Introduction
Benzidine (BZ) is a chemical compound used in the
production of dyes, and its sulfate is used predominantly in
industry (1). However, its
carcinogenic properties have been well established, particularly in
relation to bladder cancer (2).
Historical observations dating back to the late 19th century, such
as Rehn's findings in German aniline dye factories, first
highlighted the link between BZ exposure and bladder cancer
(2). Subsequent epidemiological
studies and experimental validation further confirmed the
carcinogenic nature of BZ, leading to its classification as a human
carcinogen by the International Agency for Research on Cancer in
1987 (3). Despite its ban in
commercial production, BZ continues to pose health risks as it is
still found in various products, including hair dyes, paints and
plastics (4).
Given the well-documented association between BZ
exposure and bladder cancer, concerns about its potential role in
promoting other urothelial carcinoma types, such as upper urinary
tract urothelial carcinoma (UTUC), have increased. While urothelial
carcinoma of the bladder is the most common urinary tract
malignancy, accounting for 95% of cases, UTUC accounts for the
remaining 5% of cases (5).
However, whether BZ exposure contributes to the development of UTUC
remains largely unexplored. Investigating the potential link
between BZ and UTUC could provide valuable insights into the
broader impact of BZ exposure on urothelial carcinogenesis and
inform preventive measures to mitigate its adverse health
effects.
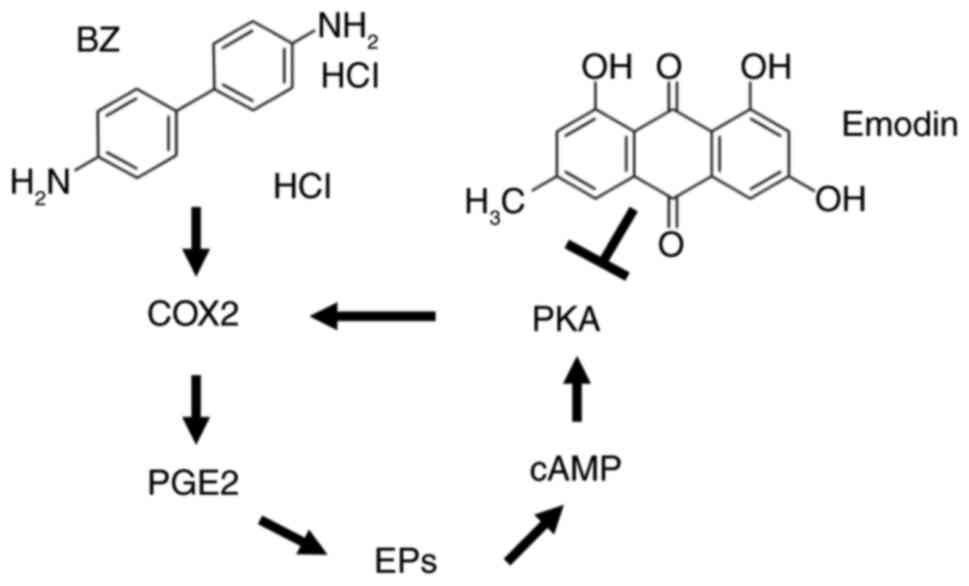
Prostaglandin E2 (PGE2), a metabolite of arachidonic
acid catalyzed by cyclooxygenase 2 (COX2), is known to promote
tumor cell proliferation, angiogenesis, invasion and metastasis
(6). A previous study has
established a close link between the occurrence and development of
UC and PGE2 (7). PGE2 exerts its
effects by binding to four types of prostaglandin E receptors on
the cell membrane, which regulate intracellular cyclic adenosine
monophosphate (cAMP) levels, calcium ion concentrations and
phosphatidylinositol activation (7). Upon activation of these receptors,
the G protein undergoes a conformational change, leading to the
separation of the Gαs subunit from the Gβγ subunit, which
subsequently activates adenylate cyclase to produce the second
messenger cAMP (8). This molecule
activates downstream regulatory elements such as protein kinase A
(PKA), further promoting tumor proliferation and invasion (9). PKA expression may be linked to the
invasive and metastatic properties of tumors, as its upregulation
can activate metalloproteinases (MMPs), such as MMP9, which are
essential for tumor infiltration (10). Additionally, PKA upregulation can
increase the expression of several angiogenic factors, including
vascular endothelial growth factor (VEGF) (11). In the context of UTUC, the
COX2/PGE2/cAMP/PKA signaling pathway plays a critical role in tumor
progression (12,13). Elevated levels of COX2 and its
downstream products, such as PGE2, can enhance UTUC cell survival
and migration. The subsequent increase in cAMP levels and
activation of PKA further facilitates these processes, contributing
to tumor growth and metastasis. Understanding the interplay between
these molecular factors is crucial for developing targeted
interventions to prevent or treat UTUC.
Rhubarb, a traditional Chinese herb, is renowned for
its ability to enhance cardiovascular function (14,15). Emodin, a vital constituent present
not only in rhubarb but also in various plants, such as Aloe vera,
He Shou Wu and Tiger Balm (16),
has garnered increasing attention for its notable antitumor,
anti-inflammatory and antibacterial properties (14,15). Notably, a study has revealed that
emodin suppresses VEGF transcription by targeting the transcription
factors, nuclear receptor corepressor 2 and seryl-tRNA synthetase,
thereby impeding triple-negative breast cancer progression
(16). Furthermore, in colon
cancer, emodin hinders angiogenesis by inhibiting the expression of
acyl-CoA synthetase long-chain family member 4 (17).
Emodin has been shown to inhibit the development of
bladder cancer (18,19). Hence, investigating whether emodin
can counteract BZ-induced UTUC progression may provide valuable
insights into mitigating the adverse health effects of BZ exposure
and further understanding the broader impact of emodin on UC.
Materials and methods
Cell culture
BFTC909 cells were purchased from Shanghai Chuanqiu
Biotechnology Co., Ltd. and incubated in DMEM/F12 (HyClone; Cytiva)
supplemented with 10% fetal bovine serum (FBS; HyClone; Cytiva) at
37°C with 5% CO2. UM-UC-14 cells were purchased from
Shanghai Fusheng Industrial Co., Ltd. and cultured in Eagle's
minimum essential medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 2 mM L-glutamine, 10% FBS, antibiotic-antimycotic
and 1% non-essential amino acids at 37°C with 5%
CO2.
Western blotting
The cells were incubated with high-efficiency RIPA
lysis buffer (Beijing Solarbio Science & Technology Co., Ltd.)
on ice for 5 min. After centrifugation at 11,000 × g for 20 min at
4°C, the supernatant was collected, and the protein concentration
was determined using a BCA kit (Beijing Solarbio Science &
Technology Co., Ltd.). Then, 20 μg/lane of sample was
separated by 12.5% SDS-PAGE and transferred to a PVDF membrane,
followed by incubation with 8% skim milk powder (Thermo Fisher
Scientific, Inc.) at room temperature for 2 h. After washing with
PBST (0.1% Tween) three times, the PVDF membrane was incubated with
the following primary antibodies: Anti-PKA (cat. no. ab75991;
1:1,000), anti-COX2 (cat. no. ab179800; 1:1,000), anti-MMP9 (cat.
no. ab76003; 1:1,000), anti-VEGF (cat. no. ab32152; 1:1,000) and
anti-GAPDH (cat. no. ab8245; 1:3,000) (all primary antibodies were
purchased from Abcam) at 4°C overnight. After three washes with
PBST, the PVDF membranes were incubated with HRP-labeled secondary
antibodies [1:5,000; cat. nos. SE131 (anti-mouse) and SE134
(anti-rabbit); Beijing Solarbio Science & Technology Co., Ltd.]
at room temperature for 2 h. Subsequently, the protein expression
signals were detected with ECL Western Blotting Substrate (Beijing
Solarbio Science & Technology Co., Ltd.). GAPDH was used as an
internal reference and ImageJ software (version 1.53a; National
Institutes of Health; https://imagej.nih.gov/ij/) was used for analysis.
Cell Counting Kit-8 (CCK-8) assay
BFTC909 and UM-UC-14 cells were inoculated into
96-well cell culture plates at 1,000 cells per well and incubated
at 37°C overnight. Subsequently, the cells were divided into the
control, BZ (Sigma-Aldrich; Merck KGaA; 1, 5, 10 and 50 nM), and
emodin (Sigma-Aldrich; Merck KGaA; 12.5, 25, 50, 100 and 200
μM) groups and incubated at 37°C for 48 h. Then, 10
μl CCK-8 solution (Beijing Solarbio Science & Technology
Co., Ltd.) was added to each well and the cells were incubated at
37°C for 4 h. Finally, the absorbance was measured at 450 nm, and
three replicate wells were set up for each group.
Determination of cAMP concentration and
lactate dehydrogenase (LDH) leakage
BFTC909 and UM-UC-14 cells were lysed by sonication
in an ice bath and centrifuged at 5,000 × g for 20 min at 4°C.
After the supernatant was collected, the intracellular cAMP and LDH
concentrations were analyzed using the cAMP Assay Kit (cat. no.
ab65355; Abcam) and LDH Assay Kit (cat. no. ab102526; Abcam)
according to the manufacturer's instructions.
Wound healing assay
BFTC909 and UM-UC-14 cells were inoculated at
5×105 cells/well in 6-well plates and incubated
overnight. The next day, the cells were gently scraped with a 200
μl pipette tip, then washed well with PBS (this was noted as
0 h). Next, the BFTC909 and UM-UC-14 cells were treated with 10 nM
BZ or 50 μM emodin at 37°C for 24 h in medium containing 2%
serum. Representative images at 0 and 24 h were collected using a
light microscope (Olympus Corporation), and the wound healing area
was recorded (S1 represented the initial wound area, and S2
represented the wound area after 24 h of treatment). The cell
migration rate (%) was calculated as [(S1-S2)/S1] ×100%, and each
set of experiments was repeated three times. ImageJ software
(version 1.53a; National Institutes of Health; https://imagej.nih.gov/ij/) was used for analysis.
Transwell assay
In brief, both BFTC909 and UM-UC-14 cells were
seeded at a concentration of 1×104 in the upper chamber
with 200 μl serum-free DMEM. Then, 600 μl DMEM
containing 10% FBS was added to the lower chambers at 37°C for 24
h. Subsequently, the cells that migrated to the lower chamber were
fixed with 4% paraformaldehyde (Beijing Solarbio Science &
Technology Co., Ltd.) at room temperature for 20 min and stained
with 0.1% crystal violet for 20 min at room temperature.
