medium (cat. no. 31985070; Gibco; Thermo Fisher Scientific, Inc.)
was added to each of two DNase/RNase-Free Eppendorf (EP) tubes. One
tube had 4 μl of Lipofectamine™ 2000 (cat. no. 11668030;
Thermo Fisher Scientific, Inc.) and the other had 8 μl of
RNA primers. After incubation for 5 min at room temperature, the
contents of the two tubes were mixed and then left to stand for 20
min at room temperature. After the cells were washed with PBS, 1600
μl of serum-free medium was added to each well and the
mixture was then added. Cells were cultured in a CO2
incubator at 37°C for 6 h and subsequently changed to serum-added
medium while the transfection time was recorded. The time interval
between transfection and subsequent experiments was ~24 h.
Co-immunoprecipitation (Co-IP)
The Pierce™ Classic Magnetic IP/Co-IP Kit (cat. no.
88804; Thermo Fisher Scientific, Inc.) was used to detect the
presence of interactions between CDK12 and IGFBP3. When the density
of PC3 cells in the 6-well plate reached 80%, the medium was
aspirated and 1 ml of lysis solution was added to one well after
two washes with prechilled PBS for 5 min on ice. The cells were
scraped off with a cell scraper and the collected cell suspension
was added to an EP tube and placed on ice. The samples were
centrifuged at 15,000 × g for 10 min at 4°C. After centrifugation,
the supernatant was transferred to a new centrifuge tube, which
contained the cell lysate. To reduce the amount of non-specific
proteins in the lysate mixture and to reduce the amount of proteins
that may cross-react with the protein A/G beads, 1 μl of
Goat Anti-Rabbit IgG Antibody (H+L), HRP-conjugated (1:1,000; cat.
no. bs-0295G; BIOSS) and 20 μl of Protein A/G beads were
added to the cell lysates. The samples were incubated for 30-60 min
at 4°C with rotation and then centrifuged for 1 min at 15,000 × g
at 4°C. The supernatant was transferred to a new EP tube and was
set aside. Subsequently, 500 μl of lysate was added to the
new EP tube. Anti-IGFBP3 antibody (1:70; cat. no. ab193910; Abcam)
was added, mixed gently and then incubated overnight at 4°C. After
the incubation, 20 μl Protein A/G magnetic beads were added,
mixed gently and then incubated overnight at 4°C. Subsequently, the
supernatant was discarded after centrifugation at 11,000 × g for 3
min (50 μl of supernatant was retained as 'input') and the
immunoprecipitate was retained. The precipitate was washed with 200
μl of lysate, centrifuged at 11,000 × g for 1 min and
retained. The sample was then washed once with 100 μl of 1X
conditioning buffer (100X for dilution). The precipitate was
resuspended in 20 to 40 μl of 2X SDS sample buffer and
vortexed. The samples were placed in a metal bath at 100°C for 5
min and they were finally placed in a centrifuge at 15,000 × g for
1 min. For western blotting, less than 10 μl of sample per
well was sufficient.
Western blotting
Proteins were extracted from the cells with RIPA
buffer (cat. no. P0013D; Beyotime Institute of Biotechnology) and
then separated on 7.5% SDS-polyacrylamide gels (cat. no. P2011; New
Cell & Molecular Biotech Co., Ltd.). The samples were then
transferred onto PVDF membranes (cat. no. 10600029; Cytiva) or NC
membranes (cat. no. 10600002; Cytiva). Non-specific binding was
blocked with 5% non-fat milk powder for 2 h at room temperature.
Primary antibodies were incubated for 12 h at 4°C: Anti-β-actin
(1:10,000; cat. no. T0022; Affinity Biosciences, Ltd.), anti-CDK12
(1:1,000; cat. no. 11973s; Cell Signaling Technology, Inc.),
anti-IGFBP3 (1:1,000; cat. no. ab193910; Abcam), anti-Akt (1:1,000;
cat. no. 9272; Cell Signaling Technology, Inc.), anti-p-Akt
(1:1,000; cat. no. T40067; Abmart Pharmaceutical Technology Co.,
Ltd.) and anti-VEGFA (1:1,000; cat. no. 19003-1-AP; ProteinTech
Group, Inc.). After incubation with the corresponding secondary
antibody (HRP-conjugated Affinipure Goat Anti-Mouse IgG; 1:10,000;
cat. no. SA00001-1; ProteinTech Group, Inc. and HRP-conjugated
Affinipure goat Anti-Rabbit IgG; 1:10,000; cat. no. SA00001-2;
ProteinTech Group, Inc.) for 1 h at room temperature, the protein
bands were visualized by an EZ-ECL Kit (cat. no. 20-500-120;
Biological Industries) using a gel imaging system (cat. no.
AL600RGB; GE Healthcare). Protein band intensities were normalized
to that of β-actin. ImageJ software (v.1.8; National Institutes of
Health) was used for densitometric analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells of the sgNT
group and sgCDK12 group using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.). The extracted mRNAs were then
subjected to reverse transcription to cDNA (37°C for 15 min, 85°C
for 5 sec, 4°C to cool down) by using Evo M-MLV RT Premix for qPCR
from Accurate Biotechnology Co., Ltd. (cat. no. AG11706). The
SYBR® Green Premix Pro Taq HS qPCR kit (cat. no.
AG11701; Accurate Biotechnology Co., Ltd.) assisted in quantifying
obtained cDNAs, and 7500 Software v.2.3 on Applied Biosystems™ 7500
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.) was adopted to detect quantified cDNAs. The qPCR conditions
were as follows: Initial denaturation at 95°C for 30 sec; followed
by 40 cycles of denaturation at 95°C for 5 sec; annealing at 60°C
for 30 sec and finally a system default dissociation stage. The
2-ΔΔCq method was used to quantify the fold change in
gene expression (19). Primers
were provided by Sangon Biotech Co., Ltd. The sequences of the
primers were as follows: CDK12 forward, 5′-CTA ACA GCA GAG AGC GTC
ACC-3′ and reverse, 5′-AAA GGT TTG ATA ACT GTG CCC A-3′ (20); IGFBP3 forward, 5′-TGT GGC CAT GAC
TGA GGA AA-3′ and reverse, 5′-TGC CGA CCT TCT TGG GTT T-3′
(21); and GAPDH forward, 5′-CAT
GAG AAG TAT GAC AAC AGC CT-3′ and reverse 5′-AGT CCT TCC ACG ATA
CCA AAG T-3′ (22).
Cell proliferation assay
Cell proliferation was detected using Cell Counting
Kit-8 (CCK-8; cat. no. C0005; TopScience) and EdU assays (cat. no.
C0071S; Beyotime Institute of Biotechnology). Cell suspensions of
the sgNT group, sgCDK12 group and siIGFBP3/sgCDK12 group were
spread in five 96-well plates with 5×103 cells per well
for the C4-2 cell line and 2×103 cells per well for the
PC3 cell line. CCK-8 reagent was added at 10:1 at 0, 24, 48, 72 and
96 h and incubated in the dark at 37°C for 2 h. Absorbance was
measured at 450 nm wavelength using a microplate reader
(PerkinElmer, Inc.). Cell suspensions of the sgNT group, sgCDK12
group and siIGFBP3/sgCDK12 group were spread overnight in 24-well
plates at 8×104 cells per well. Cells were labeled with
EdU, and 100 μl of click reaction solution was added to each
well and incubated at room temperature for 30 min in the dark.
Finally, the cells were stained with Hoechst 33342 for nuclear
staining. EdU labeling and Hoechst staining were examined using
fluorescence microscopy (Carl Zeiss AG). The fluorescence intensity
of the EdU-labeling proliferation assay was quantified by ImageJ
software (v.1.8; National Institutes of Health).
Colony formation assay
Cells from the sgNT group, sgCDK12 group and
siIGFBP3/sgCDK12 group were digested and spread into 6-well plates
at 5×103 cells per well for the C4-2 cell line and
1×103 cells per well for the PC3 cell line. The medium
was changed every 2-3 days. After 14 days at 37°C, the medium was
discarded and the cell colonies were fixed with 4% paraformaldehyde
at room temperature for 30 min and stained with crystal violet
staining solution (cat. no. C0121; Beyotime Institute of
Biotechnology) at room temperature for 15 min. Colonies consisting
of >50 cells were counted with ImageJ software (v.1.8; National
Institutes of Health).
