1. Introduction
As a lethal disease, cancer poses a challenge to the
entire human race. Despite the progress made in modern medicine,
cancer continues to be a leading cause of mortality worldwide.
Moreover, the number of oncological diseases exhibits a steady
growth rate due to poor ecology worldwide, particularly when
considering the effect of microplastics on the epithelial barrier
of mucous membranes (1), as well as
the negative impact of agricultural pesticides (2), the nitrate poisoning of air (3)and drinking water (4), the consumption of red meat (5), and residing in proximity to industrial
facilities (6).
Research is focused on socially important forms of
cancer, including lung cancer, breast cancer and colorectal cancer
(CRC), which is one of the most prevalent oncological diseases
(7). Colonoscopy (8) with a subsequent morphological analysis
of colon biopsy samples does not always suffice for the effective
prevention of CRC (9-11).
Additional pathomorphological tests on colon biopsy samples,
involving molecular medicine methods, can significantly enhance the
scope of diagnostics. However, the removal of precancerous lesions
only does not solve the issue of effectively preventing CRC in
high-risk patients. One of the possible solutions could be the
development of pharmacological treatment protocols to regulate
molecular mechanisms, responsible for the regeneration of cellular
elements of the colon mucous membrane. In this context, special
focus could be placed on decreasing β-catenin level that is one of
the most critical indicators of the functional activity of
neoplastic cells.
The present review discusses the methods of the
pharmacological regulation of β-catenin in patients with
precancerous colorectal lesions who are at a high risk of
developing CRC.
2. Current state of affairs
Among the socially significant types of cancer, CRC
stands out. At 60-70 years of age, the risk of developing the
disease is highest and increases as one ages (12). The risk factors include hereditary
genetic syndromes (Table I),
communicable and non-communicable diseases, lifestyle risk factors
(Table II) and inflammatory bowel
disease (IBD).
 | Table IThe most common genetic hereditary
syndromes of colorectal cancer. |
Table I
The most common genetic hereditary
syndromes of colorectal cancer.
| Name of hereditary
syndrome | Type of
inheritance | (Refs.) |
|---|
| Familial
adenomatous polyposis (FAP) syndrome |
Autosomal-dominant | (113) |
| Mutated MMR gene
syndrome |
Autosomal-dominant | (114) |
| Lynch syndrome |
Autosomal-dominant | (115) |
| Muir-Torre
syndrome |
Autosomal-dominant | (116) |
| Peutz-Jeghers
syndrome |
Autosomal-dominant | (117) |
| Cowden
syndrome |
Autosomal-dominant | (118) |
| Juvenile polyposis
syndrome |
Autosomal-dominant | (119) |
| Serrated polyposis
syndrome (SPS) | Unknown | (120) |
| Cronkhite-Canada
syndrome | No reliable data
available | |
| Table IICommon lifestyle factors of
colorectal cancer. |
Table II
Common lifestyle factors of
colorectal cancer.
| Risk factors | (Refs.) |
|---|
| Low socioeconomic
status | (121,122) |
| Overweight and
obesity | |
| Sedentary
lifestyle | |
| Smoking
tobacco | |
| Alcohol abuse | |
| Low fiber, high fat
diet | |
| Consumption of red
and overcooked meat | |
| Insulin-resistant
diabetes mellitus | |
| Acromegaly | |
| Organ
transplantation with long-term immunosuppression | |
| Long-term androgen
deprivation | |
A family history of CRC is one of the key risk
factors, as well as the presence of syndromes, such as familial
adenomatous polyposis (FAP), mismatch repair (MMR) gene mutations,
Lynch, Muir-Torre, Peutz-Jeghers, Cowden, juvenile polyposis with
autosomal dominant type of inheritance, serrated polyposis syndrome
and Cronkhite-Canada syndrome with an unknown type of inheritance
(Table I).
The results of multiple studies have demonstrated
that patients with a positive family history have a lower risk of
mortality due to CRC. A possible explanation for the link between a
family history of CRC and improved survival rates is that those
with a cancer-related family may make more conscious lifestyle
choices, such as quitting smoking and increasing their physical
activity. Another contributor could be more frequent and thorough
screening that allows for the discovery of cancer at the early
stages and, therefore, improves the chances of survival. Genetic
differences between patients with CRC and those with no family
history may be one more explanation for the existing contrast in
survival rates. Studies have shown that patients with CRC and a
positive family history are more likely to have heightened levels
of microsatellite instability, which may lead to improved survival
rates. Determining the influence, that family history has on the
prognosis of CRC, is not an easy feat, since both genetic and
environmental factors are to be considered; therefore, further
research is required (13).
Among individuals aged 20-49 years, the incidence of
CRC has increased in nine countries (Germany, USA, Australia,
Canada, New Zealand, Great Britain, Denmark, Slovenia and Sweden),
while only three countries have exhibited a decrease in its
incidence (Italy, Austria and Lithuania) (14), and more than half of patients do not
have the disease in their family history (15).
There is a consistent rate of morbidity among the
elderly in North America, Europe and Oceania. A growing trend of
the prevalence of CRC has been observed among young individuals
with a high income in nine different countries on three different
continents (14).
At the same time, the frequency of right-sided CRC
makes it objectively more difficult to make a diagnosis during the
endoscopic examination (16), as is
the case with late-developing symptoms of colon obstruction due to
a larger diameter of the right portion of the colon (16).
Colonoscopy is a gold standard for the diagnosis of
colorectal neoplasms (16).
Precancerous lesions are frequently discovered in patients aged ≥50
years during colorectal screening. The overwhelming majority of
cases exhibit no symptoms, apart from occasional stomach aches and
other signs of dyspepsia (17).
The diagnosis of precancerous lesions during
colonoscopy is relatively easy; however, neoplasms in the proximal
regions of the colon are poorly visible in a routine endoscopy
(16) due to the larger diameter of
the right-sided segments of the colon. This results in the later
manifestation of intestinal obstruction and other CRC symptoms
(16). The removal of such
precancerous lesions is a crucial step in preventing CRC.
3. Clinical and morphological features of
precancerous lesions in the colon
The European Society of Gastrointestinal Endoscopy
recommends the cold snare method for the removal of polyps ≤9 mm in
size. Diathermocoagulation together with mucous membrane lifting
are used to remove sessile polyps ≤20 mm in size. If a polyp is
pedunculated and >20 mm in size, those with a large head or a
stem with >10 mm in diameter should be treated with a
combination of diathermocoagulation and adrenaline injection into
the stem or with one of the preventive mechanical techniques of
hemostasis. However, larger size, visual signs of invasion and
diagnostic extent of endoscopic ultrasonography are not always
conclusive enough to select the only right method of polyp
elimination (10); thus, an urgent
morphological analysis may be required.
The World Health Organization (2019) (18) distinguishes CRC with serrated
histoarchitecture, tubular and tubulovillous adenomas, focal
intraepithelial neoplasia with underlying chronic inflammation
(Table III). Moreover, as
demonstrated in clinical practice, precancerous colorectal lesions
often have mixed histological patterns with both serrated and
tubular components, and there are no data either on what type of
adenoma they should be classified as, or their molecular nature.
Precancerous lesions with a serrated architecture appear due to the
impaired proliferation in the cambium layer of intestinal
epithelium in the crypt base, resulting in relocation of
proliferating cells into the apical sections and development of
serrated pattern (Fig. 1A) due to
the competition for a place on a basal lamina (18).
 | Figure 1(A) A hyperplastic polyp, represented
by a neoplasm with a serrated histoarchitecture, which develops as
a result of impaired proliferation of the epithelium of the base of
the crypts, which leads to the migration of proliferating cells
into the apical parts of the intestinal crypts and the development
of a serrated pattern. The tissue was obtained from a female
patient, 70 years of age. The excision of the colon tumor was
performed on May 22, 2023. A section of the mucous membrane of the
colon measuring ~5.0 mm was prepared with further standard
histological examination of the biopsy specimen and embedding in
paraffin. Cutting was carried out using a microtome, with a section
thickness of 4 µm. Standard hematoxylin and eosin staining was then
performed. The magnification of the microscope cannot be reliably
determined, since a series of images were taken with a
magnification of x10 and subsequent stitching of the
microphotographs. (B) Sissle serrated adenoma, represented by a
neoplasm with basal localization of proliferation zones in the
intestinal crypts and horizontal displacement of the proliferative
zone, as well as the presence of dysplasia. The tissue was obtained
from a female patient, 52 years of age. The excision of the colon
tumor was performed on February 27, 2023. A flat section of the
colon mucosa with a diameter of ~1.7 mm, with serial sections was
prepared, with further standard histological processing of the
biopsy specimen, and embedding in paraffin. Cutting was carried out
using a microtome, with a section thickness of 4 µm. Standard
hematoxylin and eosin staining was then performed. The
magnification of the microscope cannot be reliably determined,
since a series of images were taken with a magnification of x10 and
subsequent stitching of the microphotographs. (C) Tubular adenoma,
represented by a neoplasm with the presence of a classic tubular
pattern with dysplastic changes, elongation, an increase in the
number of tubules and the development of finger-like outgrowths.
The tissue was obtained from a female patient, 49 years of age. The
excision of the colon tumor was performed on June 1, 2023. The
image depicts an exophytic neoplasm of the colon mucosa,
pedunculated, 1.4 and 1.0 cm in size, with serial sections; further
standard histological examination of the biopsy specimen and
embedding in paraffin were performed. The section was cut was
carried using a microtome, with a section thickness of 4 µm.