Representative images were observed under a light microscope
(Olympus Corporation). ImageJ software (version 1.53a; National
Institutes of Health; https://imagej.nih.gov/ij/) was used for analysis.
Small interfering RNA (siRNA)
transfection
BFTC909 and UM-UC-14 cells were inoculated at a
density of 5×105 cells/well in 6-well plates and
incubated overnight. Next, 10 μl siRNA targeting COX2
(sense, 5′-AAAUUUGAACAAUAAUUUGGU-3′; antisense,
5′-CAAAUUAUUGUUCAAAUUUAG-3′) or negative control (NC; sense,
5′-UUCUCCGAACGUGUCACGUTT-3′; antisense,
5′-ACGUGACACGUUCGGAGAATT-3′) (Shanghai GenePharma Co., Ltd.) was
added to serum-free medium to achieve a final concentration of 20
nM. Then, 10 μl HiPerFect Transfection Reagent (Qiagen GmbH)
was added to the siRNA solution. The mixture was incubated at room
temperature for 15 min to form siRNA-HiPerFect complexes. The
complexes were carefully added to the respective wells and
incubated at 37°C in a 5% CO2 incubator for 24 h. After
the incubation period, the cells were collected for subsequent
experiments.
Sp-8-CPT-cAMPS treatment
The cells were divided into four groups for
treatment: i) Control (Con) group, cells were treated with 10 nM BZ
alone for 48 h; ii) emodin group, cells were pretreated with 50
μM emodin for 1 h, followed by the addition of 10 nM BZ for
48 h; iii) Sp-8-CPT-cAMPS group, cells were pretreated with 10
μM Sp-8-CPT-cAMPS (cat. no. HY-120994B; MedChemExpress) for
1 h, followed by the addition of 10 nM BZ for 48 h; and iv) emodin
+ Sp-8-CPT-cAMPS group, cells were pretreated with a combination of
10 μM Sp-8-CPT-cAMPS and 50 μM emodin for 1 h,
followed by the addition of 10 nM BZ for 48 h. Then, the cells were
collected for further assay.
Detection of intracellular
malondialdehyde (MDA), superoxide dismutase (SOD), reactive oxygen
species (ROS) and total antioxidant capacity
Intracellular MDA, SOD, ROS and total antioxidant
capacity were analyzed using a Lipid Peroxidation MDA Assay Kit
(cat. no. S0131M; Beyotime Institute of Biotechnology), Reactive
Oxygen Species Assay Kit (cat. no. S0033S; Beyotime Institute of
Biotechnology), Total Superoxide Dismutase Assay Kit (cat. no.
S0101S; Beyotime Institute of Biotechnology) and Total Antioxidant
Capacity Assay Kit (cat. no. S0121; Beyotime Institute of
Biotechnology), respectively. All operations were carried out in
strict accordance with the instruction manuals.
Intracellular Ca2+ assay
Detection of intracellular calcium ion levels was
accomplished using an Intracellular Ca2+ Calcium Ion
Assay Kit (cat. no. HR8227; Beijing Biolab Technology Co., Ltd.).
Briefly, BFTC909 and UM-UC-14 cells were collected, and F04
staining solution was incubated with the cells at 37°C for 30 min,
after which the intracellular Ca2+ content was detected
(excitation wavelength of 488-495 nm and emission wavelength of 516
nm).
JC-1 staining
The cells were initially divided into four groups:
The control, emodin (50 μM), BZ (10 nM) and BZ + emodin
groups. BFTC909 and UM-UC-14 cells were then treated with the
indicated drugs for 24 h. Subsequently, the cells were co-incubated
with a final concentration of 1 μM JC-1 dye at 37°C for 30
min. After staining, the cells were washed with PBS and observed
under a fluorescence microscope (Olympus Corporation) to assess
changes in the mitochondrial membrane potential. At low membrane
potentials, JC-1 emits green fluorescence as a monomer, whereas at
higher potentials, JC-1 forms 'J-aggregates', emitting red
fluorescence.
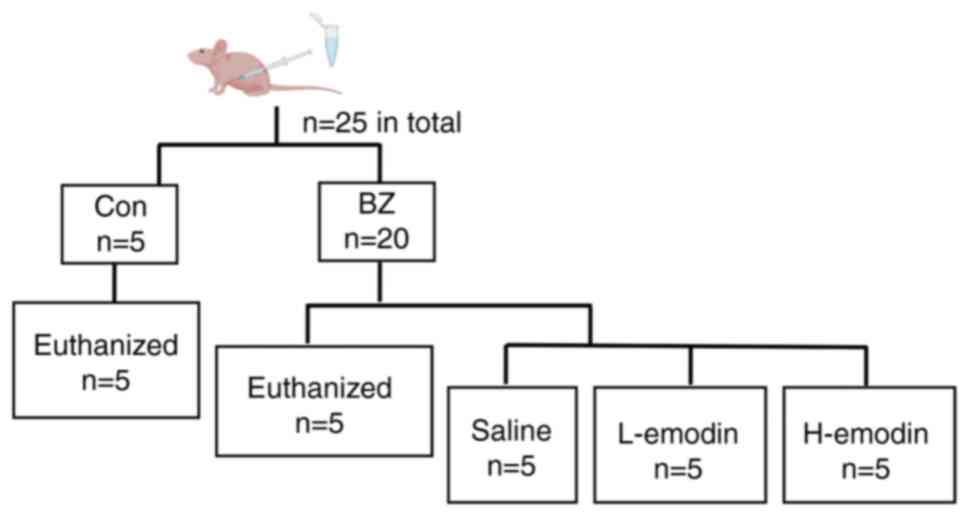
Nude mouse tumor assay
SPF male BALB/c-nu nude mice (6-8 weeks old; n=25)
weighing 15-20 g were obtained from Jinzhou Medical University
Experimental Animal Center (Jinzhou, China). All mice were kept in
housing with free access to food and water, in an environment with
a temperature of 20-24°C and a humidity level of 45-55% throughout
a 12 h light/dark cycle. In the experiment, a BFTC909 cell
suspension was prepared at a density of 5×107 cells/ml,
0.1 ml was then subcutaneously inoculated into the dorsal region of
the nude mice to construct UTUC transplantation tumors. The tumor
formation time was 14 days. Subsequently, the nude mice were
divided into two groups: The control (n=5) and BZ (n=20) groups
(Fig. 1). In the BZ group, nude
mice were administered BZ at a dose of 22 mg/kg body weight in 1 ml
of water per dose via gavage for 5 consecutive days, based on
previous studies (20,21). The control group was administered
an equal volume of water. Subsequently, nude mice in the control
and BZ groups (n=5 for each group) were sacrificed, and the tumor
volume was evaluated as follows: tumor volume=length ×
width2/2. After evaluation, the remaining 15 nude mice
in the BZ group were randomly divided into three groups: the
Control (Con) vehicle group, the Low-dose emodin (L-emodin) group,
and the High-dose emodin (H-emodin) group. The Control vehicle
group received 1 ml of 2% DMSO in saline by gavage once daily. The
L-emodin group was administered emodin at a dose of 40 mg/kg
intraperitoneally in 1 ml of PBS once daily, while the H-emodin
group received emodin at a dose of 80 mg/kg intraperitoneally in 1
ml of PBS once daily. The entire intervention process lasted for 4
weeks.
The humane endpoints used to determine when animals
should be euthanized were severe behavioral abnormalities, such as
persistent self-mutilation or inability to move normally. The
entire intervention process lasted for 4 weeks, and the tumor
formation time was 14 days. Thus, the total duration of the
experiment was 6 weeks. No animals were euthanized before the end
of the study. There were also no instances of animal death due to
the experimental procedures. Animal health and behavior were
monitored once daily. For anesthesia, the mice were anesthetized
using isoflurane, with an induction concentration of 2-3% and a
maintenance concentration of 1.5-2%. Deep anesthesia was confirmed
by the absence of a response to a paw pinch and the absence of a
corneal reflex, following ethical guidelines for animal
experimentation. Once deep anesthesia was achieved, ~0.6 ml of
cardiac blood was collected via cardiac puncture. Death was
verified by confirming the absence of a response to a paw pinch and
the absence of a corneal reflex. After blood collection, the
subcutaneous xenograft tumors were harvested for further
experiments, including the determination of cAMP and PGE2 levels,
western blotting and immunohistochemistry (IHC).
Detection of alanine aminotransferase
(ALT), aspartate aminotransferase (AST), creatinine and urea
levels
The blood samples were centrifuged at 1,500 × g for
10 min at 4°C to separate the serum. An automated biochemical
analyzer (Hitachi High-Technologies, Japan) was used to measure the
ALT, AST, creatinine and urea levels in the serum, using the
respective detection kits: ALT Detection Kit, AST Detection Kit,
Creatinine Detection Kit and Urea Detection Kit (all from Zybio
Inc.).
IHC
Tumor tissues were fixed using 4% paraformaldehyde
(Beijing Solarbio Science & Technology Co., Ltd.) for 24 h at
room temperature. After fixation, the tissues were dehydrated with
gradient alcohol, cleared with xylene and finally embedded in
paraffin. Specifically, the embedded tissues were cut into 5
μm-thick sections, sequentially placed in xylene (three
times, 10 min each), gradient alcohol (100, 95, 85 and 70%, 5 min
each), and finally washed with distilled water. The tissue sections
were autoclave-treated in sodium citrate buffer (pH 6.0; Beijing
Solarbio Science & Technology Co., Ltd.) for 15 min. After
deparaffinization and rehydration, sections were incubated with 3%
hydrogen peroxide (cat. no. H1009; MilliporeSigma) in methanol for
10 min at room temperature to quench endogenous peroxidase
activity. Afterwards, the sections were blocked with a solution
containing 5% BSA (Thermo Fisher Scientific, Inc.) for 30 min at
room temperature. A Ki-67 (1:50; cat. no. 9449, CST Biological
Reagents Co., Ltd.) primary antibody was added dropwise to the
tissue sections and incubated overnight at 4°C in a wet box. The
sections were subsequently washed three times with PBS for 5 min
each. Biotinylated secondary antibodies (1:50; cat. no. SHB131;
Beijing Solarbio Science & Technology Co., Ltd.) were added to
the sections and incubated for 60 min at room temperature. The
sections were washed three times with PBS for 5 min each. DAB color
development solution (OriGene Technologies, Inc.) was added
dropwise to the sections and incubated for 3 min at room
temperature, then the reaction was terminated with distilled water.