Cell migration assay
Cell migration was detected using Transwell
migration assays and wound healing assays. Cell suspensions of the
sgNT group, sgCDK12 group and siIGFBP3/sgCDK12 group were spread
equally into Petri dishes. When the cell density reached the same
level, the medium was discarded in the dish and then three straight
lines of equal thickness on each dish were scratched with a
10-μl pipette tip. After PBS washes, serum-free medium was
added. Images were captured at ×40 magnification using a light
microscope (Olympus Co.) at 0, 24, 48, 72 and 96 h after scratching
to compare the wound healing ability of different cell lines. The
wound closures were calculated by ImageJ software. Transwell
migration assays were performed using a 24-well Transwell system
(cat. no. 3495; Corning, Inc.). Cells from the sgNT group, sgCDK12
group and siIGFBP3/sgCDK12 group were digested and then resuspended
in serum-free medium. A total of 1×105 cells were added
into each upper chamber. Subsequently, 600 μl of
serum-containing medium was added to the lower chambers. The cells
were incubated in a CO2 incubator at 37°C for 24 h. The
cells crossing the membrane were fixed with 4% paraformaldehyde at
room temperature for 30 min and then stained with 100% crystal
violet staining solution at room temperature for 15 min. Cells
crossing the membrane were counted by ImageJ software after taking
pictures under a light microscope (magnification, ×40 and ×100;
Olympus Corporation).
Tube formation assay
The cell supernatant from the sgNT group, sgCDK12
group and siIGFBP3/sgCDK12 group was taken as the conditioned
medium in advance. The materials that would come in contact with
the Matrigel matrix (cat. no. 356234; Corning, Inc.) were
precooled. When HUVECs grew to 70-90% confluence, the medium was
changed to serum-free medium. A total of 50 μl of Matrigel
matrix per well was added to a 96-well plate and then placed into
the incubator for 30 min to solidify the Matrigel matrix. HUVECs
were resuspended in ~1 ml of conditioned medium to adjust their
density to 3×105 cells/ml. A total of 100 μl of
cell suspension was placed in each well and the time was recorded
as 0 h after plating. Images were captured using a light microscope
(Olympus Corporation) at ×40 magnification at 2, 4 and 6 h. The
total tubule length was calculated using ImageJ software.
Immunohistochemistry (IHC)
Specimens were fixed in 10% neutral formalin at room
temperature for 48 h, paraffin-embedded and ultramicro-sectioned to
3-μm thick. Slices were dewaxed with xylene, rehydrated with
graded alcohol and incubated with 3% Peroxidase Closure Solution
(cat. no. ZLI-9311; Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.) at room temperature for 30 min to block endogenous
peroxidase activity. Slices were repaired with citrate repair
solution (cat. no. ZLI-9064; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.) at high pressure to repair antigen and
incubated overnight in an IHC wet box at 4°C with anti-CDK12 (1:50;
cat. no. ab246887; Abcam), anti-IGFBP3 (1:80; cat. no. ab193910;
Abcam), anti-VEGFA (1:200; cat. no. 19003-1-AP; ProteinTech Group,
Inc.) and anti-CD31 (1:2,000; cat. no. ab182981; Abcam) antibodies.
The next day, slices were washed with PBS and incubated with
HRP-labeled anti-rabbit IgG (100%; cat. no. DS-0004; Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd.) at room
temperature for 1 h. Immunoreactivity was detected with the DAB kit
(cat. no. ZLI-9018; Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.), and the sections were then observed under a light
microscope (magnification, ×100 and ×400; Olympus Corporation).
Scores for CDK12, IGFBP3, and VEGFA staining intensity were defined
as follows: negative, 0 points; weakly positive, 1 point; moderate,
2 points; and strongly positive, 3 points. The frequency of
positive cells was defined as follows: less than 5%, 0 points;
5-25%, 1 point; 26-50%, 2 points; 51-75%, 3 points and more than
75%, 4 points. CD31 IHC staining was used to determine the
microvessel density (MVD) by first finding the densest area of
microvessels with a ×10 objective and subsequently to assess the
number of microvessels in the ×20 objective field.
Hematoxylin-eosin (H&E) staining
Specimens were fixed in 10% neutral formalin at room
temperature for 48 h, paraffin-embedded and ultramicro-sectioned to
3-μm thick. Slices were dewaxed with xylene and rehydrated
with graded alcohol. Subsequently, slices were stained with H&E
using standard procedures. Hematoxylin staining was used for 5 min
at room temperature and eosin staining was used for 2 min at room
temperature. The sections were then observed under a light
microscope (magnification, ×100 and ×400; Olympus Corporation).
LinkedOmics database
The LinkedOmics database (http://www.linkedomics.org.) is a publicly available
portal that includes multiomics from all 32 cancer types in The
Cancer Genome Atlas (TCGA) and 10 from Clinical Proteomics Tumor
Analysis Consortium (CPTAC) cancer cohort data. After
identification of the 'prostate adenocarcinoma' type, the following
analysis was performed on September 27, 2022, according to the
'Manual and Tutorial' on the website: Pearson correlation
coefficient between CDK12 and IGFBP3, Pearson correlation
coefficient between CDK12 and VEGFA, Pearson correlation
coefficient between IGFBP3 and VEGFA, and the relationship between
CDK12, IGFBP3 and overall survival (OS) in patients with PCa.
In vivo tumor assays
The cells of the PC3/sgNT group and PC3/sgCDK12
group were suspended in serum-free medium. Six-week-old male BALB/c
nude mice (weight, 25-30 g; 3 mice/group; Jiangsu Huachuang Xinnuo
Pharmaceutical Technology Co, Ltd.) were injected subcutaneously
with 300 μl and 1×107 cells each. The cells were
placed on ice at all times during the injection. The protocol for
the animal experiments was approved by the Experimental Animal
Ethics Committee of Anhui Medical University (approval no.
LLSC20190458). Mice were housed under specific pathogen-free
conditions at 25°C with a 12 h light/dark cycle and free access to
food and water. Tumor size and mouse weight were measured every 3
days after feeding until the tumor was visible to the naked eye and
started to be recorded as Day 0. Tumor growth was calculated using
the following formula: Volume =0.52× length × width2.
When the tumors were nearly 15 mm in diameter, the mice were
sacrificed using the spinal dislocation method. The tumors were
taken from the mice, weighed and preserved.
Statistical analysis
All experiments had three independent replicates,
and the data were presented as the mean ± SD. ImageJ software
(v.1.8; National Institutes of Health) was used for densitometric
analysis. Statistical analyses were performed using GraphPad Prism
8.0.2 software (GraphPad Software; Dotmatics). Comparisons between
two groups were performed using a two-tailed unpaired Student's
t-test, and multiple groups were analyzed by one-way ANOVA followed
by Tukey's post hoc test for further pairwise comparisons.
Pearson's correlation coefficient was used to study the
relationship between variables. In all cases, P<0.05 was
considered to indicate a statistically significant difference.