Standard hematoxylin and eosin staining was performed. The
magnification of the microscope cannot be reliably determined,
since a series of images were taken with a magnification of x10 and
subsequent stitching of the microphotographs. (D) Dysplasia in
inflammatory bowel disease is characterized by a peculiar
arrangement of dysplastic epithelial cells occupying the entire
crypt. The tissue was obtained from a male patient, 66 years of
age, with an established diagnosis of inflammatory bowel disease,
namely ulcerative colitis. A colon biopsy was performed on November
30, 2022. A biopsy of the colon mucosa, 0.5 mm in size, with
further standard histological examination of the biopsy and
embedding in paraffin were performed. Cutting was then carried out
using a microtome, with a section thickness of 4 µm. Standard
hematoxylin and eosin staining was performed. The image is
presented as x10 magnification with stitching of the
microphotographs. |
| Table IIIPrecancerous lesions and subtypes of
the colon. |
Table III
Precancerous lesions and subtypes of
the colon.
| Name of
lesions | Types | (Refs.) |
|---|
| Polypoid lesion
with serrated histoarchitecture | HP/hyperplastic
polyp | (18) |
| | MVHP/microvesicular
hyperplastic polyp | |
| |
GCHP/goblet-cell-rich hyperplastic
polyp | |
| | SSL/sessile
serrated lesion with dysplasia or not | |
| | TSA/traditional
serrated adenoma | |
| Tubulovillous
adenomas |
TA/tubular/conventional adenoma | |
| |
TVA/villous/tubulovillous adenoma | |
| Focal
intraepithelial neoplasia in chronic inflammatory bowel
disease | Inflammatory bowel
isease | |
The localization of proliferation zones in basal
sections of colonic crypt without dysplasia indicators is typical
for hyperplastic polyp, in contrast to which serrated adenoma is
characterized by the horizontal displacement of a proliferative
zone along its own muscularis mucosae, spread of serrated pattern
into the crypt base, dilatation of its basal parts, asymmetric
proliferation with dysplasia signs, formation of slit-like serrated
structures and ectopic crypts similar to intussusception (Fig. 1B) (18).
The morphology of tubular adenomas shows the
presence of glandular crypts, having mostly a typical architecture,
exhibiting elongation, and increase in their number. The epithelium
has enlarged hyperchromatic nuclei with varying degrees of
stratification, spindle formation and loss of polarity, as well as
a further overall decrease in the number of goblet cells. The
specific feature of tubulovillous adenoma is the presence of
finger-like projections, appearing as a result of crypt elongation
(Fig. 1C).
A separate subtype of precancerous colorectal
lesions is intraepithelial neoplasia in the case of IBD that
creates additional complications for the differential diagnosis of
serrated and conventional adenomas that are not IBD-related. The
primary pathomorphological variation between IBD dysplasia and
other precancerous lesions is considered to be the unique
localization of dysplastic cells, which typically occurs in the
upper crypts during sporadic adenomas and in the entire crypts
during colitis (18,19) (Fig.
1D). In reality, morphologists usually cannot assess the exact
localization of dysplastic cells due to the nature of tangential
section, the incorrect placement of the sample in paraffin-embedded
blocks, etc. One of the ways to solve this issue is using molecular
genetic diagnostics.
4. Molecular markers of CRC
Reduced DNA methylation and associated epigenetic
disorders of gene expression (20,21) play
a particularly critical pathophysiological role in the development
of CRC. The disruption of DNA methylation is caused by mutations in
DNA methyltransferase (DNMT) genes responsible for the
genome-wide methylation patterns (22); in particular, the methylation status
of the promoter regions of the methylguanine-DNA-methyltransferase
gene (23,24) he disruption of which is predominantly
associated with mutations in the KRAS and BRAF genes
(25), which are found in 22% of
hyperplastic polyps, 25% of sessile serrated adenomas, and 50% of
serrated adenocarcinomas (24).
The proteins, bone morphogenetic protein 3, N-myc
downstream-regulated gene 4, annexin A10 (25-27),
Runt-related transcription factor 3, suppressor of cytokine
signaling 1, neurogenin 1, calcium channel, voltage-dependent, T
type, alpha 1G subunit, insulin-like growth factor 2, p16, human
mutL homolog 1 and MSX2-interacting nuclear target protein
(28-30)
have been described as markers of impaired genomic methylation
status for CRC. The impairment of the DNA methylation status is
typical for right-sided location of CRC, female patients,
individuals of an advanced age (31), and high levels of genomic instability
(32-34).
Genomic instability is a natural result of mutation
accumulation and a driving force of the neoplastic process. A
central role in the development of this phenomenon belongs to
epigenetic failures in the expression of MMR genes, which function
as a system of correcting improperly paired nucleotides, deletions
or inclusions of incorrect bases, occurring during DNA replication.
The proliferation of cells with an unstable genome is accompanied
by the development of aneuploidy, which is observed in 65-85% of
sporadic colorectal tumors (35-38),
and is often accompanied by mutations in the adenomatous polyposis
coli (APC), tumor protein p53 (TP53), catenin beta-1
and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic
subunit alpha genes, in 20% of cases (39). APC gene mutation can initiate
both tubulovillous (40-42)
and serrated adenomas (43,44).
Among the first markers to exhibit an increase in
expression, are the non-coding RNAs, CCAT1 and HOTAIR, in the
plasma of patients with CRC; their expression was found to be
significantly higher in patients with CRC than in those of the
healthy controls. Other circulating IncRNAs that have been
described as potential biomarkers for CRC detection include
LOC285194, RP11-462C24.1 and Nbla12061, 91H, PVT-1 and MEG3, NEAT1
and NEAT2(45).
The serum levels of insulin-like growth factor
binding protein-2 are elevated in patients with colon cancer, which
is associated with neoplastic changes in the colon and
carcinoembryonic antigen concentrations (45), as well as the overactivation of
pyruvate kinase M2.
Dbf4-dependent kinase genes, which inhibit the
Wnt-signaling pathway, are epigenetically silenced in CRC cells due
to promoter hypermethylation. The activation of DKKS through small
interfering RNA promotes the growth and invasion of cancer cells
in vitro (45).
When precancerous lesions transform into CRC, a
number of molecular mechanisms are activated, resulting in an
increased level of β-catenin in pathologically altered cells. The
Wnt signaling pathway plays a crucial role in this process.
5. Role of β-catenin in the transformation
of precancerous lesions into CRC
β-catenin plays a crucial role in the Wnt signaling
pathway, which regulates the balance between the symmetric and
asymmetric division of both normal stem cells and cancer stem
cells. β-catenin binds to the multiprotein destruction complex,
which includes APC, Axin, casein kinase 1 and glycogen synthase
kinase (GSK)-3, without the presence of a Wnt ligand. The secondary
consequence is that GSK-3 phosphorylates β-catenin, leading to its
ultimate degradation in the proteasome.
Wnt-ligands uses the transmembrane receptors,
Frizzled and low-density lipoprotein receptor-related protein 4/5,
to affect the cells. This complex activates the cytoplasmic protein
Dishevelled, stops β-catenin degradation and inactivates GSK-3β,
leading to accumulation of β-catenin in the cytoplasm and creation
of conditions, allowing β-catenin to enter the nucleus, activate
T-cell factor/lymphoid enhancer-binding factor and triggers the
expression of C-MYC, cyclin-dependent kinase 4,
WNT1-inducible signaling pathway protein-1, peroxisome
proliferator-activated receptor γ (PPAR-γ) and other
genes.
As an alternative mechanism for increasing β-catenin
synthesis in precancerous lesions cells, both the direct activation
of the phosphatidylinositol-3-kinase (PI3K)/protein kinase B
(AKT)/mammalian target of rapamycin (mTOR) signaling pathway, which
is the predominant signaling pathway that inhibits apoptosis, and
the direct activation of intracellular AKT have been described.
PI3Ks are intracellular lipid kinases involved in
the regulation of cell proliferation, differentiation and survival,
that promote cell growth by inhibiting apoptosis in colorectal
cancer cells and influences the effectiveness of chemotherapeutic
agents (46).
AKT (AKT proto-oncogene), which plays
a key role in several types of cell death, as well as in the
destruction of extracellular signaling molecules, oxidation,
osmotic stress and ischemic shock, phosphorylates GSK-3Β and
increases the content of β-catenin in the cancer stem cells. In
turn, mTOR forms two multiprotein complexes: mTorc1 and mTorc2,
which are capable of antagonistically regulating each other's
activity, while the first reduces and the second increases the
content of β-catenin (Fig. 2).