The nuclei were lightly stained with hematoxylin (Beijing Solarbio
Science & Technology Co., Ltd.) for 30 sec at room temperature
and washed with tap water. Subsequently, the sections were
dehydrated with gradient alcohol, cleared with xylene, and blocked
with neutral gum (Beijing Solarbio Science & Technology Co.,
Ltd.). The sections were finally visualized with a light microscope
(BZ-X; Keyence Corporation). ImageJ software (version 1.53a;
National Institutes of Health; https://imagej.nih.gov/ij/) was used for analysis. The
Ki-67 positive cell rate=(Ki-67 positive cells/total cells)
×100%.
Immunofluorescence (IF)
The cells were fixed in 4% paraformaldehyde for 15
min at room temperature. The cells were subsequently washed three
times with PBS for 5 min each, then permeabilized with 0.3% Triton
X-100 (in PBS) for 10 min at room temperature. The cells were
blocked with 5% BSA (Thermo Fisher Scientific, Inc.) for 1 h at
room temperature, followed by overnight incubation with a COX2
primary antibody (1:50; cat. no. ab179800; Abcam) in a wet box at
4°C. The cells were subsequently washed three times with PBS for 5
min each. The cells were incubated with goat anti-rabbit IgG/Cy3
(1:200; cat. no. K1034G-Cy3; Beijing Solarbio Science &
Technology Co., Ltd.) in a wet box protected from the light for 1 h
at room temperature. The cells were incubated with Hoechst (Beijing
Solarbio Science & Technology Co., Ltd.) for 10 min at room
temperature, then washed three times with PBS for 5 min each time.
The coverslips were sealed with a sealer containing an
anti-fluorescence attenuation reagent (Beijing Solarbio Science
& Technology Co., Ltd.). The cells were observed using a
fluorescence microscope (Keyence Corporation).
Statistical analysis
The data are presented as the mean ± standard
deviation and were analyzed using Prism 9.0 (Dotmatics).
Comparisons between two groups were made using unpaired Student's
t-test, and comparisons between more than two groups were made
using one-way ANOVA followed by Tukey's HSD test. P<0.05 was
considered to indicate statistically significant difference.
Results
BZ promotes UTUC tumor growth
The impact of BZ on the viability of BFTC and
UM-UC-14 cells was initially investigated. As shown in Fig. 2A and B, BZ significantly enhanced
the viability of both cell lines at concentrations of 1, 5, 10 and
50 nM. Furthermore, the wound healing and Transwell assays
demonstrated that BZ facilitated the migration of BFTC and UM-UC-14
cells (Fig. 2C-F). Subsequent
in vivo experiments further validated these findings,
indicating that BZ promoted subcutaneous tumor growth in nude mice
(Fig. 2G). IHC staining revealed
that BZ increased the percentage of Ki-67-positive cells compared
with the control group (Fig.
2H).
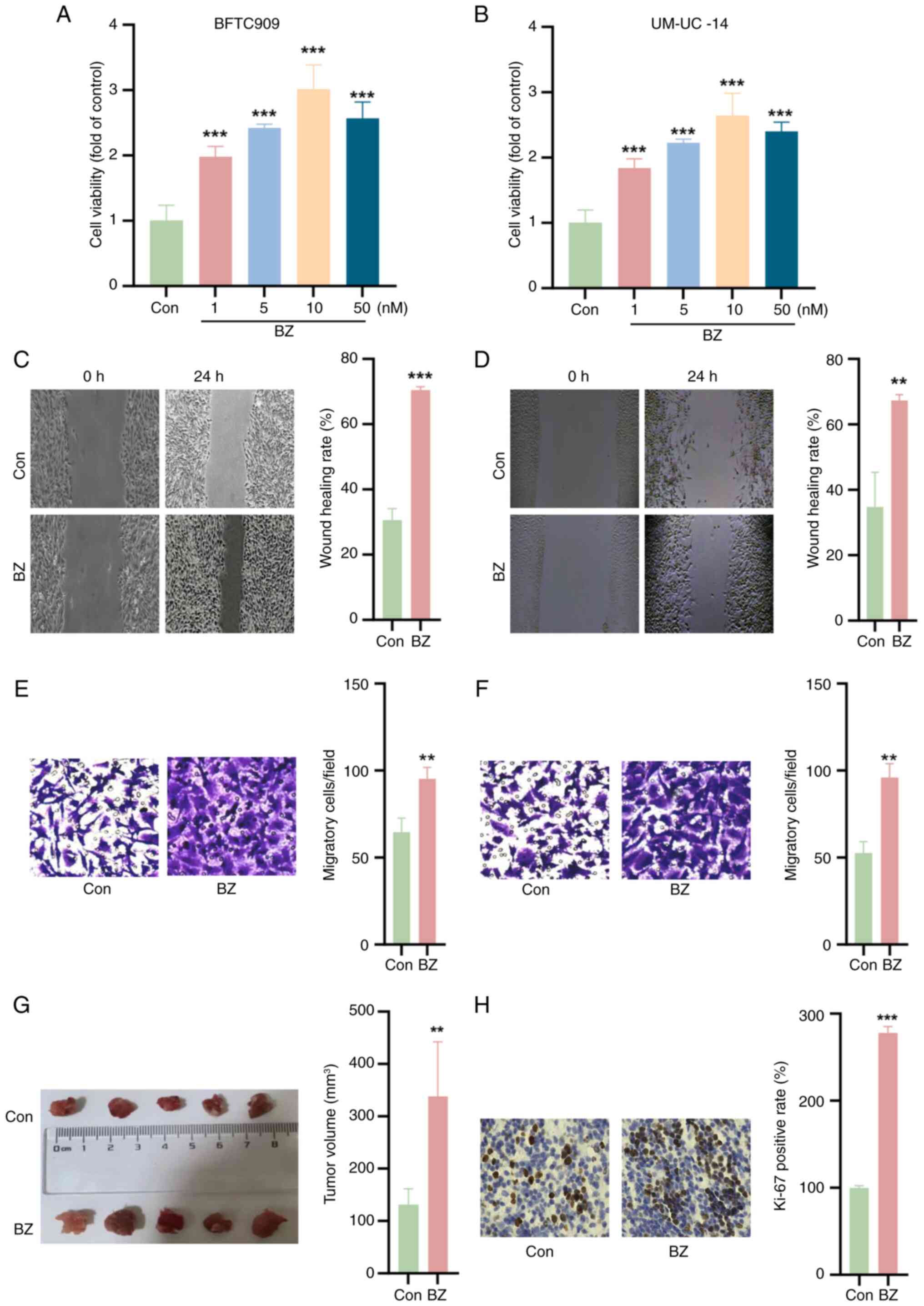 | Figure 2BZ promotes upper urinary tract
urothelial carcinoma cell growth. Cell Counting Kit-8 assays were
used to explore the effects of BZ (1, 5, 10 and 50 nM for 24 h) on
the viability of (A) BFTC and (B) UM-UC-14 cells. Wound healing
assays demonstrating the migratory capacity of (C) BFTC and (D)
UM-UC-14 cells upon BZ treatment (10 nM BZ at 37°C for 48 h);
magnification, ×4. Transwell assays demonstrating the migratory
capacity of (E) BFTC and (F) UM-UC-14 cells upon BZ treatment (10
nM BZ at 37°C for 48 h); magnification, ×20. (G) In vivo
experiments confirming the promotion of subcutaneous tumor growth
in nude mice following BZ administration (22 mg/kg body weight for
5 consecutive days). (H) Immunohistochemistry staining showing
that, compared with Con, BZ increased the percentage of
Ki-67-positive cells; magnification, ×20. **P<0.01,
***P<0.001 vs. Con. BZ, benzidine; Con, control. |
Emodin decreases cell viability and
mitochondrial membrane potential in BZ-Induced malignant cells
It was next evaluated whether emodin could inhibit
BZ-induced malignant cell survival in vitro. Compared with
BZ alone (10 nM), emodin significantly reduced the viability of
both BFTC and UM-UC-14 cells (at concentrations of 12.5, 25, 50,
100, and 200 μM) in a concentration-dependent manner
(Fig. 3A and B). The LDH leakage
rate, a critical indicator of cell damage, was significantly lower
in the BZ-treated group than in the control group; however, emodin
treatment increased the LDH leakage rate (Fig. 3C and D). Changes in mitochondrial
membrane potential were assessed using the JC-1 staining method.
JC-1 is a commonly used fluorescent probe for evaluating
mitochondrial membrane potential; it forms aggregates and emits red
fluorescence at high membrane potential, while at low membrane
potential, it remains in the monomeric form and emits green
fluorescence. In this experiment, strong red fluorescence was
observed in the untreated control group, indicating a high
mitochondrial membrane potential. Compared with the control, cells
treated with emodin showed a significant decrease in red
fluorescence and an increase in green fluorescence in both BFTC and
UM-UC-14 cells, indicating a decrease in mitochondrial membrane
potential. Even in the presence of BZ, emodin effectively lowered
the mitochondrial membrane potential, as evidenced by the reduced
red and increased green fluorescence (Fig. 3E and F).