Results
Role of CDK12 in the regulation of
IGFBP3
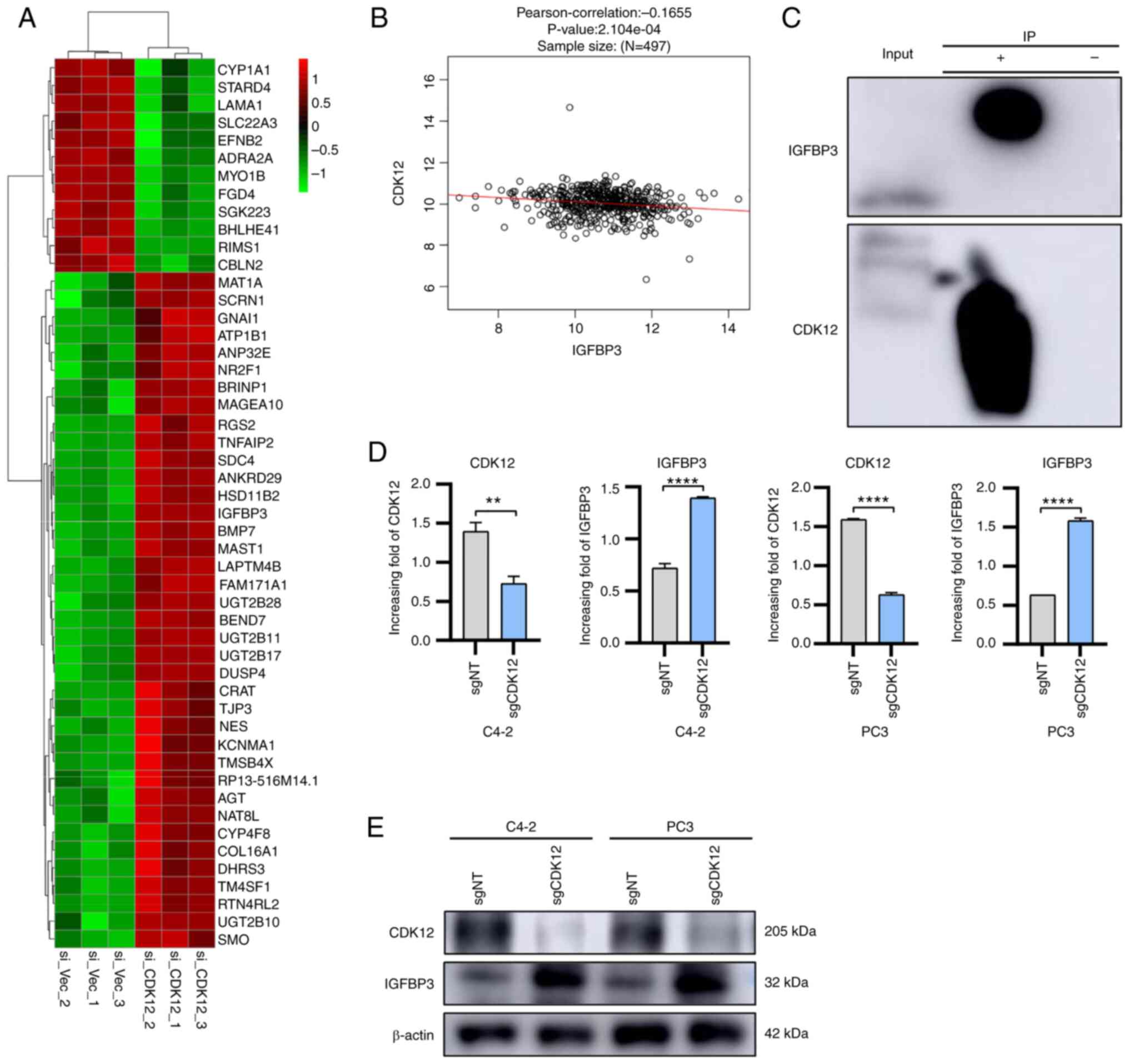
After knocking down CDK12 in C4-2 PCa cells, mRNA of
the C4-2 cells from the knockdown group and control group was
sequenced and the results are illustrated in a heatmap. The heatmap
revealed that IGFBP3 was significantly upregulated after CDK12 was
knocked down (Fig. 1A).
Subsequently, the Pearson correlation coefficient between CDK12 and
IGFBP3 was analyzed in the LinkedOmics database (http://www.linkedomics.org): There was a negative
correlation between CDK12 and IGFBP3 (Fig. 1B). Therefore, it was hypothesized
that CDK12 can inhibit the expression of IGFBP3 in anterior
adenocarcinoma cells. To further verify this conjecture, the
interaction between IGFBP3 protein and CDK12 protein by co-IP was
assessed (Fig. 1C). Next, the
CRISPR/CAS9 technique was used to stably knock out CDK12 in PCa
cells to further observe the relationship between CDK12 and IGFBP3.
RNA and protein were extracted from the sgNT group and sgCDK12
group for RT-qPCR and western blotting, respectively. The assay
results demonstrated that CDK12 was knocked down in the sgCDK12
group of cells and it was revealed that IGFBP3 in the sgCDK12 group
was higher at both the RNA and protein levels than that in the sgNT
group (Fig. 1D and E). In
summary, these findings indicated that CDK12 plays a negative
regulatory role on IGFBP3 in PCa cells.
CDK12 promotes the proliferation, colony
formation and migration of PCa cells by inhibiting IGFBP3
To clarify the role of CDK12/IGFBP3 in PCa cells,
IGFBP3 was knocked down on the basis of knocking out CDK12,
resulting in three groups of cells: i) sgNT group, ii) sgCDK12
group and iii) siIGFBP3/sgCDK12 group. These three groups of cells
were used for CCK-8, EdU and colony formation assays. CCK-8 assays
demonstrated that the proliferation ability of PCa cells decreased
after the knockout of CDK12; on the basis of knocking out CDK12,
the cell proliferation of the IGFBP3 knockdown group was restored
(Fig. 2A and B). EdU assays
revealed that the EdU positive ration decreased after the knockout
of CDK12; on the basis of knocking out CDK12, the EdU positive
ration of the IGFBP3 knockdown group was restored (Fig. 2C and D). Colony formation assays
showed that the number of cell colonies was reduced after the
knockout of CDK12; on the basis of knocking out CDK12, the number
of the IGFBP3 knockdown group was restored (Fig. 2E and F). Transwell migration and
wound healing assays were subsequently performed. Transwell
migration assays revealed that the intensity of cells through a
porous membrane decreased after CDK12 was knocked out; on the basis
of knocking out CDK12, the intensity of IGFBP3 knockdown group was
restored (Fig. 3A and B). Wound
healing assays demonstrated that the migration of PCa cells
decreased after CDK12 was knocked out; on the basis of knocking out
CDK12, the cell migration of IGFBP3 knockdown cells was restored
(Fig. 3C and D). In addition, the
relationship between CDK12, IGFBP3 and OS in PCa patients was
analyzed separately in the LinkedOmics database. The results
indicated that patients with CDK12 above the median had shorter OS
(P=0.9739) (Fig. S1A) while
patients with IGFBP3 above the median had longer OS (P=0.679)
(Fig. S1B). Therefore, it was
inferred that CDK12 promotes the proliferation, cloning and
migration of PCa cells and that IGFBP3 inhibits the proliferation,
cloning and migration of PCa cells. Furthermore, the aforementioned
cell behavioral effects of CDK12 on PCa are accomplished through
the inhibition of IGFBP3 expression.
 | Figure 2Effect of CDK12/IGFBP3 on cell
proliferation in advanced prostate cancer. Cells (C4-2 and PC3)
from the sgNT group, sgCDK12 group and siIGFBP3/sgCDK12 group were