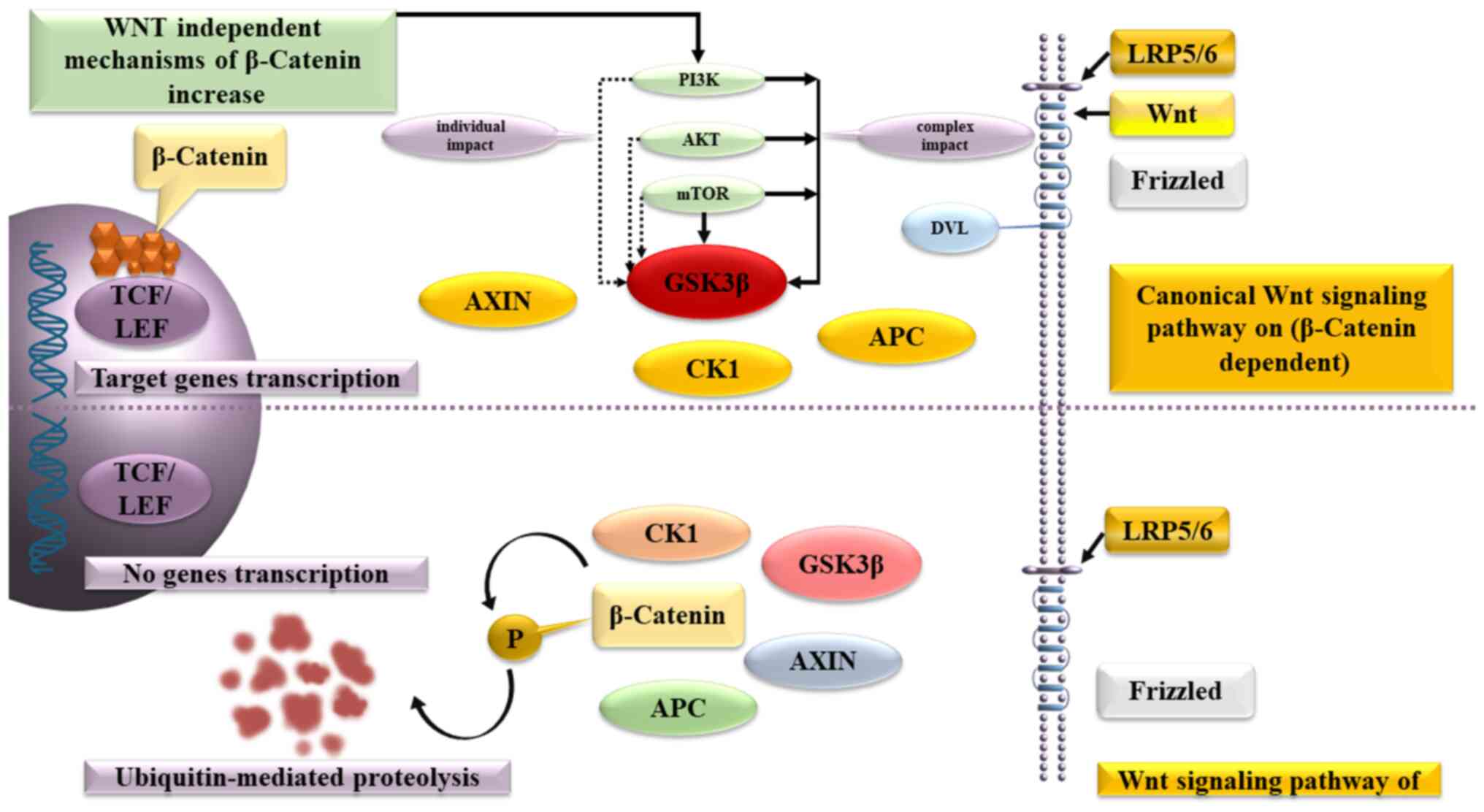 | Figure 2Pathogenesis of β-catenin
accumulation in the canonical and non-canonical WNT signaling
pathway, as well as alternative mechanisms of nuclear β-catenin
accumulation in the nucleus with a predominant effect on GSK-3β.
GSK-3, glycogen synthase kinase-3; TCF/LEF, T-cell factor/lymphoid
enhancer factor; AXIN 1/2, AXIS inhibition protein; CK1, casein
kinase 1; APС; PI3K, phosphatidylinositol-3-kinase; AKT, protein
kinase B; mTOR, mammalian target of rapamycin; DVL, Dishevelled;
LRP5/6, low-density lipoprotein receptor-related protein. |
A higher β-catenin level activates telomerases
(tert), causing their elongation, the immortalization of
pathologically altered cells and the increased production of
transforming growth factor β (TGFβ), that uses the SMAD and DAXX,
death-associated protein 6 signaling pathways to activate the
PI3K/AKT/mTOR axis. This results in the accumulation of β-catenin
(47) and the triggering of
epithelial-mesenchymal transition (EMT) in precancerous lesion
cells (48-50),
accompanied by activation of Snail, Twist, Slug, zinc finger E-box
binding homeobox (ZEB)1, ZEB2, lymphoid enhancer-binding factor 1
and other transcription factors.
The most critical part of EMT is the suppression of
E-cadherin synthesis, involved in the formation of tight junctions
between epitheliocytes, the increased expression of vimentin,
smooth muscle actin, fibronectin and genes, responsible for the
mesenchymal phenotype of epitheliocytes. The increased synthesis of
extracellular matrix metalloproteinases completes the
transformation of precancerous lesions into CRC and distinguishes
the cells, capable of invasion into surrounding tissues and
penetration into distant organs. In this respect, β-catenin
accumulation in the cells of precancerous lesions gives a strong
indication that transformation into CRC has been activated
(51); however, the existing methods
for diagnosing precancerous lesions and CRC, as aforementioned, do
not include a standardized immunohistochemical assessment of the
nuclear expression of β-catenin, despite the fact that the
literature data on chemoprevention of colorectal cancer focuses on
reducing the level of β-catenin.
6. Inflammatory bowel disease and CRC
Certain difficulties in the differential diagnosis
of precancerous lesions and CRC are associated with IBD, involving
mucous damage due to infection, allergic reactions or medication,
that adversely affects the ability of goblet cells to produce
mucin, functioning as a physical and chemical barrier between gut
microbiota and mucous membrane (51).
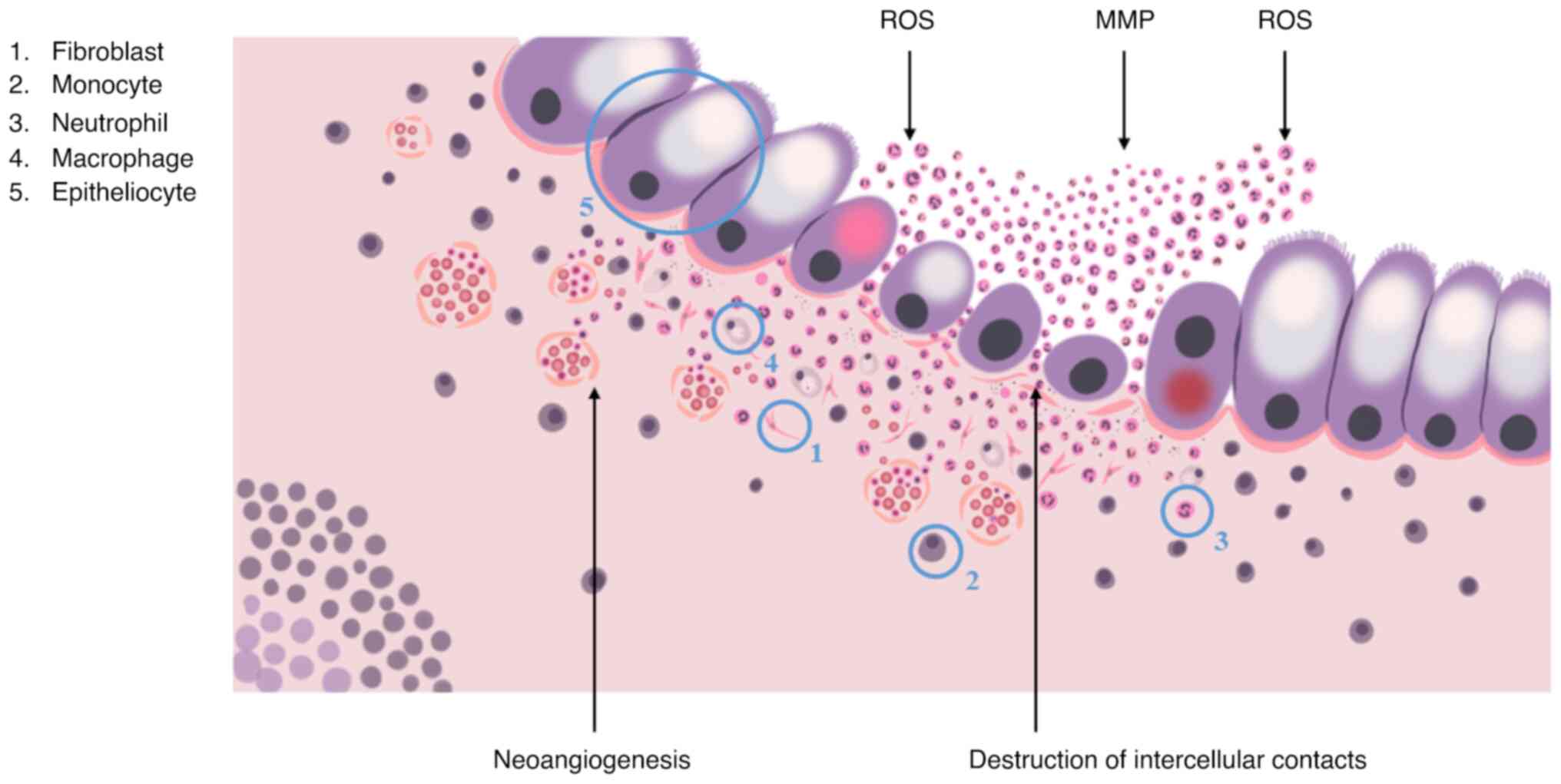
Neutrophils and monocytes are the first to react to
the disruption by migrating towards the damaged area with help of
chemotactic gradients, formed by interleukin (IL)-1β, IL-6, tumor
necrosis factor (TNF)-α cytokines, chemokine (C-C motif) ligand 8,
C-X-C motif chemokine ligand 10 chemokines, macrophage inflammatory
protein 2, granulocyte-macrophage colony-stimulating factor
(GM-CSF), and granulocyte colony-stimulating factor (52).