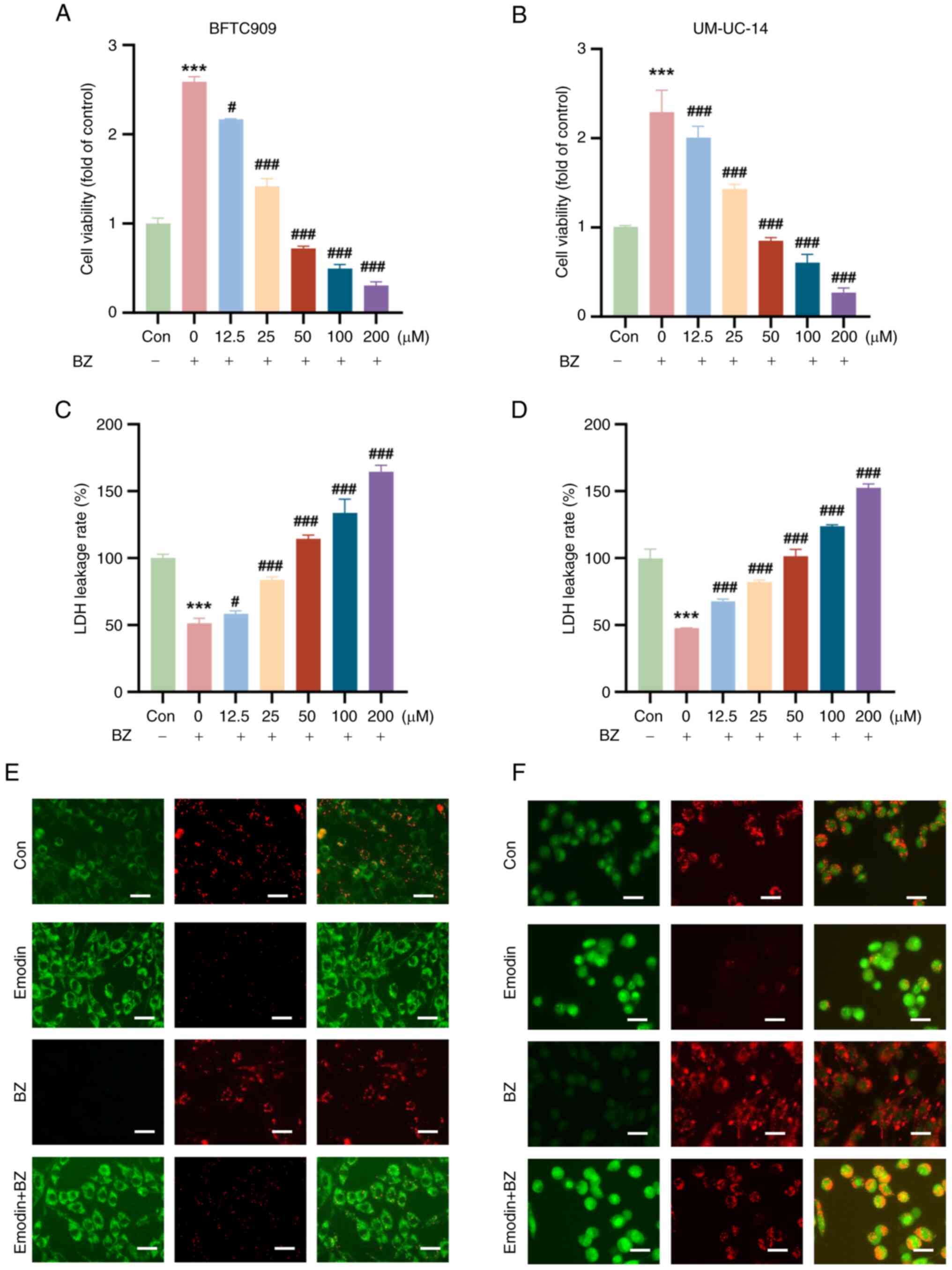 | Figure 3Emodin suppresses BZ-increased
viability and induces cellular damage in bladder cancer cells. BFTC
and UM-UC-14 cells were preincubated with varying concentrations of
emodin (12.5, 25, 50, 100 and 200 μM) at 37°C for 1 h. Then,
the cells were further treated with 10 nM BZ at 37°C for another 48
h in the presence of emodin at the indicated concentrations. The
dose-dependent inhibitory effects of emodin on the viability of (A)
BFTC and (B) UM-UC-14 cell lines treated with varying
concentrations of emodin (12.5, 25, 50, 100 and 200 μM)
compared with cells treated with BZ alone. Effect of emodin
treatment on LDH leakage in (C) BFTC and (D) UM-UC-14 cell lines.
JC-1 staining showed changes in the mitochondrial membrane
potential in (E) BFTC and (F) UM-UC-14 cells.
***P<0.001 vs. Con; #P<0.05,
###P<0.001 vs. BZ alone. BZ, benzidine; Con, control;
LDH, lactate dehydrogenase. |
BZ activates COX2/PKA signaling in
UTUC
The effect of BZ on the COX2/PKA signaling pathway
was further investigated. It was observed that BZ significantly
upregulated the protein expression of PKA and COX2 in BFTC and
UM-UC-14 cells transfected with si-NC (Fig. 4A and B). To investigate the role
of COX2 in BZ-induced PKA signaling activation, siRNAs specifically
targeting COX2 were screened. si-COX2 effectively downregulated
COX2 expression and subsequently reduced PKA expression.
Furthermore, COX2 knockdown reversed the BZ-induced increase in PKA
expression. These findings indicated that BZ-mediated activation of
PKA signaling was achieved through the induction of COX2 (Fig. 4A and 4B). Additionally, BZ treatment led to
increased levels of cAMP and PGE2 in cell lysates, but si-COX2
reversed these effects with or without BZ treatment (Fig. 4C-F). In vivo, BZ also
elevated the protein expression of PKA and COX2 in tumor tissues
from nude mice (Fig. 4G) and
increased the levels of cAMP and PGE2 within these tumors (Fig. 4H and I).
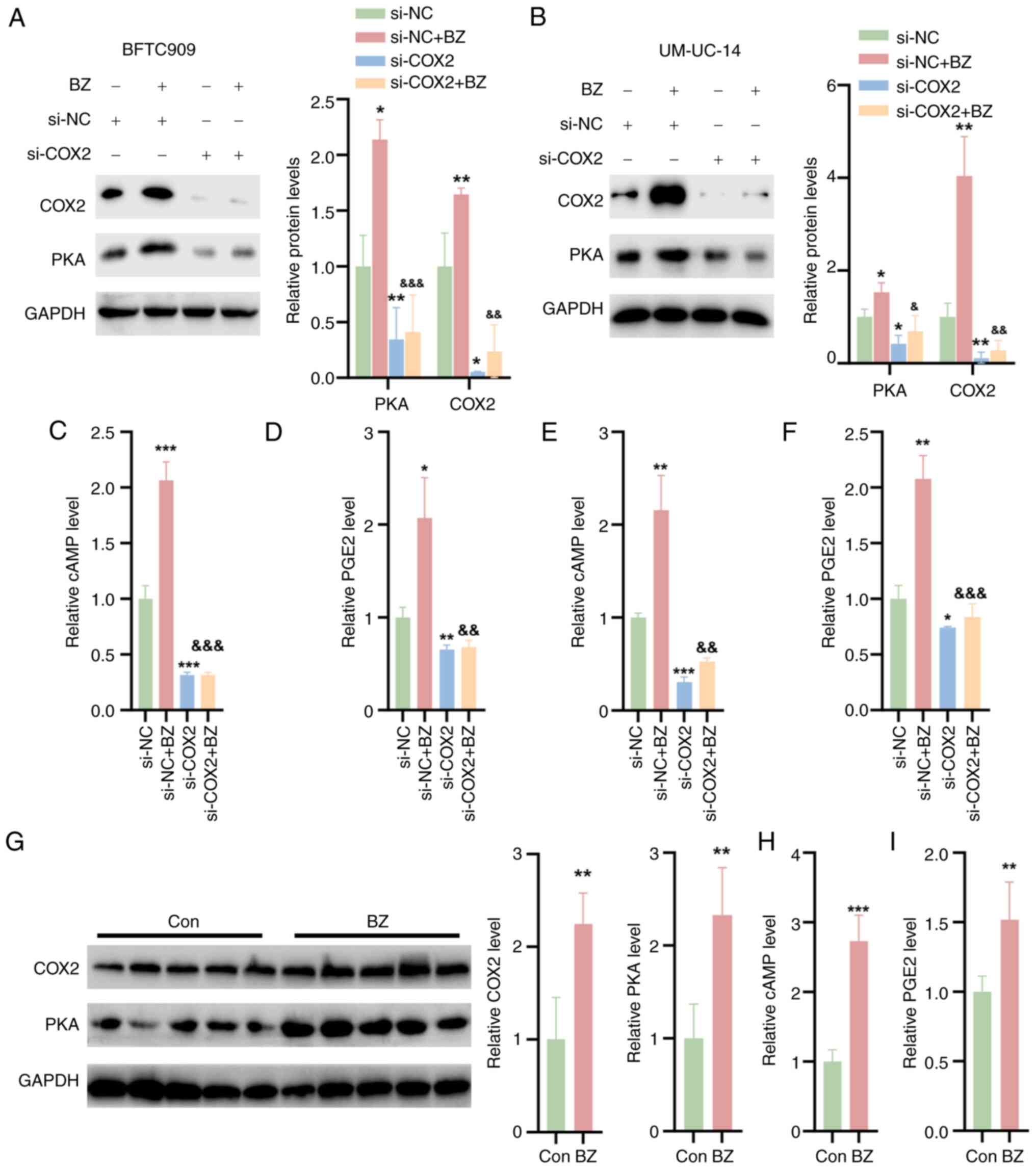 | Figure 4BZ activates COX2/PKA signaling in
upper urinary tract urothelial carcinoma cells and tumor tissues.
The BFTC and UM-UC-14 cells were divided into four groups as
follows: Control group, cells directly transfected with NC siRNA;
BZ group, cells treated with 10 nM BZ for 48 h; si-COX2 group,
cells transfected with si-COX2 for 48 h; and si-COX2 + BZ group,
cells first transfected with si-COX2 for 1 h, followed by
coincubation with 10 nM BZ at 37°C for 48 h. Western blotting
analysis indicating the protein expression of PKA and COX2 in (A)
BFTC and (B) UM-UC-14 cells treated as aforementioned. The (C) cAMP
and (D) PGE2 levels in cell lysates of BFTC cells. The (E) cAMP and
(F) PGE2 levels in cell lysates of UM-UC-14 cells. (G) Western
blotting analysis indicating the protein levels of PKA and COX2 in
tumor tissues from nude mice treated with BZ. The (H) cAMP and (I)
PGE2 levels in the tumor tissues of BZ-treated nude mice.
*P<0.05, **P<0.01,
***P<0.001 vs. si-NC; &P<0.05,
&&P<0.01,
&&&P<0.001 vs. si-NC + BZ. BZ, benzidine;
cAMP, cyclic adenosine monophosphate; Con, control; COX2,
cyclooxygenase 2; PGE2, prostaglandin E2; PKA, protein kinase A;
NC, negative control; si, small interfering RNA. |
Emodin inhibits cAMP/PKA/COX2 signaling
in UTUC cells in the presence of BZ
It was also investigated whether emodin inhibits the
cAMP/PKA/COX2 signaling pathway in UTUC cells in the presence of
BZ. The findings indicated that emodin combined with BZ markedly
decreased cAMP levels in BFTC909 and UM-UC-14 cells compared with
BZ treatment alone (Fig. 5A and
B). Additionally, western blotting analysis revealed that
emodin significantly suppressed the expression of PKA and the
downstream effector COX2 in these cells at concentrations of 25 and
50 μM, compared with BZ treatment alone (Fig. 5C and D). Additionally, IF
experiments confirmed that BZ increased COX2 expression in BFTC909
and UM-UC-14 cells, while emodin treatment reduced the BZ-induced
increase in COX2 expression (Fig.