cultured simultaneously in culture dishes of different
specifications. (A and B) Cell proliferation was analyzed by Cell
Counting Kit-8 assays, (C and D) EdU and (E and F) colony formation
assays. (C and D) Fluorescence microscopy of EdU and Hoechst
stains. Magnification, ×40. Two-tailed unpaired Student's t-test
was employed. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. IGFBP3,
insulin like growth factor binding protein 3; si, small
interfering; sg, single guide; sgNT group, group without knockdown
of CDK12 and IGFBP3 in cells; sgCDK12 group, group with knockdown
of CDK12 using CRISPR technology; siIGFBP3/sgCDK12 group, group
with knockdown of CDK12 using technology and knockdown of IGFBP3 by
transfection with siRNA; ns, not significant. |
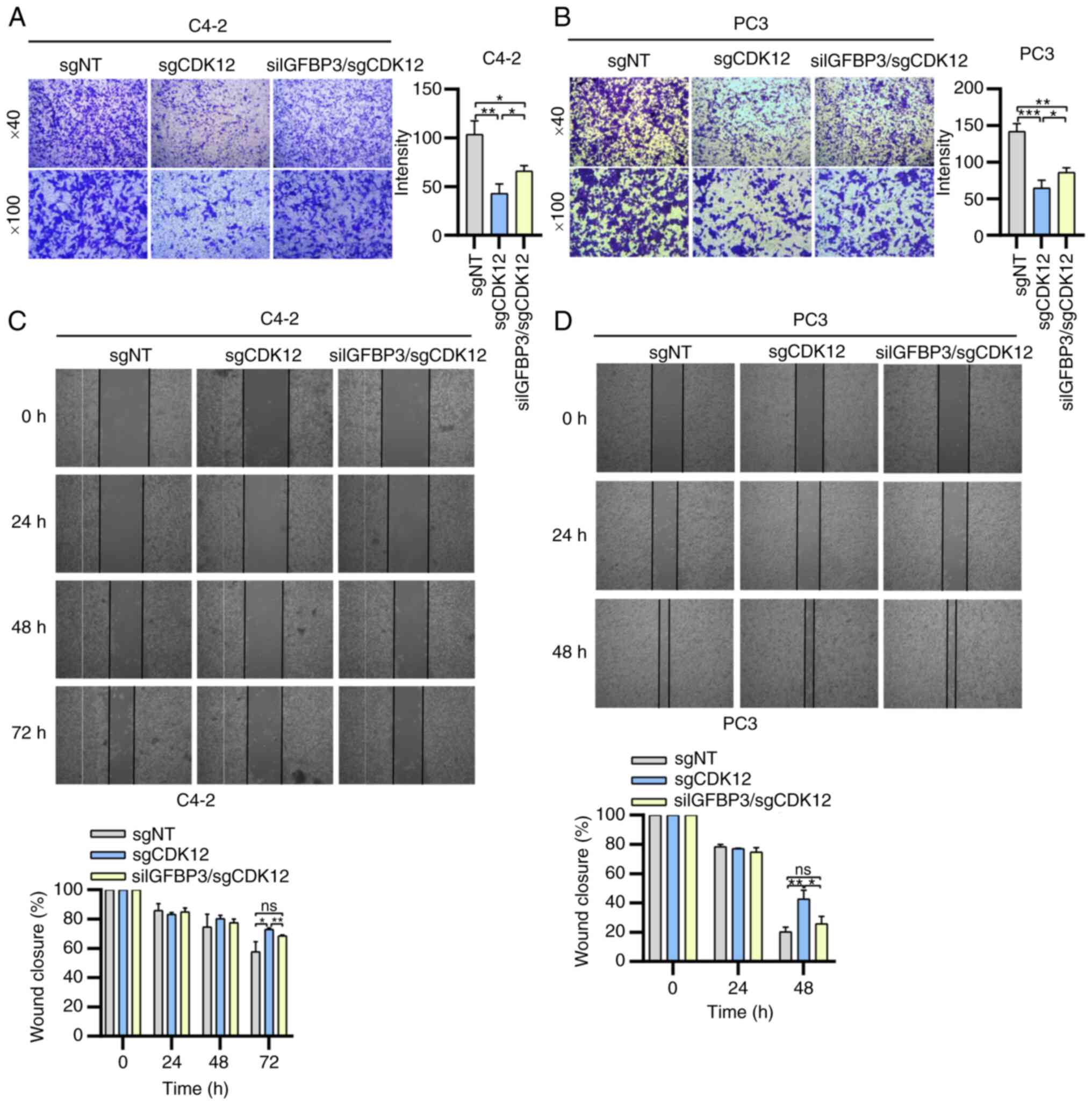 | Figure 3Effect of CDK12/IGFBP3 on cell
migration in advanced prostate cancer. The migration of cells (C4-2
and PC3) from the sgNT group, sgCDK12 group and siIGFBP3/sgCDK12
group was examined by (A and B) Transwell migration assays and (C
and D) wound healing assays using light microscopy. Magnification,
×40 and ×100 for Transwell migration assays. Magnification, ×40 for
wound healing assays. Two-tailed unpaired Student's t-test was
employed. *P<0.05; **P<0.01 and
***P<0.001. IGFBP3, insulin like growth factor
binding protein 3; si, small interfering; sg, single guide; sgNT
group, group without knockdown of CDK12 and IGFBP3 in cells;
sgCDK12 group, group with knockdown of CDK12 using CRISPR
technology; siIGFBP3/sgCDK12 group, group with knockdown of CDK12
using CRISPR technology and knockdown of IGFBP3 by transfection
with siRNA; ns, not significant. |
CDK12 promotes angiogenesis and
expression of related signal molecules in PCa by inhibiting
IGFBP3
After initially elucidating the effects of
CDK12/IGFBP3 on the cellular biological behavior of PCa, the
authors investigated the effects of CDK12/IGFBP3 on PCa
angiogenesis. The Pearson correlation coefficients were analyzed
between CDK12, IGFBP3 and VEGFA in patients with PCa patients
separately in the LinkedOmics database: CDK12 was positively
correlated with VEGFA, while IGFBP3 was negatively correlated with
VEGFA (Fig. 4A). Then, the
regulatory effects of CDK12/IGFBP3 on Akt (protein kinase B)
activity and VEGFA expression were analyzed using western blot
analysis. CDK12 was found to activate Akt and upregulate VEGFA in
the sgNT group vs. the sgCKD12 group; IGFBP3 was found to inhibit
Akt activity and downregulate VEGFA in the sgCDK12 group vs. the
siIGFBP3/sgCKD12 group (Fig. 4B).
The phosphorylation level of Akt and the expression level of VEGFA
in the siIGFBP3/sgCKD12 group were both increased compared with
those of the sgCDK12 group (Fig.
4B), thus, the authors inferred that CDK12 activates Akt and
promotes VEGFA expression by inhibiting IGFBP3. The results of the
tube formation assay demonstrated that the total tubule length
decreased after CDK12 knockdown, while the total tubule length
increased again after knockdown of CDK12 and IGFBP3 to a level that
was not significantly different from that of the sgNT group
(Fig. 4C and D). This finding
further validated that CDK12 can promote PCa angiogenesis by
inhibiting IGFBP3.
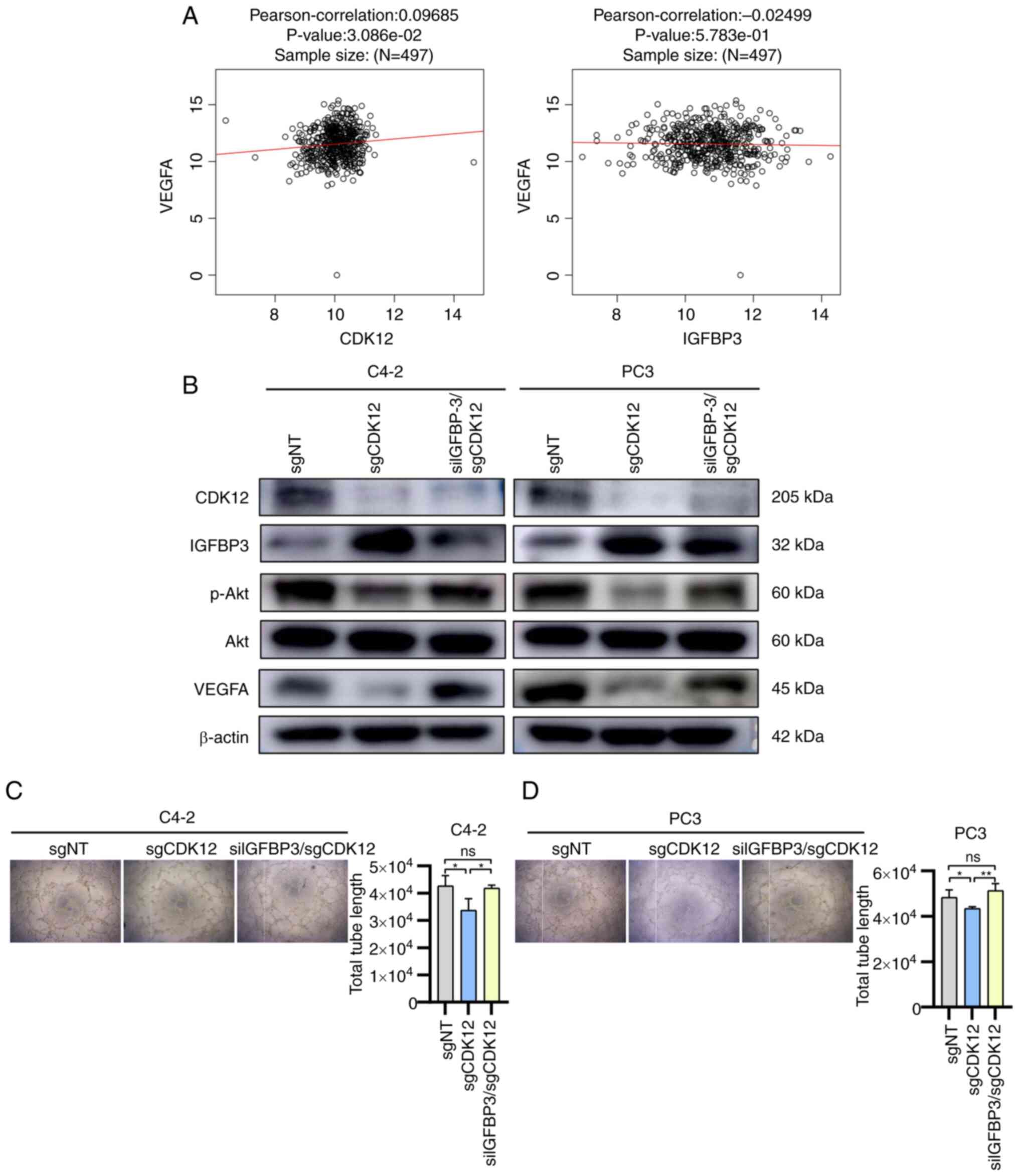 | Figure 4Effect of CDK12/IGFBP3 on
angiogenesis and expression of related signal molecules in advanced
prostate cancer. (A) Pearson correlation coefficients between CDK12
and VEGFA and between IGFBP3 and VEGFA were analyzed in the
LinkedOmics database. (B) Total proteins of cells (C4-2 and PC3)
from the sgNT group, sgCDK12 group and siIGFBP3/sgCDK12 group were
extracted and protein levels of CDK12, IGFBP3 and
angiogenesis-related signals were measured by western blotting.