Neutrophils provide the elimination of pathogens via
phagocytosis, the formation of reactive oxygen species and the
release of matrix metalloproteinases, elastase and neutrophil
extracellular traps. It is generally accepted that once neutrophils
complete their task, they should immediately undergo apoptosis,
reducing inflammation, promoting the restoration of surrounding
tissues and the return to normal tissue homeostasis. Normally, the
regulation of local neutrophil activity is performed by
immunosuppressive cytokines, in particular, IL-10 and TGF-β, that
are produced by M2-activated macrophages, a result of
transformation, undergone by monocytes that migrated to the
phlogogenic area. In the case of IBD, this does not take place due
to excessive phlogogenic stimulation (52).
The accumulation of activated neutrophils,
macrophages and dendritic cells promotes changes in the crypt
structure, the disruption of intercellular contacts and integrity
of the basal lamina, that leads to the formation of crypt abscesses
and is characterized by the increased synthesis of the
pro-inflammatory cytokines, TNF-α and IL-1β, and the secretion of
non-cytokine inflammatory molecules, attracting T-cells and
neutrophils to the area of inflammation (Fig. 3). In turn, M1-activated macrophages
in collaboration with intestinal fibroblasts induce fibrotic
process, which is a disproportionate synthesis and the accumulation
of extracellular matrix components, causing the disruption of
mucosal histological architecture, the formation of ulcers and
scars of the colonic wall, which significantly complicates
endoscopic and morphological diagnosis of CRC (52).
In light of the above, molecular markers indicating
the systemic nature of the pathological process are of particular
diagnostic value. There is no consensus on such markers, but it is
known that neutrophils, monocytes and, to a certain extent,
dendritic cells are formed in the bone marrow from a common
multipotent progenitor cell of the megakaryocytic lineage that is a
direct descendant of a hematopoietic stem cell. It is reasonable to
assume that genetic mutations in the stem cell or progenitor cell
may prevent immunocytes from performing their regulatory functions,
lead to their immortalization and cause disease progression.
A similar scenario was described (53) as one of the possible manifestations
of the phenomenon, known as the clonal hematopoiesis of
indeterminate potential (CHIP) that is frequently observed during
the normal process of ageing (54).
The phenomenon is based on the fact that in by the age of 70,
hematopoietic stem cells of bone marrow accumulate from 350,000 to
1,400,000 mutations, including DNMT3A, Tet methylcytosine
dioxygenase 2 (TET2), additional sex combs like-1, protein
phosphatase 1D, Janus kinase 2 (JAK2), splicing factor 3b
subunit 1, serine/arginine-rich splicing factor 2, TP53,
guanine nucleotide binding protein, alpha stimulating activity
polypeptide and guanine nucleotide-binding protein. If even one of
the mutations appears to be able to guarantee the dominance of
corresponding hematopoietic stem cell clone over other clones, a
state of clonal expansion appears, which, on the one hand,
significantly increases the risk of hematological cancer and, on
the other hand, promotes the appearance of mutant immunocyte
clones, responsible for a number of chronic inflammatory diseases
(54).
Mutations, typical for CHIP, are found in
hematopoietic stem cells, monocytes and granulocytes. Individuals
with the DNMT3A mutation have a high level of IL-6 in their
blood; mouse macrophages with TET2 mutation, when stimulated
by bacterial endotoxin, exhibit a significantly increased
production of IL-1β, -2, -6 and -8. The JAK2 mutation is
always accompanied by a much stronger activation of granulocytes
and the intensified production of IL-6 and -18, without an increase
in the C-reactive protein level. Based on this, it is possible to
make an accurate assessment of the local processes in the colon,
taking into consideration the existing molecular and genetic
damage, found in the stem cells and immunocytes (53).
7. Chemoprophylaxis
IBD is an unmodifiable risk factor for the
development of CRC (54). One of the
significant causes of CRC in IBD-associated individuals is chronic
inflammation, which is widely acknowledged to promote CRC. The
mechanisms responsible for this are not yet fully understood
(55). While the suppression of
inflammation could potentially lower the risk of IBD-related CRC,
there is limited evidence to suggest that anti-inflammatory agents,
which are commonly used to treat IBD, have chemopreventive effects
on patients with cancer (55).
Decreasing the level of β-catenin, as a key
component of the Wnt signaling pathway, plays a crucial role in the
prevention of precancerous lesions that are not associated with
chronic inflammation. Genes, proteins involved in the signaling
pathways of CRC and inhibitors are listed in Table IV (56-58).
| Table IVGenes, proteins and inhibitors. |
Table IV
Genes, proteins and inhibitors.
| Genes | Proteins | Pathways in colon
cancer | Inhibitors | Source/(Refs.) |
|---|
| KRAS | KRAS | ERK, PI3K/AKT/mTOR,
RAS, regulating pluripotency of stem cells, WNT signaling
pathways | Glecirasib | MedChemExpress |
| | | | Rineterkib | |
| | | | Sotorasib | |
| | | | Adagrasib | |
| BRAF | BRAF | ERK, PI3K/AKT/mTOR,
RAS, WNT signaling pathways | Uplarafenib | MedChemExpress |
| | | | Lifirafenib | |
| | | | Tinlorafenib | |
| | | | Anticancer agent
124 | |
| | | | Exarafenib | |
| | | | Avutometinib | |
| | | | Belvarafenib | |
| APC | APC/DP2.5 | WNT signaling
pathways | 4-APC
hydrobromide | MedChemExpress |
| | | | Eftilagimod
alfa | |
| | | | Drotrecogin alfa
activated | |
| | | | Danicopan | |
| TP53 | p53/TRP53 |
Mutation-inactivated TP53 to
transcription | Kevetrin
hydrochloride | MedChemExpress |
| | | | PRIMA-1 | |
| | | | Eprenetapopt | |
| | | | Rebemadlin | |
| CTNNB1 | Catenin beta-1/β-
catenin | PI3K/AKT/mTOR, WNT
signaling pathways | SSTC3 | MedChemExpress |
| | | | 5-aminosalicylic
acid | |
| | | | Aspirin | |
| | | | Nimesulide | |
| | | | Troglitazone | |
| | | | Bezafibrate | |
| PIK3CA | P110α | PI3K/AKT.mTOR, ERK,
TNF, PD-L1 expression and PD-1 checkpoint pathways in cancer | Alpelisib | MedChemExpress |
| | | | Vevorisertib | |
| | | | Taselisib | |
| | | | Vevorisertib | |
| | | |
Trihydrochloride | |
| C-MYC | bHLH | Wnt, JAK-STAT
signaling pathways | Idarubicin | MedChemExpress |
| | | | hydrochloride | |
| | | | Agrimol B | |
| | | | Mollugin | |
| CYCD | CDKs | Wnt, Hippo,
JAK-STAT signaling pathways | Alvocidib | MedChemExpress |
| | | | Seliciclib | |
| | | | Palbociclib | |
| | | | Ribociclib | |
| | | | Abemaciclib | |
| WISP1 | WISP1 | Wnt signaling
pathways | Cabazitaxel | MedChemExpress |
| | | | Docetaxel | |
| | | | Paclitaxel | |
| JAK2 | JAK2 | PD-L1 expression
and PD-1 checkpoint, JAK-STAT, PI3K/AKT/mTOR signaling
pathways | Tofacitinib | MedChemExpress |
| | | | Baricitinib | |
| | | | Ruxolitinib | |
| | | | Upadacitinib | |
| | | | Fedratinib | |
| | | | Abrocitinib | |
| GNB1 | GNB1 | PI3K/AKT/mTOR, RAS,
MAPK signaling pathways | BEZ235 | (56) |
Non-steroidal anti-inflammatory drugs
(NSAIDs)
Sulfasalazine and medication with 5-aminosalicylic
acid (5-ASA) are considered to be anti-inflammatory preventative
drugs. Treatment with sulfasalizine for at least 3 months is
considered to have a protective effect, despite the disease
activity (59).
A recent meta-analysis confirmed the chemopreventive
effects of 5-ASA drugs at a dose of 1.2 g/day in patients with IBD,
particularly those with ulcerative colitis, while success was only
achieved in prevention of CRC, but not dysplasia (60). Another study demonstrated that
mesalamine was associated with risk reduction at the same dosage
(61). 5-ASA has chemopreventive
effect, enabled via various pathways (62). Induction of S-phase by 5-ASA reduces
the frequency of DNA mutations (63,64).
Synthase activity and reactive oxygen species formation are also
reduced (65,66). The suppression of the EGF and c-Myc
pathways induces apoptosis (66,67). The
Wnt/β-catenin pathway is modulated and PPAR-γ is activated by
5-ASA, resulting in the inhibition of cell proliferation (68). Finally, 5-ASA inhibits the NF-κB
pathway and blocks both the cyclooxygenase (COX)-2-dependent and
COX-2 independent growth of cancer cells (69,70).