5E).
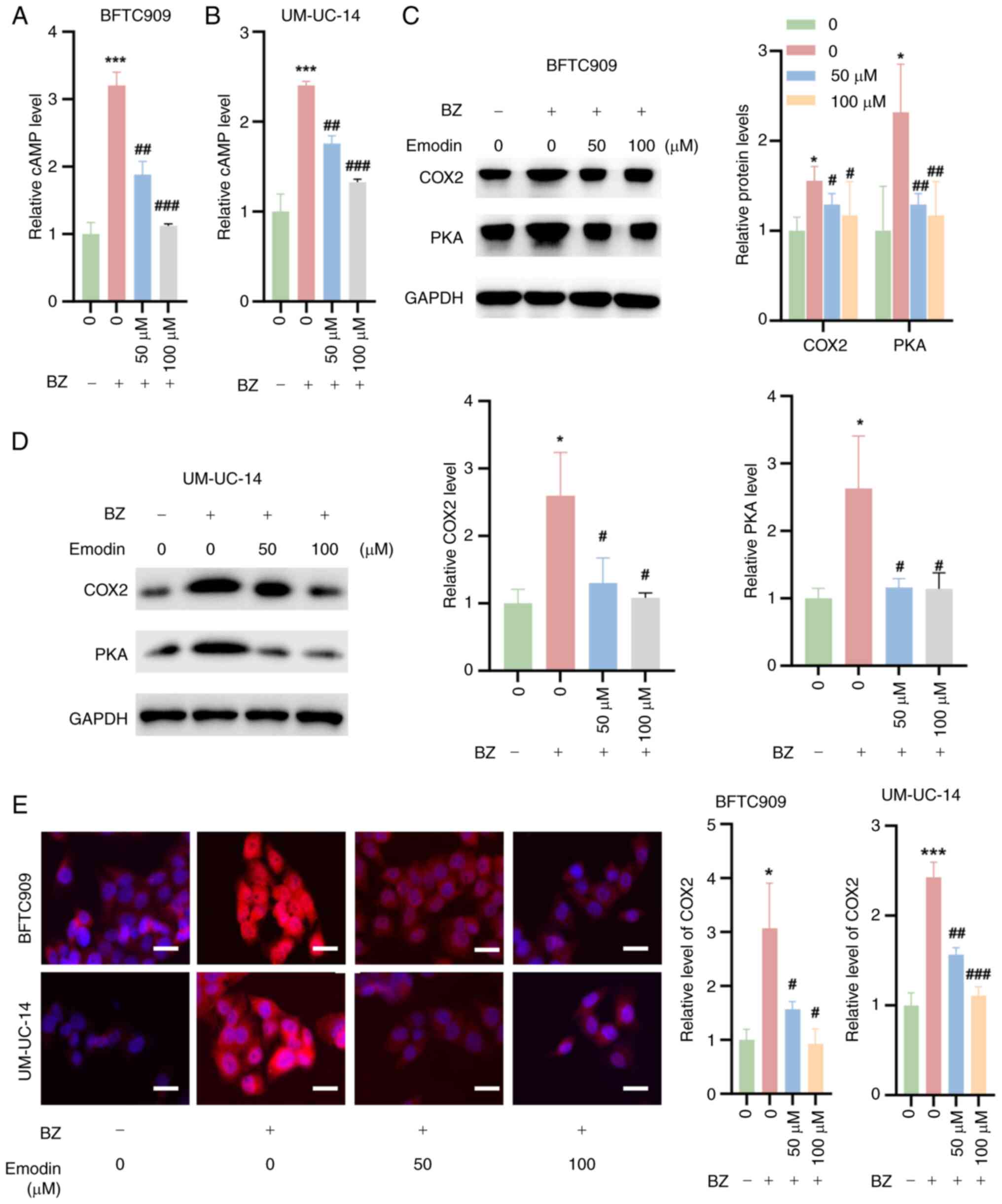 | Figure 5Inhibition of cAMP/PKA/COX2 signaling
by emodin in upper urinary tract urothelial carcinoma cells. BFTC
and UM-UC-14 cells were preincubated with 50 or 100 μM
emodin at 37°C for 1 h. Then, the cells were further treated with
10 nM BZ at 37°C for another 48 h in the presence of emodin at the
indicated concentrations. cAMP levels in (A) BFTC909 and (B)
UM-UC-14 cells treated as aforementioned. Western blotting analysis
demonstrating the expression levels of PKA and the downstream
signaling molecule, COX2, in (C) BFTC909 and (D) UM-UC-14 cells
treated as aforementioned. (E) Immunofluorescence staining
indicating COX2 expression in BFTC909 and UM-UC-14 cells treated as
aforementioned. *P<0.05, ***P<0.001 vs.
0 μM emodin; #P<0.05,
##P<0.01,###P<0.001 vs. BZ + 0
μM emodin. BZ, benzidine; cAMP, cyclic adenosine
monophosphate; Con, control; COX2, cyclooxygenase 2; PKA, protein
kinase A. |
Emodin inhibits the survival of UTUC
cells by inhibiting PKA signaling when combined with BZ
To further investigate the capacity of emodin to
suppress the malignant phenotype of UTUC cells by inhibiting PKA
signaling, the BFTC909 and UM-UC-14 cells were preincubated with BZ
before further treatment. Subsequent exposure to Sp-8-CPT-cAMPS, a
PKA activator, resulted in a significant increase in PKA and COX2
protein levels, as demonstrated by western blotting analysis
(Fig. 6A and B). Notably, the
emodin-induced downregulation of PKA and COX2 expression was
partially reversed by Sp-8-CPT-cAMPS. Additionally, while emodin
significantly reduced the PGE2 levels, treatment with
Sp-8-CPT-cAMPS led to an increase in PGE2 production and it
significantly reversed the reduction caused by emodin (Fig. 6C and D). In terms of cell
viability, compared with the control, Sp-8-CPT-cAMPS notably
enhanced the survival of BFTC909 and UM-UC-14 cells, and it notably
mitigated the emodin-induced decrease in cell viability (Fig. 6E and F). These findings suggest
that emodin effectively inhibits the viability of UTUC cells
predominantly through the inhibition of PKA signaling.
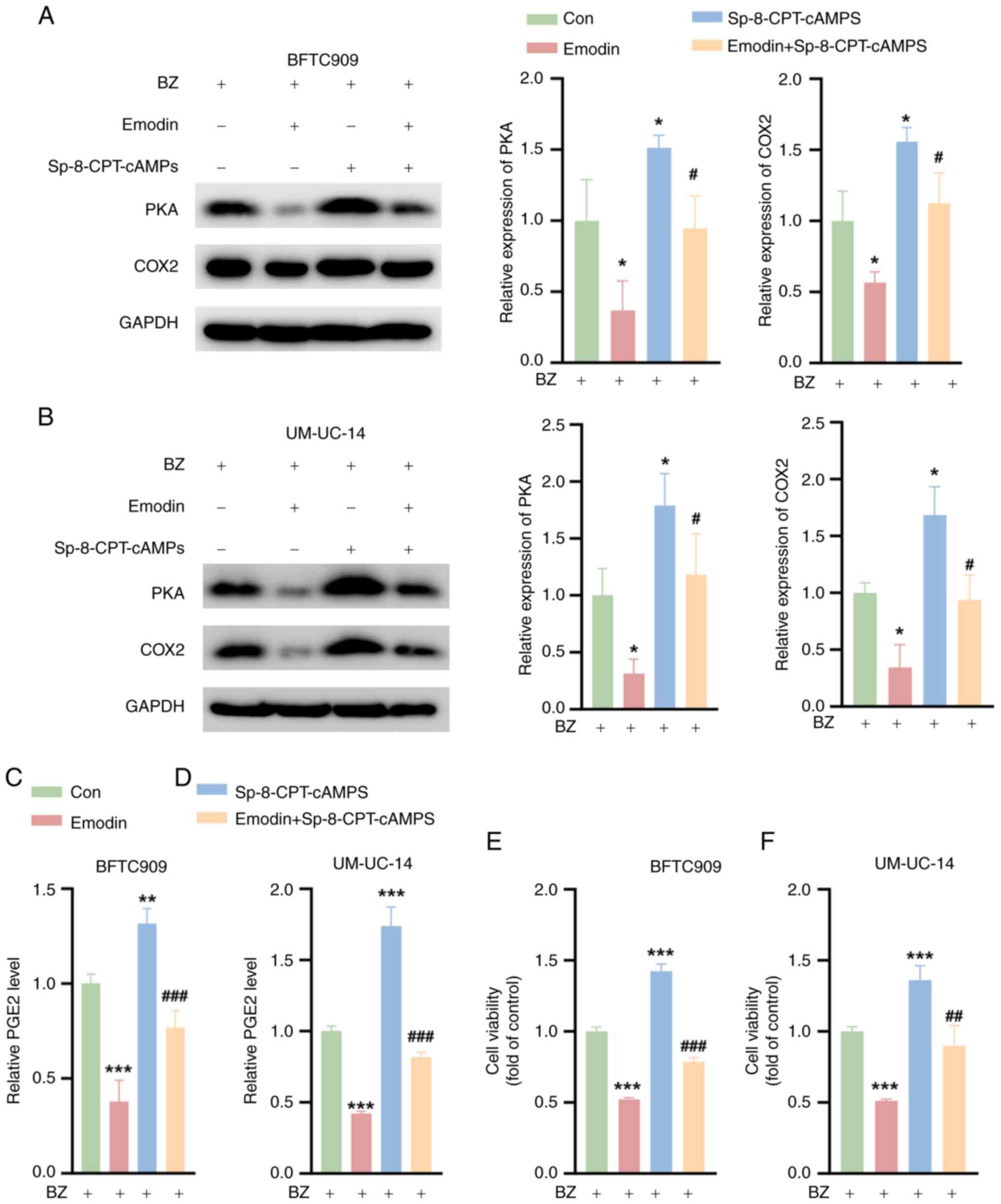 | Figure 6Emodin inhibits the survival of upper
urinary tract urothelial carcinoma Cells by inhibiting PKA
signaling. The cells were divided into four groups. Con group,
cells were treated with 10 nM BZ alone for 48 h; Emodin group,
cells were pretreated with 50 μM emodin for 1 h, followed by
the addition of 10 nM BZ for 48 h; Sp-8-CPT-cAMPS group, cells were
pretreated with 10 μM Sp-8-CPT-cAMPS for 1 h, followed by
the addition of 10 nM BZ for 48 h; and Emodin + Sp-8-CPT-cAMPS
group, cells were pretreated with a combination of 10 μM
Sp-8-CPT-cAMPS and 50 μM emodin for 1 h, followed by the
addition of 10 nM BZ for 48 h. Western blotting analysis showing
the PKA and COX2 expression levels in (A) BFTC909 and (B) UM-UC-14
cells treated as aforementioned. The PGE2 levels in (C) BFTC909 and
(D) UM-UC-14 cells treated as aforementioned. Cell Counting Kit-8
assays indicating the viability of (E) BFTC909 and (F) UM-UC-14
cells treated as aforementioned. *P<0.05,
**P<0.01, ***P<0.001 vs. Con;
#P<0.05, ##P<0.01,
###P<0.001 vs. emodin. BZ, benzidine; Con, control;
COX2, cyclooxygenase 2; PGE2, prostaglandin E2; PKA, protein kinase
A. |
Emodin reduces UTUC cell migration by
inhibiting MMP9 and VEGF
Previous studies have shown that PGE2 enhances tumor
cell infiltration and metastasis by upregulating MMP9 and VEGF
(22,23). In the present study, the effects
of emodin on the expression of these critical proteins was
investigated. The BFTC909 and UM-UC-14 cells were preincubated with
10 nM BZ before further treatment. It was observed that emodin
significantly reduced the expression of MMP9 and VEGF in both
BZ-treated BFTC909 and UM-UC-14 cells compared with the control
cells. Conversely, treatment with Sp-8-CPT-cAMPS alone led to an
increase in the expression of these proteins (Fig. 7A and B). Notably, Sp-8-CPT-cAMPS
also counteracted the suppressive effect of emodin on MMP9 and VEGF
expression (Fig. 7A and B).