β-actin was used as an internal control. (C and D) Supernatant of
sgNT group, sgCDK12 group and siIGFBP3/sgCDK12 group cells (C4-2
and PC3) were collected. Equal amounts of HUVECs were resuspended
with the supernatant for culture. Using light microscopy after 6 h.
Magnification, ×40. Two-tailed unpaired Student's t-test was used.
*P<0.05 and **P<0.01. IGFBP3, insulin
like growth factor binding protein 3; si, small interfering; sg,
single guide; sgNT group, group without knockdown of CDK12 and
IGFBP3 in cells; sgCDK12 group: group with knockdown of CDK12 using
CRISPR technology; siIGFBP3/sgCDK12 group, group with knockdown of
CDK12 using CRISPR technology and knockdown of IGFBP3 by
transfection with siRNA; HUVECs, human umbilical vein endothelial
cells; ns, not significant. |
Results of tumorigenic experiments in
nude mice
After transplanting equal amounts of sgNT and sgCDK2
group cells into nude mice subcutaneously, the tumor size and nude
mouse body weight were recorded every three days after tumor
formation. The transplanted tumors in the sgNT group gradually
increased in size, while those in the sgCDK12 group grew slowly
with minimal volume changes (Fig.
5B). Nude mice were sacrificed to remove the tumors when the
largest tumor was close to 150 mm in diameter. Visual observation
revealed that the tumor volume of nude mice in the sgNT group was
larger than that in the sgCDK12 group; weighing of the transplanted
tumors revealed that the weight of the sgNT group was larger than
that of the sgCDK12 group (Fig. 5A
and B). The transplanted tumors were embedded into wax blocks
and then sliced for H&E and IHC, which showed lower CDK12
expression in the sgCDK12 group (Fig.
5C and D). The MVD of the tissues was counted after incubation
with anti-CD31 antibodies and the MVD was 17, 29 and 19/field of
view at ×200 magnification for the sgNT group and 7, 8 and 9/field
of view at ×200 magnification for the sgCDK12 MVD (Fig. 5F). After incubation with
anti-IGFBP3 antibodies and anti-VEGFA antibodies, the results
revealed that IGFBP3 expression in the sgNT group was lower than
that in the sgCDK12 group (Fig.
5E) and VEGFA expression was higher than that in the sgCDK12
group (Fig. 5G).
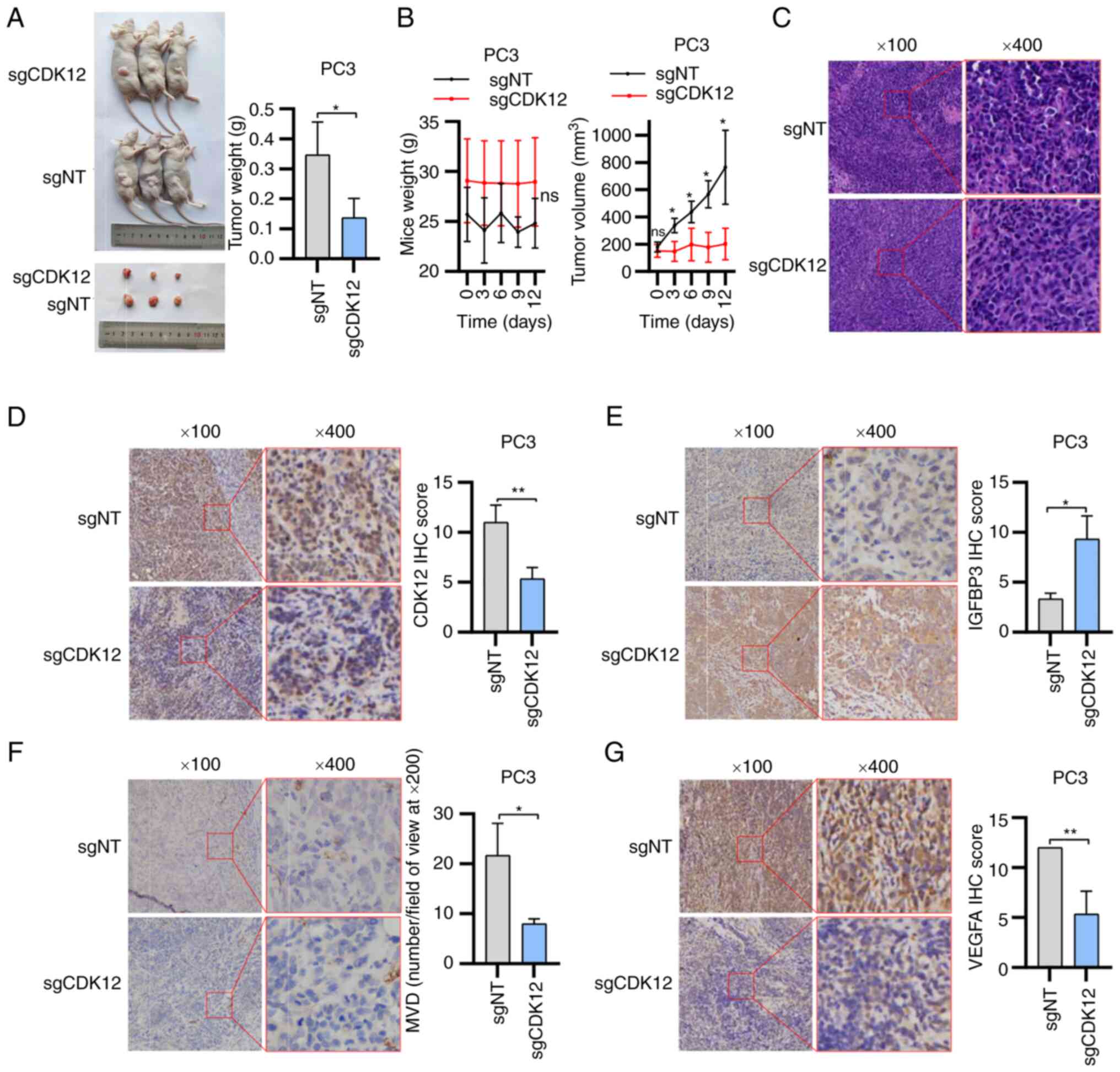 | Figure 5Results of tumorigenic experiments in
nude mice. (A) Nude mice and subcutaneous xenograft tumor images.
(B) Tumor volume change in mice and weight comparison after
removal. (C) Tumor blocks were sectioned and stained for
hematoxylin-eosin staining. Immunohistochemistry was used to detect
the expression of (D) CDK12, (E) IGFBP3, (F) CD31 and (G) VEGFA in
sgNT group tissues and in sgCDK12 group tissues by using light
microscopy. Magnification, ×100 and ×400. Two-tailed unpaired
Student's t-test was used. *P<0.05 and
**P<0.01. IGFBP3, insulin like growth factor binding
protein 3; si, small interfering; sg, single guide; sgNT group,
group without knockdown of CDK12 and IGFBP3 in cells; sgCDK12
group, group with knockdown of CDK12 using CRISPR technology;
siIGFBP3/sgCDK12 group, group with knockdown of CDK12 using CRISPR
technology and knockdown of IGFBP3 by transfection with siRNA; ns,
not significant; MVD, microvessel density. |
Discussion
CDKs are a family of serine/threonine kinases. Based
on their regulatory functions, CDKs can be functionally subdivided
into CDKs related to cell cycle processes and CDKs related to
transcription (23). CDK12 is
located on chromosome 17, contains 14 exons and encodes a protein
of ~164 kDa consisting of 1,490 amino acids (24). CDK12 consists of different
functional structural domains: a centrally located kinase domain,
several RS (arginine/serine) patterns near the N-terminus and a
proline-rich pattern, which can act as a binding site for other
proteins (25). CDK12 is
considered to regulate gene transcription by phosphorylating RNA
polymerase II, which can directly regulate transcription by
phosphorylating serine residues within the C-terminal structural
domain of RNA polymerase II (YSPTSPS), which is essential for
transcriptional elongation (25,26). In the present study, significant
changes were first observed in IGFBP3 after CDK12 was knocked down,
as shown by RNA-seq, and several studies have reported a strong
association between IGFBP3 and angiogenesis (21,27-29). Subsequently, the study of
CDK12/IGFBP3 in PCa was initiated. The human IGFBP3 gene is located
on the short arm of chromosome 7 and has five exons. In the
N-terminal region of IGFBP3, there are post-translational
modification sites; in addition, there are many phosphorylation
sites and three glycosylation sites (30). Despite the high long-term survival
rates of localized PCa, metastatic PCa remains largely incurable,
even after intensive multimodal therapy (3). Therefore, in the present study, a
series of in vivo and in vitro experiments were
conducted to study the biological behavior of mid-to-late stage PCa
cells from Chinese men and their proteins and RNAs were extracted
to observe the alteration of the target molecules. The C4-2 cells
demonstrate CRPC characteristics and PC3 cells demonstrate NEPC
characteristics. Lack of the employment of more cell lines
constituted a limitation of the present study. Proteins from PC3
cells were extracted for co-IP and the results revealed that CDK12
interacts with IGFBP3 in a protein-protein mechanism. However, this
finding did not yet indicate a direct regulatory relationship
between CDK12 and IGFBP3, this is also a limitation of the present
study, while there may be a third interacting protein between them
(31). Western blot analysis was
also employed and revealed that CDK12 inhibits the protein
expression of IGFBP3. RT-qPCR results revealed that CDK12 also
inhibits the mRNA synthesis of IGFBP3. The correlation between
CDK12 and IGFBP3 mined from the LinkedOmics database is consistent
with the aforementioned experimental results.