There is evidence to indicate that the inhibitory effect of
β-catenin is suppressed (71).
Aspirin is effective for the prevention and
treatment of CRC associated with IBD due to inhibition of COX
enzymes, since they are the most well-studied targets of aspirin
(72). The risk was 20% lower after
5 years of use and 30% lower after 10 years of use, while
increasing the dosage up to 100 mg/day decreased the risk by 10%,
and up to 325 mg/day, by 35% (73).
Aspirin also locally reduces the expression of β-catenin (74).
Presently, to the best of our knowledge, there is no
evidence of the protective effect of aspirin against IBD-induced
CRC, since patients with IBD exhibit complications due to the
induced damage to the colon mucous membrane, with the subsequent
development of ulcers, rendering the risk of its use more serious
than its potential benefits (75).
Other NSAIDs, such as sulindac, have been proven to
induce the apoptosis of colon cancer cells, indicating the class
effect of COX inhibitors in the treatment or prevention of CRC that
is not related to IBD. The inhibitor of СОХ-2, nimesulide, and the
PPARγ ligand, troglitazone, effectively suppress colon
carcinogenesis, with nimesulide having a more potent inhibitory
effect than troglitazone (76).
Chemoprophylactic medication for non-IBD-related CRC
also includes aminosalicylates due to their ability to inhibit COX,
lipoxygenases, platelet-activating factor, IL-1β and eliminate
reactive oxygen species (77).
According to scientific publications, the use of 400
mg Celecoxib daily reduces the incidence of adenoma relapses by
34%, lowers the risk of advanced adenomas development by 55% and
provides a 7-fold lower chance of developing fatal outcomes
(78).
Antiparasitic drugs
Bezafibrate also reduces the incidence of colorectal
adenocarcinoma, although to a lesser extent than nimesulide and
troglitazone. In colorectal adenocarcinoma, the use of nimesulide,
troglitazone and bezafibrate has been shown to suppress cell
proliferation activity, induce apoptosis, and decrease β-catenin,
COX-2, inducible nitric oxide synthase and nitrotyrosine
immunoreactivity (76). Niclosamide
is also able to reduce β-catenin expression by disrupting the
metastasis-associated in colon cancer 1-β-catenin-S100A4 axis
(79).
Probiotic bacteria
The use of probiotic bacteria is a novel method used
for the prevention of carcinogenic exposure. Conjugated linoleic
acid production in mouse models of CRC activates PPAR-γ, which
inhibits COX-2 and induces apoptosis (80).
Glucocorticosteroids (GCs)
GCs, which include endogenous substances, such as
cortisol, cortisone and corticosterone, are secreted by the
hypothalamic-pituitary-adrenal (HPA) axis, which is affected by
various factors, such as circadian rhythm, stress and inflammatory
stimuli. When IL-1, TNF and IL-6 activate the HPA axis, the
production of GCs and the secretion of GCs is stimulated (78).
Inflammation is indirectly regulated by the HPA
axis, which inhibits the activation, migration and proliferation of
immune cells of both the innate and adaptive immune systems,
mediated by the glucocorticoid receptor (GR) located in the
cytoplasm. The glucocorticoid receptor is associated with heat
shock proteins, immunophilins, kinases and phospholipases
(receptorosomes), which form a complex. Following spatial changes
and GC/GR interaction, GR dissociates from receptorosomes and
translocates to the nucleus (78).
An enzyme known as activated GR exerts a genomic
effect caused by a DNA-binding sequence that contains two zinc
finger motifs. Specific glucocorticoid response elements are at the
core of this sequence, which affect various genes, including
proinflammatory mediators and transcription factors (e.g.,
activator protein-1 and NF-κB). At the same time, GC/GR trigger an
increase in the expression of IL-1 receptor antagonists, Iκ-B and
lipocortin-1. These transcriptional effects are responsible for the
immunoregulatory and anti-inflammatory effects of GR.
Glucocorticoid-induced leucine zipper is a significant target of
GC/GR transcriptional activity, as it affects the mitogen-activated
protein kinase pathway and NF-κB transcriptional activity,
ultimately leading to the regulation of immune-mediated and
inflammatory responses (78).
Budesonide and other GCs are associated with the suppression of the
function of the HPA axis and endogenous cortisol levels. The
immunosuppressive effects of corticosteroids (budesonide,
hydrocortisone, prednisolone and methylprednisolone) are reduced by
decreasing the pathological production of IL-1, -2, -3, -4, -5, -6,
-8, -10 and -12, and TNF-α, interferon-γ and GM-CSF (81).
The reduced synthesis of anti-inflammatory cytokines
leads to remission in patients with active IBD. However, the
medications are not recommended for long-term use due to severe
side-effects (82), including peptic
ulcers, cataracts, hypertension, adrenal atrophy, amenorrhea, type
II diabetes, hyperglycemia, Cushing's syndrome, a high risk of
infection, osteoporosis and avascular necrosis. According to the
study by Lichtenstein et al (83), the use of steroids is associated with
both progression and a risk of mortality in patients with IBD, when
compared to immunomodulators and biological therapies. Between 1994
and 2008, Targownik et al (84) discovered that steroids only had an
effective efficacy of 50% in patients with IBD within the first 5
years of diagnosis, which increased to 62% in the initial 10 years,
despite progress in biotherapy.
Immunomodulators
Immunomodulators can be used to achieve lasting
remission and are commonly prescribed in patients who are not
responsive to aminosalicylates and corticosteroids, or as a
supplement to anti-TNF therapy for antibody prevention,
particularly with infliximab (85,86).
Biological therapy
Bioengineered antibodies target specific molecules
or proteins that cause inflammation or participate in inflammatory
process; among these, there is an adhesion molecule antagonist
(vedolizumab, natalizumab), a drug targeting IL-23/IL-12
(ustekinumab) (87). The effects of
ustekinumab treatment may be determined by the modulation of IL-23
expression and the levels of miR-29. For example, for miR-126 and
vedolizumab, the potential is the same; Harris et al
(88) explained how endogenous
miR-126 inhibits leukocyte adhesion through the regulation of
vascular cell adhesion molecule-1, an adhesion molecule expressed
by endothelial cells.
TNF-α inhibitors
During bowel inflammation, TNF is produced by
different immune cells, including macrophages, T-cells and
dendritic cells, in the intestine of patients with IBD (89), to induce neoangiogenesis (90). In addition, different immune cells of
mucous membrane are activated to produce pro-inflammatory cytokines
and stimulate the death of Paneth cells via necroptosis (91) or to induce the apoptosis of colon
epithelial cells (92). Therefore,
inhibiting TNF can suppress colon inflammation. Anti-TNF drugs
induce and sustain the healing of mucous membranes in cases of
moderate and severe IBD and, as a result, may have
chemoprophylactic advantages by reducing long-term chronic
inflammation (93). TNF-α has been
reported to promote inflammation and IBD-CRC, facilitating DNA
damage, stimulating angiogenesis and inducing COX-2 expression that
also induces angiogenesis, resulting in tumor growth. TNF-α
expression, as demonstrated using mouse models, is associated with
development of colon cancer, while the inhibition of TNF-α reduces
inflammation and tumor growth; the effect is particularly
especially visible in mice, having received the anti-TNF agents,
infliximab and etanercept (94,95).
Modern studies have not proven that TNF inhibitors prevent
dysplasia development (93).
Some studies have shown that tofacitinib inhibits
JAK-1, JAK-2 and JAK-3, thus blocking the signaling pathways of
cytokines, containing γ-chain, mostly IL-2, IL-4, IL-7, IL-9, IL-3,
IL-5 and IL-21. Notably, JAK inhibition has been found to be
effective in suppressing T-cells, natural killer cells and
modulating pro-inflammatory cytokines; this opens up the
possibility of simultaneously blocking the activity of several
pro-inflammatory cytokines (96).
Thiopurines
Thiopurines have been used to maintain remission in
patients with IBD in order to avoid a long-term use of steroids
(97). However, a connection has
been reported between the use of thiopurines and a higher risk of
developing lymphoproliferative malignant neoplasms in 5% of cases
(98). A previous meta-analysis
revealed an association between treatment with thiopurine and the
risk of CRC development in patients with IBD, particularly the ones
with ulcerative colitis (99). Some
data have been published, indicating the reduction of a high degree
dysplasia and CRC both in case-control studies, and cohort ones
(78). A more significant
chemoprophylactic effect was recorded in patients who are at a high
risk of developing CRC, having the disease for >8 years.
However, this study did not discover any protective effect in
patients with IBD or extensive colitis (99). Although thiopurines are known to
decrease the risk of developing CRC in patients with IBD, they may
have carcinogenic properties. As a result, the 2017 European
Crohn's and Colitis Organisation (ECCO) consensus did not recommend
chemoprophylaxis with thiopurines (100).
Statins
Specifically, statins target the
3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG-CoA
reductase) enzyme, which in turn reduces cholesterol production and
promotes the elimination of low-density lipoproteins. A number of
patients with hyperlipidemia use these drugs. For prophylaxis and
the treatment of CRC, the use of statins is also critical (101). Researchers have indicated that
statins can exert a chemoprophylactic effect on CRC by targeting
the inflammation-induced proliferation of colon cancer and
potentially affecting intracellular oxidative stress, apoptosis and
vascular endothelial growth factor inhibitors (102). However, studies on the impact that
statins have on CRC in patients with IBD are limited; therefore,
the chemoprophylactic use of statins for patients with IBD remains
a controversial topic (103-106).