Furthermore, emodin decreased the migration of BFTC909 and UM-UC-14
cells relative to that of control cells, while Sp-8-CPT-cAMPS
promoted cell migration (Fig. 7C and
D). Most notably, Sp-8-CPT-cAMPS reversed the emodin-induced
decrease in migration in these BZ-treated cell lines (Fig. 7C and D).
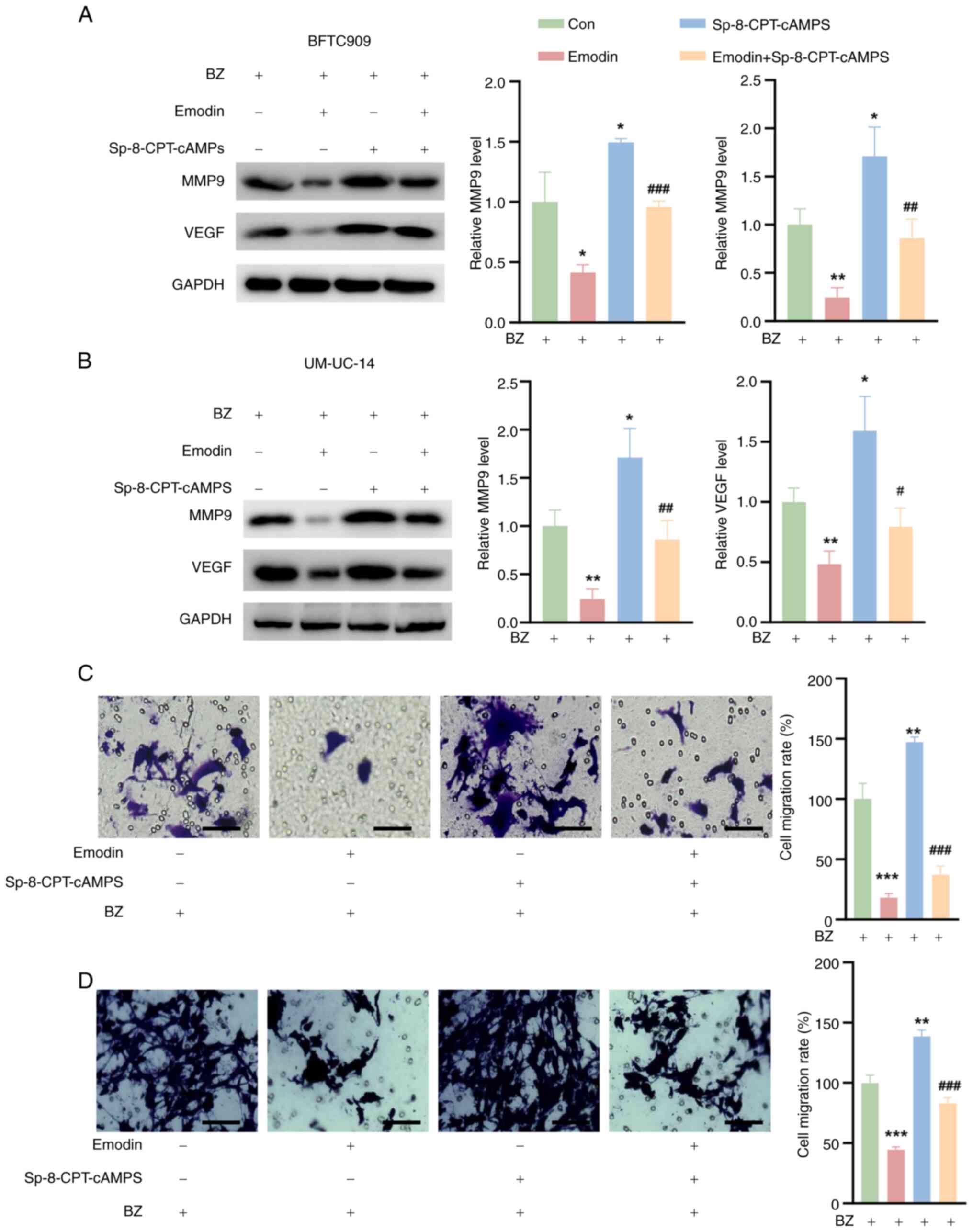 | Figure 7Effects of emodin and Sp-8-CPT-cAMPS
on MMP9 and VEGF expression and cell migration in BFTC909 and
UM-UC-14 cells. The cells were divided into four groups. Con group,
cells were treated with 10 nM BZ alone for 48 h; Emodin group,
cells were pretreated with 50 μM emodin for 1 h, followed by
the addition of 10 nM BZ for 48 h; Sp-8-CPT-cAMPS group, cells were
pretreated with 10 μM Sp-8-CPT-cAMPS for 1 h, followed by
the addition of 10 nM BZ for 48 h; and Emodin + Sp-8-CPT-cAMPS
group, cells were pretreated with a combination of 10 μM
Sp-8-CPT-cAMPS and 50 μM emodin for 1 h, followed by the
addition of 10 nM BZ for 48 h. MMP9 and VEGF levels in (A) BFTC909
and (B) UM-UC-14 cells treated as aforementioned. Transwell assay
results of (C) BFTC909 and (D) UM-UC-14 cells treated as
aforementioned. *P<0.05, **P<0.01,
***P<0.001 vs. Con; #P<0.05,
##P<0.01, ###P<0.001 vs. emodin. BZ,
benzidine; Con, control; MMP9, matrix metalloproteinase 9; VEGF,
vascular endothelial growth factor. |
Emodin inhibits tumor growth in nude mice
in vivo
In vivo experiments involving nude mice
pretreated with BZ confirmed that emodin could effectively decrease
the weight and volume of tumors (Fig.
8A-C). Additionally, emodin reduced the levels of cAMP and PGE2
in tumor tissues (Fig. 8D and E).
The in vivo safety of emodin was also evaluated. The
experimental results showed that emodin did not have any
significant toxic effects on the liver or kidneys of the treated
mice. Liver function tests, including the measurement of ALT and
AST enzymes, revealed no significant differences between the
emodin-treated group and the control group (Fig. 8F and G). Similarly, kidney
function markers, such as creatinine and UREA, remained within the
normal ranges, indicating no renal toxicity (Fig. 8H and I). By contrast, emodin
decreased the LDH leakage rate in tumor tissues compared with the
control tissues (Fig. 8J). Ki-67
staining also showed that L-emodin and H-emodin decreased the
percentage of Ki-67-positive cells (Fig. 8K). Furthermore, there was a
notable decrease in the expression of MMP9 and VEGF in the tumor
tissues of these nude mice (Fig.
8L). These findings collectively indicate that emodin exerts a
notable inhibitory effect on tumor development in vivo.