A number of studies have revealed that patients with
CDK12 deletion mutations have shorter OS than patients with
wild-type CDK12 (32-34). However, a different study used a
CRISPR screen to find that CDK12 is required for PCa cell survival.
CDK12 accelerates the progression of PCa to some extent. Some
researchers verified the dependence of PCa cells on CDK12 from the
Project Score; the mRNA level of CDK12 was found to be
significantly higher in PCa than in normal prostate tissue from
Tomlins; in addition, researchers found from TCGA data that
patients with PCa with lower CDK12 mRNA levels had slightly longer
disease-free survival (35). In a
study conducted by Wu et al (25), cell proliferation was observed to
be decreased following CDK12 knockdown by kinetic imaging
confluence measurements conducted at 3-h intervals. Furthermore, Wu
et al (25) suggested that
CDK12 deficiency may result in elevated T cell infiltration. It is
possible that inconsistent findings regarding CDK12 are due to
various factors, including the absence of patient-derived T cells
in culture dishes or in nude mice, which may necessitate further
investigation. Regarding IGFBP3, this protein inhibits cell
proliferation and stimulates apoptosis (36). IGFBP3 overexpression has been
demonstrated to induce G1 phase cell cycle arrest and inhibit PCa
metastasis in breast cancer (37). The antitumor activity and
mechanism of action of IGFBP3 have also been extensively validated
in various preclinical model systems (38-40). A previous in vitro study in
PCa have also revealed that IGFBP3 independently inhibits PCa cell
growth and promotes apoptosis (41). In addition, many of the roles of
IGFBP3, including inhibition of the NF-kB pathway, appear to be
independent of its ability to isolate IGF. Low IGFBP3 in serum is
associated with a risk of more aggressive PCa (16). To confirm the essential role of
CDK12 in PCa, CRISPR/CAS9 was used to knock down CDK12 in the two
PCa cell lines aforementioned. Preliminary studies by the authors
have confirmed that CDK12 negatively regulates IGFBP3. Therefore,
the expression of IGFBP3 in cells was knocked down using siRNA in
addition to the knockdown of CDK12 so that it could be analyzed
whether the effect of CDK12 on PCa cells is related to IGFBP3. Both
the CCK-8 assay and EdU assay results indicated that CDK12 promoted
PCa cell proliferation by inhibiting IGFBP3. Because the expression
of IGFBP3 was upregulated after CDK12 was knocked down, the
proliferation of PCa cells was diminished at this time and after
knocking down CDK12, the expression of IGFBP3 was simultaneously
knocked down and it was found that the cell proliferation was
restored. The EdU assay revealed that there was no longer a
significant difference in the proliferative capacity of PC3 cells
between the sgNT group and the siIGFBP3/sgCDK12 group. In parallel,
a colony formation assay was performed as a corroboration of the
first two proliferation assays. The results reinforced the
conclusion that CDK12 promotes the proliferation of PCa cells by
inhibiting IGFBP3. The effect of CDK12/IGFBP3 on the migration of
PCa cells was then investigated by Transwell migration and wound
healing assays. The knockdown of CDK12 reduced the number of cells
crossing the membrane and slowed the rate of wound closure. The
cell migration ability was restored after simultaneous knockdown of
IGFBP3. The wound healing assay demonstrated that there was no
longer a significant difference in the migratory capacity of PC3
cells between the sgNT group and the siIGFBP3/sgCDK12 group. The
authors thus concluded that CDK12 promotes PCa cell migration by
inhibiting IGFBP3.
Blood vessels form the largest network in the human
body, and when dysregulated, the formation of new blood vessels can
lead to many malignant, ischemic, inflammatory, infectious and
immune diseases (10,42). Angiogenesis is a key feature in
tumorigenesis and remains a potential target for antiangiogenic
therapy (43). Angiogenesis
divides the development of any solid tumor into two stages: The
avascular stage and the vascular stage. Because of this,
angiogenesis plays a key role in the biology of solid tumors. If
this vascular growth can be stopped by interrupting the tumor
signals that trigger capillary proliferation, the small tumor
population will remain dormant, similar to normal cells, relying on
the diffusion principle to absorb nutrients and release
catabolites. If this phenomenon can be achieved, it may become an
effective therapy in its own right or a powerful adjunct to other
treatments (44). However,
antiangiogenic therapies have also revealed that resistance to
antiangiogenic drugs readily arises and may involve alterations in
vascular signaling mechanisms (45). There is still a gap in the reports
of CDK12 in relation to angiogenesis, but there are many vascular
reports on IGFBP3 and angiogenesis. IGFBP3, a serum component
associated with angiogenesis, has been reported to have pro- and
antiangiogenic effects (46).
IGFBP3 has been suggested to promote cell migration and
angiogenesis in endothelial precursor cells (47). IGFBP3 also enhanced the mRNA
expression and protein secretion of VEGF in HUVECs (48). IGFBP3 reduced cellular capillary
formation in HUVECs and reduced angiogenesis in chick embryonic
chorionic villus assays and xenografts of IGFBP3 transfectants
significantly reduced tumor growth and angiogenesis (21). The farnesyltransferase inhibitor
(FTI) SCH66336 has been demonstrated to have antitumor activity
against head and neck squamous cell carcinoma (HNSCC) in
vitro and in vivo. A previous study suggested that
IGFBP3 may be a major target for the antitumor activity of FTIs and
that IGFBP3 is an effective treatment for HNSCC angiogenesis
(18). In addition, IGFBP3
inhibited the adhesion of HNSCC cells and HUVECs to the
extracellular matrix, in part by negatively regulating integrin β4
expression in an IGF-dependent and IGF-independent manner. These
data explain how IGFBP3 regulates cancer cell metastasis and tumor
angiogenesis (49). The
LinkedOmics database was investigated and it was observed that
IGFBP3 was negatively correlated with VEGFA, which is consistent
with the aforementioned findings of others; CDK12 was also found to
be positively correlated with VEGFA, which is consistent with the
authors' expectations. VEGF is a growth factor with important
proangiogenic activity and has mitogenic and antiapoptotic effects
on endothelial cells. VEGFA, also known as VEGF, is the most
important and potent angiogenesis stimulating factor (50). Akt is closely linked to VEGF, and
Akt, also known as protein kinase B (PKB), is a serine/threonine
kinase involved in several key cellular pathways, including
proliferation, invasion, apoptosis and angiogenesis (51,52). Akt1 is essential for VEGF-induced
angiogenesis (53). Akt can also
promote tumor angiogenesis by activating nitric oxide in the
endothelium (54). Akt and VEGF
form an autocrine circuit in cells that regulates angiogenesis,
where VEGF activates the Akt signaling pathway, which in turn
regulates the expression of VEGF and its receptors. Activation of
Akt also induces the expression of HIF-1α, which plays a key role
in the regulation of genes such as VEGF and heme oxygenase 1
(55). The western blot analysis
results of the present study suggested that CDK12 activates Akt and
upregulates VEGFA expression, while IGFBP3 has the opposite effect
of CDK12. Increased p-Akt and VEGFA levels were observed in
siIGFBP3/sgCDK12 group cells relative to sgCDK12 group cells or a
convergence of p-Akt and VEGFA content in siIGFBP3/sgCDK12 group
cells to sgNT group cells. Therefore, it was concluded that CDK12
inhibits IGFBP3 and thus activates Akt and upregulates VEGFA.