Ursodeoxycholic acid
Patients with IBD and primary sclerosing cholangitis
(PSC) have a 5-9-fold higher risk of CRC than patients with IBD
(107,108). High level of bile acids in the
colon may produce carcinogenic effects, leading to the
proliferation of colonic epithelial cells, ultimately leading to
the development of dysplasia or CRC (109,110).
Researchers have suggested that ursodeoxycholic acid may reduce
colonic dysplasia in patients with ulcerative colitis and PSC
(110), although these data are
controversial (109).
Folic acid
Patients with IBD may suffer from foliate deficiency
due to inadequate nutrition, the competitive inhibition of
intestinal absorption of sulfasalazine and excessive luminal losses
(111,112). Chemoprophylaxis with folic acid is
feasible due to its low cost, good tolerability and safety. Further
studies are required however, to determine the chemopreventive
effects of folic acid.
8. Conclusion and future perspectives
Despite the advances of modern pharmacological
industry, the chemoprophylaxis of CRC for patients with
precancerous lesions remains rather ineffective. Precancerous
colorectal lesions are detected daily via endoscopic methods and
are confirmed pathomorphologically. Patients with IBD are also left
to constantly balance between the remission and exacerbation
stages. The key to success may be the local reduction of nuclear
β-catenin that is the main initiator of oncogenic transformation of
precancerous lesions into CRC. The immunohistochemical evaluation
of nuclear β-catenin hyperexpression in precancerous cells may aid
in the differential diagnosis of early-stage cancer and dysplasia,
and may improve the assessment of the treatment efficacy in IBD
itself and IBD-associated lesions.
Multiple data on the effectiveness of
chemoprevention remain conflicting; there are no reliable data on
the reduction of nuclear β-catenin expression in studies on most
pharmacological drugs, and in some cases, the supposed benefit is
overshadowed by existing side-effects, particularly as regards GCs
and thiopurines. There is also an issue with the cross-inhibition
of drugs. Therefore, further studies are warranted to investigate
the phenomenon of clonal hematopoiesis with uncertain potential,
which is probably a decisive factor in the progression of IBD, and
further studies of repurposed drugs are also required.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
NEO collected and examined the data from the
relevant literature for inclusion in the review, and wrote the
manuscript. ISB conceived and designed the study and contributed
data tools. KDK made critical revisions on the intellectual content
of the study. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
The Ethics Committee of the Far Eastern Federal
University (Russky Island, Russia) decided to approve the ongoing
research work ‘Prognostic significance of clinical, morphological
and molecular genetic changes in precancerous lesions of the
gastrointestinal tract’. The planned study meets the basic
bioethical requirements and complies with the legislative
requirements and regulatory documents listed above (protocol No. 4,
11/18/2022). Each of the patients signed consent to the processing
of personal data, the use of research results for scientific and
practical purposes, and the publication of research data in
scientific journals in compliance with the principles of
confidentiality.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li S, Keenan JI, Shaw IC and Frizelle FA:
Could microplastics be a driver for early onset colorectal cancer?
Cancers (Basel). 15(3323)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Matich EK, Laryea JA, Seely KA, Stahr S,
Su LJ and Hsu PC: Association between pesticide exposure and
colorectal cancer risk and incidence: A systematic review.
Ecotoxicol Environ Saf. 219(112327)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pritchett N, Spangler EC, Gray GM,
Livinski AA, Sampson JN, Dawsey SM and Jones RR: Exposure to
outdoor particulate matter air pollution and risk of
gastrointestinal cancers in adults: A Systematic review and
meta-analysis of epidemiologic evidence. Environ Health Perspect.
130(36001)2022.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Picetti R, Deeney M, Pastorino S, Miller
MR, Shah A, Leon DA, Dangour AD and Green R: Nitrate and nitrite
contamination in drinking water and cancer risk: A systematic
review with meta-analysis. Environ Res. 210(112988)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
González N, Marquès M, Nadal M and Domingo
JL: Meat consumption: Which are the current global risks? A review
of recent (2010-2020) evidences. Food Res Int.
137(109341)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
García-Pérez J, Fernández de Larrea-Baz N,
Lope V, Molina AJ, O'Callaghan-Gordo C, Alonso MH, Rodríguez-Suárez
MM, Mirón-Pozo B, Alguacil J, Gómez-Acebo I, et al: Residential
proximity to industrial pollution sources and colorectal cancer
risk: A multicase-control study (MCC-Spain). Environ Int.
144(106055)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lotfollahzadeh S, Recio-Boiles A and Cagir
B: Colon cancer. In: StatPearls [Internet].StatPearls Publishing,
Treasure Island, FL, 2023.
|
|
8
|
US Preventive Services Task Force.
Davidson KW, Barry MJ, Mangione CM, Cabana M, Caughey AB, Davis EM,
Donahue KE, Doubeni CA, Krist AH, et al: Screening for colorectal
cancer: US Preventive services task force recommendation statement.
JAMA. 325:1965–1977. 2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kahi CJ, Hewett DG, Norton DL, Eckert GJ
and Rex DK: Prevalence and variable detection of proximal colon
serrated polyps during screening colonoscopy. Clin Gastroenterol
Hepatol. 9:42–46. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Steele SR, Johnson EK, Champagne B, Davis
B, Lee S, Rivadeneira D, Ross H, Hayden DA and Maykel JA: Endoscopy
and polyps-diagnostic and therapeutic advances in management. World
J Gastroenterol. 19:4277–4288. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Quintero E, Castells A, Bujanda L,
Cubiella J, Salas D, Lanas Á, Andreu M, Carballo F, Morillas JD,
Hernández C, et al: Colonoscopy versus fecal immunochemical testing
in colorectal-cancer screening. N Engl J Med. 366:697–706.
2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: . Global Cancer Statistics
2020: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li P, Li S, Chen J, Shao L, Lu X and Cai
J: Association between family history and prognosis of patients
with colorectal cancer: A systematic review and meta-analysis.
Transl Cancer Res. 11:124–133. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wautier JL and Wautier MP: Old and new
blood markers in human colorectal cancer. Int J Mol Sci.
23(12968)2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Siegel RL, Fedewa SA, Anderson WF, Miller
KD, Ma J, Rosenberg PS and Jemal A: Colorectal cancer incidence
patterns in the United States, 1974-2013. J Natl Cancer Inst.
109(djw322)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Doubeni CA, Corley DA, Quinn VP, Jensen
CD, Zauber AG, Goodman M, Johnson JR, Mehta SJ, Becerra TA, Zhao
WK, et al: Effectiveness of screening colonoscopy in reducing the
risk of death from right and left colon cancer: A large
community-based study. Gut. 67:291–298. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Cappell MS: The pathophysiology, clinical
presentation, and diagnosis of colon cancer and adenomatous polyps.
Med Clin North Am. 89:1–42, vii. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nagtegaal ID, Odze RD, Klimstra D, Paradis
V, Rugge M, Schirmacher P, Washington KM, Carneiro F and Cree IA:
WHO Classification of Tumours Editorial Board. The 2019 WHO
classification of tumours of the digestive system. Histopathology.
76:182–188. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Shah SC and Itzkowitz SH: Colorectal
cancer in inflammatory bowel disease: Mechanisms and management.
Gastroenterology. 162:715–730.e3. 2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jung G, Hernández-Illán E, Moreira L,
Balaguer F and Goel A: Epigenetics of colorectal cancer: Biomarker
and therapeutic potential. Nature reviews. Nat Rev Gastroenterol
Hepatol. 17:111–130. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lao VV and Grady WM: Epigenetics and
colorectal cancer. Nat Rev Gastroenterol Hepatol. 8:686–700.
2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cervena K, Siskova A, Buchler T, Vodicka P
and Vymetalkova V: Methylation-Based therapies for colorectal
cancer. Cells. 9(1540)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jass JR, Baker K, Zlobec I, Higuchi T,
Barker M, Buchanan D and Young J: Advanced colorectal polyps with
the molecular and morphological features of serrated polyps and
adenomas: Concept of a ‘fusion’ pathway to colorectal cancer.
Histopathology. 49:121–131. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ogino S, Kawasaki T, Kirkner GJ, Suemoto
Y, Meyerhardt JA and Fuchs CS: Molecular correlates with MGMT
promoter methylation and silencing support CpG island methylator
phenotype-low (CIMP-low) in colorectal cancer. Gut. 56:1564–1571.
2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Weisenberger DJ, Levine AJ, Long TI,
Buchanan DD, Walters R, Clendenning M, Rosty C, Joshi AD, Stern MC,
LeMarchand L, et al: Association of the colorectal CpG island
methylator phenotype with molecular features, risk factors, and
family history. Cancer Epidemiol Biomarkers Prev. 24:512–519.
2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Imperiale TF, Ransohoff DF and Itzkowitz
SH: Multitarget stool DNA testing for colorectal-cancer screening.
N Engl J Med. 371:187–188. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kadiyska T and Nossikoff A: Stool DNA
methylation assays in colorectal cancer screening. World J
Gastroenterol. 21:10057–10061. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tahara T, Yamamoto E, Madireddi P, Suzuki
H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T,
et al: Colorectal carcinomas with CpG island methylator phenotype 1
frequently contain mutations in chromatin regulators.
Gastroenterology. 146:530–538.e5. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686.