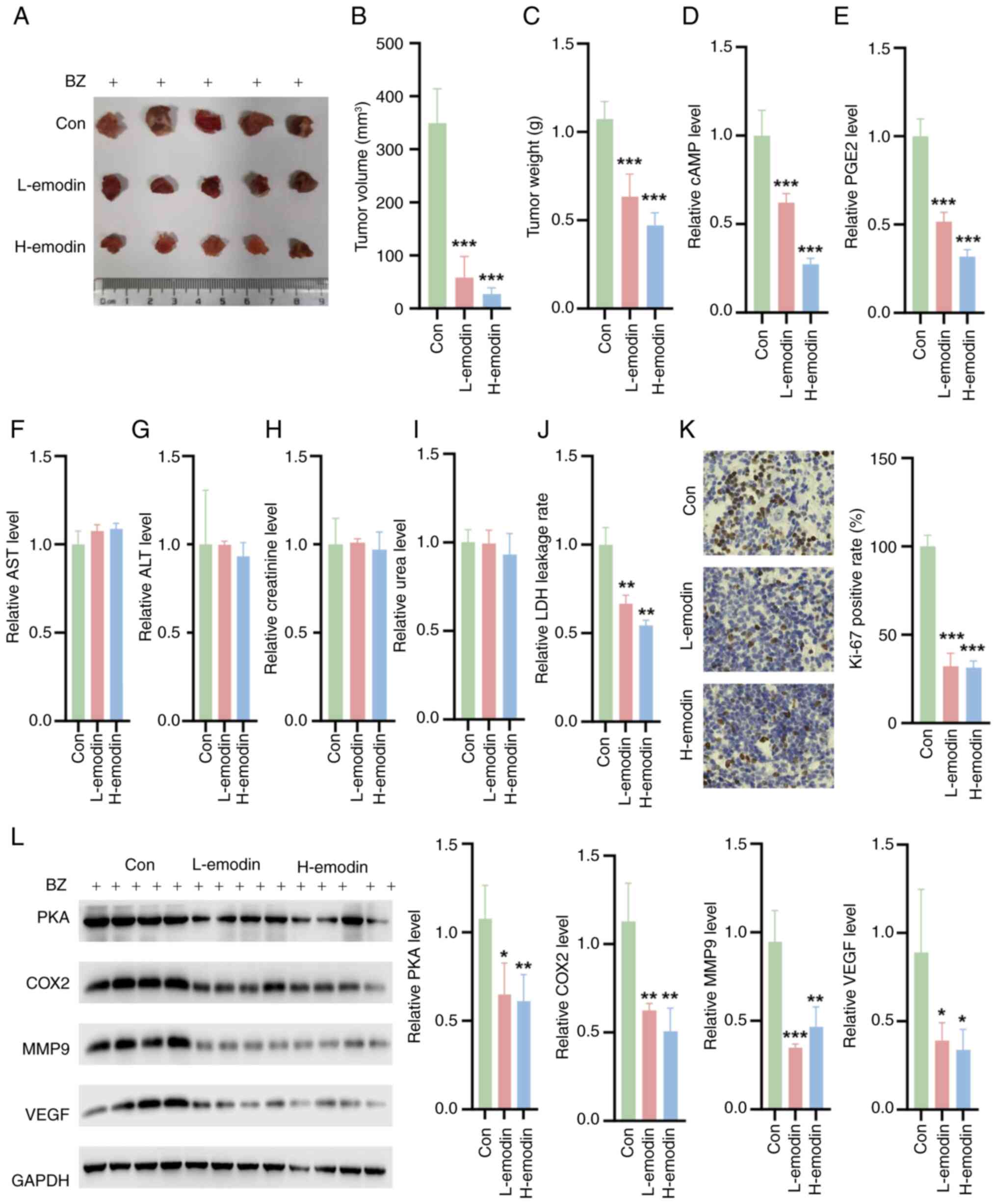 | Figure 8Impact of emodin on tumor growth and
biochemical markers in BZ-pretreated nude mice. (A) Representative
tumor images. Quantification of the (B) tumor volume and (C)
weight. (D) cAMP (D) and (E) PGE2 levels in tumor tissues. Liver
function parameters, including (F) ALT and (G) AST levels in the
treated mice. Kidney function markers, such as (H) creatinine and
(I) 0urea in the treated mice. (J) LDH leakage rate in the tumor
tissues. (K) Ki-67 staining of the tumor tissues. (L) The
expression levels of PKA, COX2, MMP9 and VEGF in the tumor tissues.
*P<0.05, **P<0.01,
***P<0.001 vs. Con. ALT, alanine transaminase; AST,
aspartate transaminase; BZ, benzidine; cAMP, cyclic adenosine
monophosphate; Con, control; COX2, cyclooxygenase 2; H-emodin, 80
mg/kg emodin; L-emodin, 40 mg/kg emodin; MMP9, matrix
metalloproteinase 9; PGE2, prostaglandin E2; PKA, protein kinase A;
VEGF, vascular endothelial growth factor. |
Discussion
Bladder cancer ranks as the most prevalent and
lethal among common malignant tumors of the urinary system. The
primary environmental contributors to bladder cancer include
tobacco smoke, arsenic in drinking water and occupational exposure
to aromatic amines (24,25). BZ, known for its carcinogenic
properties, has been banned in industrial production (26). Nevertheless, it is still detected
in certain food colors. Moreover, with the increase in tobacco use,
BZ continues to significantly contribute to the incidence of
bladder tumors (26). UTUC,
involving tumors of the renal pelvis and ureters, demands careful
clinical attention due to its recurring and highly malignant nature
(27). Early detection and
appropriate treatment can lead to clinical remission (27). A number of patients with UTUC,
particularly older adults, are asymptomatic and are often diagnosed
during routine health examinations (28). Notably, the incidence of UTUC is
markedly greater among smokers than non-smokers and poses a greater
risk to individuals working in petrochemical, plastics and other
chemical industries (28).
Research on the mechanisms of BZ-induced UTUC is still limited. To
the best of our knowledge, the present study is the first to
provide in vitro evidence that BZ promotes the survival and
migration of UTUC cells, thus supporting tumor growth. These
findings establish the significant carcinogenic role of BZ in the
progression of UTUC.
Natural drugs are increasingly recognized as vital
sources for developing therapeutic agents due to their broad
biological impacts and minimal side effects (16). Among these, Rheum
officinale, particularly its anthraquinone derivatives
extracted from its roots and rhizomes, has shown promising
potential due to its anti-inflammatory, antifibrotic, antioxidant,
antitumor and antidiabetic properties (29). Despite these known effects, the
protective role of emodin, a compound derived from R.
officinale, against BZ-induced UTUC remains unexplored. The
results of the in vitro experiments in the present study
revealed that emodin can significantly mitigate the BZ-induced
increase in UTUC cell viability and decrease the LDH leakage rate.
Additionally, emodin attenuated the increase in the mitochondrial
membrane potential induced by BZ. These findings preliminarily
indicate that emodin may have a crucial protective role in the
progression of BZ-induced UTUC, highlighting its potential as a
therapeutic agent in oncological treatments.
PGE2, the most prevalent arachidonoid lipid and a
lipid metabolite with immunomodulatory functions, has been
implicated in the development of various malignant tumors, such as
pancreatic cancer, bladder cancer and UTUC (13,30,31). Previous studies have demonstrated
that emodin inhibits PGE2 production (32,33). Therefore, the present
investigation aimed to evaluate whether BZ activates
PGE2-associated signaling pathways to facilitate the viability of
UTUC cells. Both the in vitro and in vivo experiments
confirmed that BZ upregulated the expression of PKA and COX2,
concomitantly increasing the cAMP and PGE2 levels. The present
investigation further revealed that BZ significantly upregulated
the expression of VEGF and MMP9, which are pivotal factors
implicated in tumor angiogenesis and invasion. These findings not
only elucidated the role of BZ in promoting the progression of UTUC
but also shed light on its potential carcinogenic effects on the
upper urinary tract system, extending beyond its association with
bladder cancer. In the EP2-cAMP-PKA signaling pathway, there is a
positive feedback loop that regulates the expression of PGE2 and
COX2 (34-36). Specifically, activation of PKA
leads to an increase in COX2 expression (37-40). This occurs since PKA can
phosphorylate and activate transcription factors that upregulate
the expression of COX2. Consequently, when a PKA activator is
introduced, it enhances PKA activity, which in turn elevates COX2
levels (38-40). Similarly, PGE2 and cAMP are
involved in a positive feedback mechanism. PGE2 can increase cAMP
levels by binding to its receptor, EP2, which activates adenylate
cyclase. This enzyme catalyzes the conversion of ATP to cAMP
(38-40). Elevated cAMP levels then activate
PKA, which further increases COX2 expression and subsequently PGE2
production. Hence, in the present study, BFTC909 and UM-UC-14
cells, which were pretreated with BZ, were treated with a PKA
activator, Sp-8-CPT-cAMPS, to test whether emodin exerts an
anti-UTUC effect by targeting the PKA/COX2 signaling pathway.
Compared with the control group, emodin treatment resulted in a
notable reduction in the expression levels of PKA, COX2, VEGF and
MMP9. Furthermore, the emodin-induced decreases in PKA, COX2, VEGF
and MMP9 expression were significantly reversed upon the addition
of Sp-8-CPT-cAMPS. Based on these extensive research findings, we
consider that the current data sufficiently demonstrate the
feedback regulation of COX2 by PKA. However, we recognize the
importance of directly demonstrating the role of PKA. To address
this, we plan to conduct further experiments to directly
downregulate PKA using siRNA or pharmacological inhibitors to
assess its impact on COX2 expression and the overall signaling
pathway.
Moreover, in the present study, in the in
vivo experiments using nude mice with BZ-induced subcutaneous
tumors, emodin administration led to a reduction in tumor volume.
Additionally, emodin treatment decreased PGE2 and cAMP levels and
reduced MMP9 and VEGF expression. These novel findings highlight
the crucial role of emodin in abolishing BZ-associated UTUC
development by targeting the PKA/COX2 signaling pathway, indicating
that emodin is a promising therapeutic strategy for occupational
UTUC treatment. Additionally, the experimental results showed that
emodin did not have any significant toxic effects on the liver or
kidneys of the treated mice. Liver function tests, including
measurements of ALT and AST levels, revealed no significant
differences between the emodin-treated group and the control group.
Similarly, kidney function markers, such as creatinine and UREA,
remained within normal ranges, indicating no renal toxicity. These
findings were consistent with previous studies that demonstrated
the safety profile of emodin in similar settings (16,19). The lack of hepatotoxicity and
nephrotoxicity is particularly notable given the prolonged
treatment duration and the relatively high doses used in the
experiments of the present study. This finding confirmed that the
observed therapeutic effects of emodin on inhibiting BZ-induced
cell survival and migration are not confounded by potential organ
toxicity. Future studies should continue to monitor these safety
parameters in different models and at different doses to further
confirm the non-toxic nature of emodin, thereby supporting its
potential clinical application in treating UTUC.
In conclusion, emodin inhibits the activity of the
PKA/COX2 signaling pathway, thereby suppressing the development of
UTUC induced by BZ exposure (Fig.
9). These findings lay the groundwork for a comprehensive
understanding of the molecular mechanisms underlying BZ-induced
UTUC, highlighting the potential of PKA/COX2 pathway inhibitors as
crucial targets for early intervention in UTUC. Hence, emodin has
emerged as a promising candidate for both early intervention and
therapeutic strategies in UTUC management.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YJ performed the experiments and analyzed the data.
CW, KF, XW and MT performed the animal experiments. YJ and GT
designed all the experiments, analyzed the data and gave final
approval for the version to be published. YJ and GT confirm the
authenticity of all the raw data. All the authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
This study involving animals was reviewed and
approved by Jinzhou Medical University (Jinzhou, China;
approval no. 2020-AJ-05).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
This study was supported by the Scientific Research Funding
Project of Liaoning Provincial Department of Education (grant no.
JYTJCZR2020063).
References
|
1
|
Habil MR and Hein DW: Effects of dose and
human N-acetyltransferase 1 genetic polymorphism in benzidine
metabolism and genotoxicity. Arch Toxicol. 97:1765–1772. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dietrich HG and Golka K: Bladder tumors
and aromatic amines-historical milestones from Ludwig Rehn to
Wilhelm Hueper. Front Biosci (Elite Ed). 4:279–288. 2012.