Transplanted tumors were made into sections and stained for IHC:
CDK12 expression was higher in sgNT group tissues than in the
sgCDK12 group; IGFBP3 expression was lower than in sgCDK12 group;
VEGF expression was higher than that in the sgCDK12 group and MVD
was higher than that in the sgCDK12 group. The IHC results together
with western blot analysis results supported the conclusion that
CDK12 inhibits IGFBP3 expression and promotes angiogenesis in PCa.
The results of the tube formation assay served as supporting
evidence to confirm the hypothesis that CDK12 promotes PCa
angiogenesis by inhibiting IGFBP3.
In addition to Akt and VEGF as aforementioned, the
authors consider that androgen receptor (AR) is likely involved
upstream/downstream of CDK12/IGFBP3 in mediating PCa progression,
as AR is critical to PCa pathogenesis (56). The effect of CDK12/IGFBP3 on AR
was not evaluated in the present study, which is one of the
limitations of the present study. The authors will further explore
the role of AR in this process in the future.
Given the stable knockdown of CDK12 by CRISPR/CAS9,
transplanted tumor models were constructed in nude mice using PC3
cells from the sgNT and sgCDK12 groups. Mice were sacrificed when
the largest transplanted tumor was nearly 15 mm in diameter, during
which time it was observed that transplanted tumors from mice
injected with sgNT group cells grew significantly faster than those
from mice injected with sgCDK12 group cells. After removing the
tumors, it was detected that the transplanted tumors of mice in the
sgNT group were larger in size than those in the sgCDK12 group. It
was thus demonstrated from in vivo experiments that CDK12
promotes the proliferation of PCa cells, which is consistent with
the results of the previous in vitro experiments and those
of Lei et al (35). As
supplementary material, the relationship between CDK12, IGFBP3 and
OS in PCa patients was analyzed separately after selecting the
'prostate adenocarcinoma' type in the LinkedOmics database. The
results revealed that patients with higher CDK12 expression had a
worse prognosis; those with higher IGFBP3 expression had an
improved prognosis. However, the results were not statistically
significant. Finally, the authors recognize that the limitations of
the study include the lack of clinical human tissue samples as
experiments on clinical specimens are critical.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. The datasets generated and/or
analyzed during the current study are available in the Gene
Expression Omnibus repository, (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246983).
Authors' contributions
YY and YC conceived the project and designed the
study. KZ, WL, NL, XT, YL, SY, YH, LF and WM performed the
experiments and analyzed the data. YY and KZ wrote the original
draft of the paper, with contributions from all authors. All
authors have read and approved the final version of the manuscript.
KZ and WL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The animal study was approved by the Experimental
Animal Ethics Committee of Anhui Medical University (approval no.
LLSC20190458; Hefei, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to especially thank the group
of Professor Hu at Southern University of Science and Technology
(Shenzhen, China) for sharing the results of RNA sequencing
analysis as well as Dr Wang at the First Affiliated Hospital of
Anhui Medical University (Hefei, China) for providing the cell
lines.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81972414) and the Key Natural
Science Research Project of Anhui Universities (grant no.
2023AH053332).
References
|
1
|
Daniyal M, Siddiqui ZA, Akram M, Asif HM,
Sultana S and Khan A: Epidemiology, etiology, diagnosis and
treatment of prostate cancer. Asian Pac J Cancer Prev.
15:9575–9578. 2014.
|
|
2
|
Rebbeck TR: Prostate cancer genetics:
Variation by race, ethnicity, and geography. Semin Radiat Oncol.
27:3–10. 2017.
|
|
3
|
Wang G, Zhao D, Spring DJ and DePinho RA:
Genetics and biology of prostate cancer. Genes Dev. 32:1105–1140.
2018.
|
|
4
|
Liang H, Liu Y, Guo J, Dou M, Zhang X, Hu
L and Chen J: Progression in immunotherapy for advanced prostate
cancer. Front Oncol. 13:11267522023.
|
|
5
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018.
|
|
6
|
Lassi K and Dawson NA: Emerging therapies
in castrate-resistant prostate cancer. Curr Opin Oncol. 21:260–265.
2009.
|
|
7
|
Wang W, Kong P, Feng K, Liu C, Gong X, Sun
T, Duan X, Sang Y, Jiang Y, Li X, et al: Exosomal miR-222-3p
contributes to castration-resistant prostate cancer by activating
mTOR signaling. Cancer Sci. 114:4252–4269. 2023.
|
|
8
|
Netto GJ, Amin MB, Berney DM, Compérat EM,
Gill AJ, Hartmann A, Menon S, Raspollini MR, Rubin MA, Srigley JR,
et al: The 2022 World Health Organization classification of tumors
of the urinary system and male genital Organs-Part B: Prostate and
urinary tract tumors. Eur Urol. 82:469–482. 2022.
|
|
9
|
Wang Z, Wang T, Hong D, Dong B, Wang Y,
Huang H, Zhang W, Lian B, Ji B, Shi H, et al: Single-cell
transcriptional regulation and genetic evolution of neuroendocrine
prostate cancer. iScience. 25:1045762022.
|
|
10
|
Carmeliet P: Angiogenesis in health and
disease. Nat Med. 9:653–660. 2003.
|
|
11
|
Fabian KL and Storkus WJ:
Immunotherapeutic targeting of tumor-associated blood vessels. Adv
Exp Med Biol. 1036:191–211. 2017.
|
|
12
|
Lammert E and Axnick J: Vascular lumen
formation. Cold Spring Harb Perspect Med. 2:a0066192012.
|
|
13
|
Lui GYL, Grandori C and Kemp CJ: CDK12: An
emerging therapeutic target for cancer. J Clin Pathol. 71:957–962.
2018.
|
|
14
|
Liu H, Liu K and Dong Z: Targeting CDK12
for cancer therapy: Function, mechanism, and drug discovery. Cancer
Res. 81:18–26. 2021.
|
|
15
|
Liu B, Lee KW, Anzo M, Zhang B, Zi X, Tao
Y, Shiry L, Pollak M, Lin S and Cohen P: Insulin-like growth
factor-binding protein-3 inhibition of prostate cancer growth
involves suppression of angiogenesis. Oncogene. 26:1811–1819.
2007.
|
|
16
|
Seligson DB, Yu H, Tze S, Said J, Pantuck
AJ, Cohen P and Lee KW: IGFBP-3 nuclear localization predicts human
prostate cancer recurrence. Horm Cancer. 4:12–23. 2013.
|
|
17
|
Johnson MA and Firth SM: IGFBP-3: A cell
fate pivot in cancer and disease. Growth Horm IGF Res. 24:164–173.
2014.
|
|
18
|
Oh SH, Kim WY, Kim JH, Younes MN,
El-Naggar AK, Myers JN, Kies M, Cohen P, Khuri F, Hong WK, et al:
Identification of insulin-like growth factor binding protein-3 as a
farnesyl transferase inhibitor SCH66336-induced negative regulator
of angiogenesis in head and neck squamous cell carcinoma. Clin
Cancer Res. 12:653–661. 2006.
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
20
|
Bai N, Xia F, Wang W, Lei Y, Bo J and Li
X: CDK12 promotes papillary thyroid cancer progression through
regulating the c-myc/β-catenin pathway. J Cancer. 11:4308–4315.
2020.
|
|
21
|
Shih HJ, Chen CL and Torng PL: IGFBP3
inhibits angiogenesis through intracellular regulation of THBS1
expression. Am J Cancer Res. 10:1728–1744. 2020.
|
|
22
|
He G, Li M, Fang L, Xu L, Huang X, Zheng
L, Yang L, Luo W, Cai Y, Ma W, et al: N-Myc induces the tumor
progression of prostate cancer by regulating FSCN1. Oncol Rep.
44:2265–2274. 2020.
|
|
23
|
Marciscano AE and Barbieri CE: CDK12 Gene
alterations in prostate cancer: Present, but clinically actionable?