1999.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Weisenberger DJ, Siegmund KD, Campan M,
Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D,
Buchanan D, et al: CpG island methylator phenotype underlies
sporadic microsatellite instability and is tightly associated with
BRAF mutation in colorectal cancer. Nat Genet. 38:787–793.
2006.PubMed/NCBI View
Article : Google Scholar
|
|
31
|
Snover DC: Update on the serrated pathway
to colorectal carcinoma. Hum Pathol. 42:1–10. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Nakanishi Y, Diaz-Meco MT and Moscat J:
Serrated colorectal cancer: The road less travelled? Trends Cancer.
5:742–754. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Missiaglia E, Jacobs B, D'Ario G, Di Narzo
AF, Soneson C, Budinska E, Popovici V, Vecchione L, Gerster S, Yan
P, et al: Distal and proximal colon cancers differ in terms of
molecular, pathological, and clinical features. Ann Oncol.
25:1995–2001. 2014.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tran B, Kopetz S, Tie J, Gibbs P, Jiang
ZQ, Lieu CH, Agarwal A, Maru DM, Sieber O and Desai J: Impact of
BRAF mutation and microsatellite instability on the pattern of
metastatic spread and prognosis in metastatic colorectal cancer.
Cancer. 117:4623–4632. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pino MS and Chung DC: The chromosomal
instability pathway in colon cancer. Gastroenterology.
138:2059–2072. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649.
1998.PubMed/NCBI View
Article : Google Scholar
|
|
37
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Grady WM and Carethers JM: Genomic and
epigenetic instability in colorectal cancer pathogenesis.
Gastroenterology. 135:1079–1099. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jass JR, Whitehall VL, Young J and Leggett
BA: Emerging concepts in colorectal neoplasia. Gastroenterology.
123:862–876. 2002.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Fodde R, Kuipers J, Rosenberg C, Smits R,
Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH and
Clevers H: Mutations in the APC tumour suppressor gene cause
chromosomal instability. Nat Cell Biol. 3:433–438. 2001.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sparks AB, Morin PJ, Vogelstein B and
Kinzler KW: Mutational analysis of the APC/beta-catenin/Tcf pathway
in colorectal cancer. Cancer Res. 58:1130–1134. 1998.PubMed/NCBI
|
|
42
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Futreal. Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Jass JR: Classification of colorectal
cancer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Rex DK, Ahnen DJ, Baron JA, Batts KP,
Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, et
al: Serrated lesions of the colorectum: Review and recommendations
from an expert panel. Am J Gastroenterol. 107:1315–1329; quiz 1314,
1330. 2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Zygulska AL and Pierzchalski P: Novel
diagnostic biomarkers in colorectal cancer. Int J Mol Sci.
23(852)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhong J, Ding S, Zhang X, Di W, Wang X,
Zhang H, Chen Y, Zhang Y and Hu Y: To investigate the occurrence
and development of colorectal cancer based on the PI3K/AKT/mTOR
signaling pathway. Front Biosci (Landmark Ed).
28(37)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Nusse R and Clevers H: Wnt/β-Catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Vermeulen L, De Sousa E, Melo F, van der
Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M,
Merz C, Rodermond H, et al: Wnt activity defines colon cancer stem
cells and is regulated by the microenvironment. Nat Cell Biol.
12:468–476. 2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Söreide K, Janssen EA, Söiland H, Körner H
and Baak JP: Microsatellite instability in colorectal cancer. Br J
Surg. 93:395–406. 2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz
DB, Lewis DW, Gollin SM, Gamblin TC, Geller DA and Lagasse E: A
stochastic model for cancer stem cell origin in metastatic colon
cancer. Cancer Res. 68:6932–6941. 2008.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Jaiswal S: Clonal hematopoiesis and
nonhematologic disorders. Blood. 136:1606–1614. 2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jaiswal S and Ebert BL: Clonal
hematopoiesis in human aging and disease. Science.
366(eaan4673)2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Haggar FA and Boushey RP: Colorectal
cancer epidemiology: Incidence, mortality, survival, and risk
factors. Clin Colon Rectal Surg. 22:191–197. 2009.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yoda A, Adelmant G, Tamburini J, Chapuy B,
Shindoh N, Yoda Y, Weigert O, Kopp N, Wu SC, Kim SS, et al:
Mutations in G protein β subunits promote transformation and kinase
inhibitor resistance. Nat Med. 21:71–75. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Kosianova А, Pak O and Bryukhovetskiy I:
Regulation of cancer stem cells and immunotherapy of glioblastoma
(Review). Biomed Rep. 20(24)2023.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Bryukhovetskiy I, Kosianova A, Zaitsev S,
Pak O, Sharma A and Sharma HS: Glioblastoma: What can we do for
these patients today and what will we be able to do in the future?
Prog Brain Res. 265:99–118. 2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Pinczowski D, Ekbom A, Baron J, Yuen J and
Adami HO: Risk factors for colorectal cancer in patients with
ulcerative colitis: A case-control study. Gastroenterology.
107:117–120. 1994.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Qiu X, Ma J, Wang K and Zhang H:
Chemopreventive effects of 5-aminosalicylic acid on inflammatory
bowel disease-associated colorectal cancer and dysplasia: A
systematic review with meta-analysis. Oncotarget. 8:1031–1045.
2017.PubMed/NCBI View Article : Google Scholar
|
|
61
|
OʼConnor A, Packey CD, Akbari M and Moss
AC: Mesalamine, but not sulfasalazine, reduces the risk of
colorectal neoplasia in patients with inflammatory bowel disease:
An agent-specific systematic review and meta-analysis. Inflamm
Bowel Dis. 21:2562–2569. 2015.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Lyakhovich A and Gasche C: Systematic
review: Molecular chemoprevention of colorectal malignancy by
mesalazine. Aliment Pharmacol Ther. 31:202–209. 2010.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Luciani MG, Campregher C, Fortune JM,
Kunkel TA and Gasche C: 5-ASA affects cell cycle progression in
colorectal cells by reversibly activating a replication checkpoint.
Gastroenterology. 132:221–235. 2007.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Gasche C, Goel A, Natarajan L and Boland
CR: Mesalazine improves replication fidelity in cultured colorectal
cells. Cancer Res. 65:3993–3997. 2005.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Nandi J, Saud B, Zinkievich JM, Palma DT
and Levine RA: 5-aminosalicylic acid improves indomethacin-induced
enteropathy by inhibiting iNOS transcription in rats. Dig Dis Sci.
53:123–132. 2008.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Monteleone G, Franchi L, Fina D, Caruso R,
Vavassori P, Monteleone I, Calabrese E, Naccari GC, Bellinvia S,
Testi R and Pallone F: Silencing of SH-PTP2 defines a crucial role
in the inactivation of epidermal growth factor receptor by
5-aminosalicylic acid in colon cancer cells. Cell Death Differ.
13:202–211. 2006.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Chu EC, Chai J, Ahluwalia A and Tarnawski
AS: Mesalazine downregulates c-Myc in human colon cancer cells. A
key to its chemopreventive action? Aliment Pharmacol Ther.
25:1443–1449. 2007.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Bos CL, Diks SH, Hardwick JC, Walburg KV,
Peppelenbosch MP and Richel DJ: Protein phosphatase 2A is required
for mesalazine-dependent inhibition of Wnt/beta-catenin pathway
activity. Carcinogenesis. 27:2371–2382. 2006.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Stolfi C, Pellegrini R, Franze E, Pallone
F and Monteleone G: Molecular basis of the potential of mesalazine
to prevent colorectal cancer. World J Gastroenterol. 14:4434–4439.
2008.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Song M, Xia B and Li J: Effects of topical
treatment of sodium butyrate and 5-aminosalicylic acid on
expression of trefoil factor 3, interleukin 1beta, and nuclear
factor kappaB in trinitrobenzene sulphonic acid induced colitis in
rats. Postgrad Med J. 82:130–135. 2006.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Bersuder E, Terciolo C, Lechevrel M,
Martin E, Quesnelle C, Freund JN, Reimund JM and Gross I:
Mesalazine initiates an anti-oncogenic β-catenin/MUCDHL negative
feed-back loop in colon cancer cells by cell-specific mechanisms.
Biomed Pharmacother. 146(112543)2022.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Hall DCN and Benndorf RA: Aspirin
sensitivity of PIK3CA-mutated colorectal cancer: Potential
mechanisms revisited. Cell Mol Life Sci. 79(393)2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Bosetti C, Santucci C, Gallus S,
Martinetti M and La Vecchia C: Aspirin and the risk of colorectal
and other digestive tract cancers: An updated meta-analysis through
2019. Ann Oncol. 31:558–568. 2020.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Ma J, Fan Z, Tang Q, Xia H, Zhang T and Bi
F: Aspirin attenuates YAP and β-catenin expression by promoting
β-TrCP to overcome docetaxel and vinorelbine resistance in
triple-negative breast cancer. Cell Death Dis.
11(530)2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Newman P and Muscat J: Potential role of
non-steroidal anti-inflammatory drugs in colorectal cancer
chemoprevention for inflammatory bowel disease: An umbrella review.