View Article : Google Scholar
|
|
3
|
Durgaryan R and Durgaryan N: Chemical
oxidative condensation of benzidine in non-aqueous medium:
Synthesis and investigation of oligomers and polymer with Benzidine
Diimine Units. Polymers (Basel). 14:342021. View Article : Google Scholar
|
|
4
|
Suarez-Torres JD, Orozco CA and
Ciangherotti CE: Applying Bayesian forecasting to predictive
toxicology: The probability of innate carcinogenicity to humans of
colorants synthesized from benzidine. Toxicol Lett. 351:111–134.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kenigsberg AP, Meng X, Ghandour R and
Margulis V: Oncologic outcomes of radical nephroureterectomy (RNU).
Transl Androl Urol. 9:1841–1852. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bayerl F, Meiser P, Donakonda S,
Hirschberger A, Lacher SB, Pedde AM, Hermann CD, Elewaut A, Knolle
M, Ramsauer L, et al: Tumor-derived prostaglandin E2 programs cDC1
dysfunction to impair intratumoral orchestration of anticancer
T-cell responses. Immunity. 56:1341–1358 e11. 2023. View Article : Google Scholar
|
|
7
|
Woolbright BL, Pilbeam CC and Taylor JA
III: Prostaglandin E2 as a therapeutic target in bladder cancer:
From basic science to clinical trials. Prostaglandins Other Lipid
Mediat. 148:1064092020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kissoondoyal A and Crawford DA:
Prostaglandin E2 increases neurite length and the formation of
axonal loops, and regulates cone turning in differentiating NE4C
Cells Via PKA. Cell Mol Neurobiol. 42:1385–1397. 2022. View Article : Google Scholar
|
|
9
|
Chang HH, Young SH, Sinnett-Smith J, Chou
CE, Moro A, Hertzer KM, Hines OJ, Rozengurt E and Eibl G:
Prostaglandin E2 activates the mTORC1 pathway through an
EP4/cAMP/PKA- and EP1/Ca2+-mediated mechanism in the human
pancreatic carcinoma cell line PANC-1. Am J Physiol Cell Physiol.
309:C639–C649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao P, Li XG, Yang M, Shao Q, Wang D, Liu
S, Song H, Song B, Zhang Y and Qu X: Hypoxia suppresses the
production of MMP-9 by human monocyte-derived dendritic cells and
requires activation of adenosine receptor A2b via cAMP/PKA
signaling pathway. Mol Immunol. 45:2187–2195. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fang XL, Zhang Q, Xue WW, Tao JH, Zou HD,
Lin QR and Wang YL: Suppression of cAMP/PKA/CREB signaling
ameliorates retinal injury in diabetic retinopathy. Kaohsiung J Med
Sci. 39:916–926. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeon HG, Jeong IG, Bae J, Lee JW, Won JK,
Paik JH, Kim HH, Lee SE and Lee E: Expression of Ki-67 and COX-2 in
patients with upper urinary tract urothelial carcinoma. Urology.
76:513 e7–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komatsu M, Funakoshi T, Aki T, Unuma K and
Uemura K: Aristolochic acid induces an inflammatory response with
prostaglandin E2 production and apoptosis in NRK-52E proximal
tubular cells. Toxicol Lett. 378:39–50. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liudvytska O and Kolodziejczyk-Czepas J: A
review on rhubarb-derived substances as modulators of
cardiovascular risk factors-A special emphasis on anti-obesity
action. Nutrients. 14:20532022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qin MY, Huang SQ, Zou XQ, Zhong XB, Yang
YF, Zhang YT, Mi ZC, Zhang YS and Huang ZG: Drug-containing serum
of rhubarb-astragalus capsule inhibits the epithelial-mesenchymal
transformation of HK-2 by downregulating TGF-beta1/p38MAPK/Smad2/3
pathway. J Ethnopharmacol. 280:1144142021. View Article : Google Scholar
|
|
16
|
Zou G, Zhang X, Wang L, Li X, Xie T, Zhao
J, Yan J, Wang L, Ye H, Jiao S, et al: Herb-sourced emodin inhibits
angiogenesis of breast cancer by targeting VEGFA transcription.
Theranostics. 10:6839–6853. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dai G, Wang D, Ma S, Hong S, Ding K, Tan X
and Ju W: ACSL4 promotes colorectal cancer and is a potential
therapeutic target of emodin. Phytomedicine. 102:1541492022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma L, Chen K, Jiang K, Deng G, Jiang P,
Shao J and Yu Z: Emodin inhibits the proliferation and invasion of
bladder cancer cells by downregulatingating Notch1. Int J Clin Exp
Pathol. 10:9452–9459. 2017.
|
|
19
|
Cha TL, Chuang MJ, Tang SH, Wu ST, Sun KH,
Chen TT, Sun GH, Chang SY, Yu CP, Ho JY, et al: Emodin modulates
epigenetic modifications and suppresses bladder carcinoma cell
growth. Mol Carcinog. 54:167–177. 2015. View Article : Google Scholar
|
|
20
|
Luster MI, Tucker AN, Hayes HT, Pung OJ,
Burka T, McMillan R and Eling T: Immunosuppressive effects of
benzidine in mice: Evidence of alterations in arachidonic acid
metabolism. J Immunol. 135:2754–2761. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Martin CN, Beland FA, Roth RW and Kadlubar
FF: Covalent binding of benzidine and N-acetylbenzidine to DNA at
the C-8 atom of deoxyguanosine in vivo and in vitro. Cancer Res.
42:2678–2686. 1982.PubMed/NCBI
|
|
22
|
Wong HP, Ho JW, Koo MW, Yu L, Wu WK, Lam
EK, Tai EK, Ko JK, Shin VY, Chu KM and Cho CH: Effects of
adrenaline in human colon adenocarcinoma HT-29 cells. Life Sci.
88:1108–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan X, Li J, Long L, Shi T, Liu D, Tan W,
Zhang H, Wu X, Lei X and Wang Z: Design, synthesis and biological
evaluation of N-anthraniloyl tryptamine derivatives as pleiotropic
molecules for the therapy of malignant glioma. Eur J Med Chem.
222:1135642021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Millerick-May ML, Wang L, Rice C and
Rosenman KD: Ongoing risk of bladder cancer among former workers at
the last benzidine manufacturing facility in the USA. Occup Environ
Med. 78:625–631. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Letasiova S, Medve'ova A, Sovcikova A,
Dušinská M, Volkovová K, Mosoiu C and Bartonová A: Bladder cancer,
a review of the environmental risk factors. Environ Health.
11(Suppl 1): S112012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun X, Zhang T, Deng Q, Zhou Q, Sun X, Li
E, Yu D and Zhong C: Benzidine induces epithelial-mesenchymal
transition of human bladder cancer cells through activation of ERK5
pathway. Mol Cells. 41:188–197. 2018.PubMed/NCBI
|
|
27
|
Soria F, Shariat SF, Lerner SP, Fritsche
HM, Rink M, Kassouf W, Spiess PE, Lotan Y, Ye D, Fernández MI, et
al: Epidemiology, diagnosis, preoperative evaluation and prognostic
assessment of upper-tract urothelial carcinoma (UTUC). World J
Urol. 35:379–387. 2017. View Article : Google Scholar
|
|
28
|
Farrow JM, Kern SQ, Gryzinski GM and
Sundaram CP: Nephron-sparing management of upper tract urothelial
carcinoma. Investig Clin Urol. 62:389–398. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang FY, Li RZ, Xu C, Fan XX, Li JX, Meng
WY, Wang XR, Liang TL, Guan XX, Pan HD, et al: Emodin induces
apoptosis and suppresses non-small cell lung cancer growth via
downregulation of sPLA2-IIa. Phytomedicine. 95:1537862022.
View Article : Google Scholar
|
|
30
|
Akbari B, Soltantoyeh T, Shahosseini Z,
Jadidi-Niaragh F, Hadjati J, Brown CE and Mirzaei HR: PGE2-EP2/EP4
signaling elicits mesoCAR T-cell immunosuppression in pancreatic
cancer. Front Immunol. 14:12095722023. View Article : Google Scholar
|
|
31
|
Kurtova AV, Xiao J, Mo Q, Pazhanisamy S,
Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL and Chan KS:
Blocking PGE2-induced tumor repopulation abrogates bladder cancer
chemoresistance. Nature. 517:209–213. 2015. View Article : Google Scholar
|
|
32
|
Park MY, Kwon HJ and Sung MK: Evaluation
of aloin and aloe-emodin as anti-inflammatory agents in aloe by
using murine macrophages. Biosci Biotechnol Biochem. 73:828–832.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu H, Song X, Li Y, Ma T, Bai H, Zhao M,
Wang X, Liu L and Gao L: Emodin protects knee joint cartilage in
rats through anti-matrix degradation pathway: An in vitro and in
vivo study. Life Sci. 269:1190012021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang RY and Chen GG: Cigarette smoking,
cyclooxygenase-2 pathway and cancer. Biochim Biophys Acta.
1815:158–169. 2011.
|
|
35
|
Wiktorowska-Owczarek A and Owczarek J: The
effect of hypoxia on PGE2-stimulated cAMP generation in HMEC-1.
Cell Mol Biol Lett. 20:213–221. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ye Y, Wang X, Jeschke U and von Schönfeldt
V: COX-2-PGE2-EPs in gynecological cancers. Arch Gynecol
Obstet. 301:1365–1375. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li T, Hu J, Du S, Chen Y, Wang S and Wu Q:
ERK1/2/COX-2/PGE2 signaling pathway mediates GPR91-dependent VEGF
release in streptozotocin-induced diabetes. Mol Vis. 20:1109–1121.
2014.PubMed/NCBI
|
|
38
|
Steinert D, Kuper C, Bartels H, Beck FX
and Neuhofer W: PGE2 potentiates tonicity-induced COX-2 expression
in renal medullary cells in a positive feedback loop involving
EP2-cAMP-PKA signaling. Am J Physiol Cell Physiol. 296:C75–C87.
2009. View Article : Google Scholar
|
|
39
|
Choudhary S, Kumar A, Kale RK, Raisz LG
and Pilbeam CC: Extracellular calcium induces COX-2 in osteoblasts
via a PKA pathway. Biochem Biophys Res Commun. 322:395–402. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen L, Sooranna SR, Lei K, Kandola M,
Bennett PR, Liang Z, Grammatopoulos D and Johnson MR: Cyclic AMP
increases COX-2 expression via mitogen-activated kinase in human
myometrial cells. J Cell Mol Med. 16:1447–1460. 2012. View Article : Google Scholar
|