Eur Urol. 78:680–681. 2020.
|
|
24
|
Liang S, Hu L, Wu Z, Chen Z, Liu S, Xu X
and Qian A: CDK12: A potent target and biomarker for human cancer
therapy. Cells. 9:14832020.
|
|
25
|
Wu YM, Cieślik M, Lonigro RJ, Vats P,
Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al:
Inactivation of CDK12 delineates a distinct immunogenic class of
advanced prostate cancer. Cell. 173:1770–1782.e14. 2018.
|
|
26
|
Lotan TL and Antonarakis ES: CDK12
Deficiency and the immune microenvironment in prostate cancer. Clin
Cancer Res. 27:380–382. 2021.
|
|
27
|
Dallinga MG, Habani YI, Kayser RP, Van
Noorden CJF, Klaassen I and Schlingemann RO: IGF-binding proteins 3
and 4 are regulators of sprouting angiogenesis. Mol Biol Rep.
47:2561–2572. 2020.
|
|
28
|
Li CL, Liu B, Wang ZY, Xie F, Qiao W,
Cheng J, Kuang JY, Wang Y, Zhang MX and Liu DS: Salvianolic acid B
improves myocardial function in diabetic cardiomyopathy by
suppressing IGFBP3. J Mol Cell Cardiol. 139:98–112. 2020.
|
|
29
|
Oh SH, Kim WY, Lee OH, Kang JH, Woo JK,
Kim JH, Glisson B and Lee HY: Insulin-like growth factor binding
protein-3 suppresses vascular endothelial growth factor expression
and tumor angiogenesis in head and neck squamous cell carcinoma.
Cancer Sci. 103:1259–1266. 2012.
|
|
30
|
Ranke MB: Insulin-like growth factor
binding-protein-3 (IGFBP-3). Best Pract Res Clin Endocrinol Metab.
29:701–711. 2015.
|
|
31
|
Lin JS and Lai EM: Protein-protein
interactions: Co-immunoprecipitation. Methods Mol Biol.
1615:211–219. 2017.
|
|
32
|
Antonarakis ES, Isaacsson Velho P, Fu W,
Wang H, Agarwal N, Sacristan Santos V, Maughan BL, Pili R, Adra N,
Sternberg CN, et al: CDK12-Altered prostate cancer: Clinical
features and therapeutic outcomes to standard systemic therapies,
poly (ADP-Ribose) polymerase inhibitors, and PD-1 inhibitors. JCO
Precis Oncol. 4:370–381. 2020.
|
|
33
|
Wang X, Chen H, Luo J and Xie L: CDK12
mutation in advanced prostate cancer: A marker for clinical
subtype? Eur Urol. 77:342–343. 2020.
|
|
34
|
Rescigno P, Gurel B, Pereira R, Crespo M,
Rekowski J, Rediti M, Barrero M, Mateo J, Bianchini D, Messina C,
et al: Characterizing CDK12-Mutated prostate cancers. Clin Cancer
Res. 27:566–574. 2021.
|
|
35
|
Lei H, Wang Z, Jiang D, Liu F, Liu M, Lei
X, Yang Y, He B, Yan M, Huang H, et al: CRISPR screening identifies
CDK12 as a conservative vulnerability of prostate cancer. Cell
Death Dis. 12:7402021.
|
|
36
|
Qin Z, Li X, Tang J, Jiang X, Yu Y, Wang
C, Xu W, Hua Y, Yu B and Zhang W: Association between insulin-like
growth factor-binding protein-3 polymorphism-202 A/C and the risk
of prostate cancer: A meta-analysis. Onco Targets Ther.
9:5451–5459. 2016.
|
|
37
|
Beveridge DJ, Richardson KL, Epis MR,
Brown RAM, Stuart LM, Woo AJ and Leedman PJ: The tumor suppressor
miR-642a-5p targets Wilms Tumor 1 gene and cell-cycle progression
in prostate cancer. Sci Rep. 11:180032021.
|
|
38
|
Kim WY, Kim MJ, Moon H, Yuan P, Kim JS,
Woo JK, Zhang G, Suh YA, Feng L, Behrens C, et al: Differential
impacts of insulin-like growth factor-binding protein-3 (IGFBP-3)
in epithelial IGF-induced lung cancer development. Endocrinology.
152:2164–2173. 2011.
|
|
39
|
Kim JH, Choi DS, Lee OH, Oh SH, Lippman SM
and Lee HY: Antiangiogenic antitumor activities of IGFBP-3 are
mediated by IGF-independent suppression of Erk1/2 activation and
Egr-1-mediated transcriptional events. Blood. 118:2622–2631.
2011.
|
|
40
|
Oh SH, Whang YM, Min HY, Han SH, Kang JH,
Song KH, Glisson BS, Kim YH and Lee HY: Histone deacetylase
inhibitors enhance the apoptotic activity of insulin-like growth
factor binding protein-3 by blocking PKC-induced IGFBP-3
degradation. Int J Cancer. 131:2253–2263. 2012.
|
|
41
|
Park K, Kim JH, Jeon HG, Byun SS and Lee
E: Influence of IGFBP3 gene polymorphisms on IGFBP3 serum levels
and the risk of prostate cancer in low-risk Korean men. Urology.
75:1516.e1–e7. 2010.
|
|
42
|
Dudley AC: Tumor endothelial cells. Cold
Spring Harb Perspect Med. 2:a0065362012.
|
|
43
|
Hess K, Spille DC, Adeli A, Sporns PB,
Zitta K, Hummitzsch L, Pfarr J, Stummer W, Brokinkel B, Berndt R
and Albrecht M: Occurrence of fibrotic tumor vessels in grade I
meningiomas is strongly associated with vessel density, expression
of VEGF, PlGF, IGFBP-3 and tumor recurrence. Cancers (Basel).
12:30752020.
|
|
44
|
Folkman J: Tumor angiogenesis. Adv Cancer
Res. 19:331–358. 1974.
|
|
45
|
Farnsworth RH, Lackmann M, Achen MG and
Stacker SA: Vascular remodeling in cancer. Oncogene. 33:3496–3505.
2014.
|
|
46
|
Chang KH, Chan-Ling T, McFarland EL, Afzal
A, Pan H, Baxter LC, Shaw LC, Caballero S, Sengupta N, Li Calzi S,
et al: IGF binding protein-3 regulates hematopoietic stem cell and
endothelial precursor cell function during vascular development.
Proc Natl Acad Sci USA. 104:10595–10600. 2007.
|
|
47
|
Zhao HJ, Klausen C, Zhu H, Chang HM, Li Y
and Leung PCK: Bone morphogenetic protein 2 promotes human
trophoblast cell invasion and endothelial-like tube formation
through ID1-mediated upregulation of IGF binding protein-3. FASEB
J. 34:3151–3164. 2020.
|
|
48
|
Granata R, Trovato L, Lupia E, Sala G,
Settanni F, Camussi G, Ghidoni R and Ghigo E: Insulin-like growth
factor binding protein-3 induces angiogenesis through IGF-I- and
SphK1-dependent mechanisms. J Thromb Haemost. 5:835–845. 2007.
|
|
49
|
Lee HJ, Lee JS, Hwang SJ and Lee HY:
Insulin-like growth factor binding protein-3 inhibits cell adhesion
via suppression of integrin β4 expression. Oncotarget.
6:15150–15163. 2015.
|
|
50
|
Melincovici CS, Boşca AB, Şuşman S,
Mărginean M, Mihu C, Istrate M, Moldovan IM, Roman AL and Mihu CM:
Vascular endothelial growth factor (VEGF)-key factor in normal and
pathological angiogenesis. Rom J Morphol Embryol. 59:455–467.
2018.
|
|
51
|
Shariati M and Meric-Bernstam F: Targeting
AKT for cancer therapy. Expert Opin Investig Drugs. 28:977–988.
2019.
|
|
52
|
Revathidevi S and Munirajan AK: Akt in
cancer: Mediator and more. Semin Cancer Biol. 59:80–91. 2019.
|
|
53
|
Luo Z, Fujio Y, Kureishi Y, Rudic RD,
Daumerie G, Fulton D, Sessa WC and Walsh K: Acute modulation of
endothelial Akt/PKB activity alters nitric oxide-dependent
vasomotor activity in vivo. J Clin Invest. 106:493–499. 2000.
|
|
54
|
Dimmeler S, Fleming I, Fisslthaler B,
Hermann C, Busse R and Zeiher AM: Activation of nitric oxide
synthase in endothelial cells by Akt-dependent phosphorylation.
Nature. 399:601–605. 1999.
|
|
55
|
Semenza GL: HIF-1 and tumor progression:
Pathophysiology and therapeutics. Trends Mol Med. 8(4 Suppl):
S62–S67. 2002.
|
|
56
|
Dai C, Heemers H and Sharifi N: Androgen
signaling in prostate cancer. Cold Spring Harb Perspect Med.
7:a0304522017.
|