Cancers (Basel). 15(1102)2023.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Kohno H, Suzuki R, Sugie S and Tanaka T:
Suppression of colitis-related mouse colon carcinogenesis by a
COX-2 inhibitor and PPAR ligands. BMC Cancer. 5(46)2005.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Allgayer H and Kruis W: Aminosalicylates:
Potential antineoplastic actions in colon cancer prevention. Scand
J Gastroenterol. 37:125–131. 2002.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Hsiao SW, Yen HH and Chen YY:
Chemoprevention of colitis-associated dysplasia or cancer in
inflammatory bowel disease. Gut Liver. 16:840–848. 2022.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Kortüm B, Radhakrishnan H, Zincke F,
Sachse C, Burock S, Keilholz U, Dahlmann M, Walther W, Dittmar G,
Kobelt D and Stein U: Combinatorial treatment with statins and
niclosamide prevents CRC dissemination by unhinging the
MACC1-β-catenin-S100A4 axis of metastasis. Oncogene. 41:4446–4458.
2022.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Bassaganya-Riera J, Viladomiu M, Pedragosa
M, De Simone C and Hontecillas R: Immunoregulatory mechanisms
underlying prevention of colitis-associated colorectal cancer by
probiotic bacteria. PLoS One. 7(e34676)2012.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Strehl C, Ehlers L, Gaber T and Buttgereit
F: Glucocorticoids-All-Rounders tackling the versatile players of
the immune system. Front Immunol. 10(1744)2019.PubMed/NCBI View Article : Google Scholar
|
|
82
|
An YK: Common mistakes with steroids. J
Gastroenterol Hepatol. 36 (Suppl 1):S30–S31. 2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Lichtenstein GR, Feagan BG, Cohen RD,
Salzberg BA, Diamond RH, Price S, Langholff W, Londhe A and
Sandborn WJ: Serious infection and mortality in patients with
Crohn's disease: more than 5 years of follow-up in the TREAT™
registry. Am J Gastroenterol. 107:1409–1422. 2012.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Targownik LE, Nugent Z, Singh H and
Bernstein CN: Prevalence of and outcomes associated with
corticosteroid prescription in inflammatory bowel disease. Inflamm
Bowel Dis. 20:622–630. 2014.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Melmed GY, Spiegel BM, Bressler B,
Cheifetz AS, Devlin SM, Harrell LE, Irving PM, Jones J, Kaplan GG,
Kozuch PL, et al: The appropriateness of concomitant
immunomodulators with anti-tumor necrosis factor agents for Crohn's
disease: One size does not fit all. Clin Gastroenterol Hepatol.
8:655–659. 2010.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Raine T and Kennedy NA: Immunomodulator
and biologic combination therapy in IBD: The debate that just won't
go away? J Crohns Colitis. 14:1343–1344. 2020.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Banerjee R, Ali RAR, Wei SC and Adsul S:
Biologics for the management of inflammatory bowel disease: A
review in tuberculosis-endemic countries. Gut Liver. 14:685–698.
2020.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Harris TA, Yamakuchi M, Ferlito M, Mendell
JT and Lowenstein CJ: MicroRNA-126 regulates endothelial expression
of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA.
105:1516–1521. 2008.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Neurath MF: Cytokines in inflammatory
bowel disease. Nat Rev Immunol. 14:329–342. 2014.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Rutella S, Fiorino G, Vetrano S, Correale
C, Spinelli A, Pagano N, Arena V, Maggiano N, Repici A, Malesci A
and Danese S: Infliximab therapy inhibits inflammation-induced
angiogenesis in the mucosa of patients with Crohn's disease. Am J
Gastroenterol. 106:762–770. 2011.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Günther C, Martini E, Wittkopf N, Amann K,
Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF
and Becker C: Caspase-8 regulates TNF-α-induced epithelial
necroptosis and terminal ileitis. Nature. 477:335–339.
2011.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Van den Brande JM, Koehler TC, Zelinkova
Z, Bennink RJ, te Velde AA, ten Cate FJ, van Deventer SJ,
Peppelenbosch MP and Hommes DW: Prediction of antitumour necrosis
factor clinical efficacy by real-time visualisation of apoptosis in
patients with Crohn's disease. Gut. 56:509–517. 2007.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Chapman CG and Rubin DT: The potential for
medical therapy to reduce the risk of colorectal cancer and
optimize surveillance in inflammatory bowel disease. Gastrointest
Endosc Clin N Am. 24:353–365. 2014.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Poutahidis T, Haigis KM, Rao VP, Nambiar
PR, Taylor CL, Ge Z, Watanabe K, Davidson A, Horwitz BH, Fox JG and
Erdman SE: Rapid reversal of interleukin-6-dependent epithelial
invasion in a mouse model of microbially induced colon carcinoma.
Carcinogenesis. 28:2614–2623. 2007.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Kim YJ, Hong KS, Chung JW, Kim JH and Hahm
KB: Prevention of colitis-associated carcinogenesis with
infliximab. Cancer Prev Res (Phila). 3:1314–1333. 2010.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Sandborn WJ, Ghosh S, Panes J, Vranic I,
Su C, Rousell S and Niezychowski W: Study A3921063 Investigators.
Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative
colitis. N Engl J Med. 367:616–624. 2012.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Beaugerie L, Svrcek M, Seksik P, Bouvier
AM, Simon T, Allez M, Brixi H, Gornet JM, Altwegg R, Beau P, et al:
Risk of colorectal high-grade dysplasia and cancer in a prospective
observational cohort of patients with inflammatory bowel disease.
Gastroenterology. 145:166–175.e8. 2013.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Beaugerie L, Brousse N, Bouvier AM,
Colombel JF, Lémann M, Cosnes J, Hébuterne X, Cortot A, Bouhnik Y,
Gendre JP, et al: Lymphoproliferative disorders in patients
receiving thiopurines for inflammatory bowel disease: A prospective
observational cohort study. Lancet. 374:1617–1625. 2009.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Zhu Z, Mei Z, Guo Y, Wang G, Wu T, Cui X,
Huang Z, Zhu Y, Wen D, Song J, et al: Reduced risk of inflammatory
bowel disease-associated colorectal neoplasia with use of
thiopurines: A systematic review and meta-analysis. J Crohns
Colitis. 12:546–558. 2018.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Magro F, Gionchetti P, Eliakim R,
Ardizzone S, Armuzzi A, Barreiro-de Acosta M, Burisch J, Gecse KB,
Hart AL, Hindryckx P, et al: Third European Evidence-based
consensus on diagnosis and management of ulcerative colitis. Part
1: Definitions, diagnosis, extra-intestinal manifestations,
pregnancy, cancer surveillance, surgery, and ileo-anal pouch
disorders. J Crohns Colitis. 11:649–670. 2017.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Dobrzycka M, Spychalski P, Łachiński AJ,
Kobiela P, Jędrusik P and Kobiela J: Statins and colorectal
cancer-A systematic review. Exp Clin Endocrinol Diabetes.
128:255–262. 2020.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Carrat F, Seksik P, Colombel JF,
Peyrin-Biroulet L and Beaugerie L: CESAME Study Group. The effects
of aminosalicylates or thiopurines on the risk of colorectal cancer
in inflammatory bowel disease. Aliment Pharmacol Ther. 45:533–541.
2017.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Qi XF, Kim DH, Yoon YS, Kim SK, Cai DQ,
Teng YC, Shim KY and Lee KJ: Involvement of oxidative stress in
simvastatin-induced apoptosis of murine CT26 colon carcinoma cells.
Toxicol Lett. 199:277–287. 2010.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Bergman M, Salman H, Djaldetti M and
Bessler H: Statins as modulators of colon cancer cells induced
cytokine secretion by human PBMC. Vascul Pharmacol. 54:88–92.
2011.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Ishikawa S, Hayashi H, Kinoshita K, Abe M,
Kuroki H, Tokunaga R, Tomiyasu S, Tanaka H, Sugita H, Arita T, et
al: Statins inhibit tumor progression via an enhancer of zeste
homolog 2-mediated epigenetic alteration in colorectal cancer. Int
J Cancer. 135:2528–2536. 2014.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Lim T, Lee I, Kim J and Kang WK:
Synergistic effect of simvastatin plus radiation in gastric cancer
and colorectal cancer: Implications of BIRC5 and connective tissue
growth factor. Int J Radiat Oncol Biol Phys. 93:316–325.
2015.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Singh S, Edakkanambeth Varayil J, Loftus
EV Jr and Talwalkar JA: Incidence of colorectal cancer after liver
transplantation for primary sclerosing cholangitis: A systematic
review and meta-analysis. Liver Transpl. 19:1361–1369.
2013.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Boonstra K, Weersma RK, van Erpecum KJ,
Rauws EA, Spanier BW, Poen AC, van Nieuwkerk KM, Drenth JP,
Witteman BJ, Tuynman HA, et al: Population-based epidemiology,
malignancy risk, and outcome of primary sclerosing cholangitis.
Hepatology. 58:2045–2055. 2013.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Singh S, Khanna S, Pardi DS, Loftus EV Jr
and Talwalkar JA: Effect of ursodeoxycholic acid use on the risk of
colorectal neoplasia in patients with primary sclerosing
cholangitis and inflammatory bowel disease: A systematic review and
meta-analysis. Inflamm Bowel Dis. 19:1631–1638. 2013.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Nagengast FM, Grubben MJ and van Munster
IP: Role of bile acids in colorectal carcinogenesis. Eur J Cancer.
31A:1067–1070. 1995.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Potack J and Itzkowitz SH: Colorectal
cancer in inflammatory bowel disease. Gut Liver. 2:61–73.
2008.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Swinson CM, Perry J, Lumb M and Levi AJ:
Role of sulphasalazine in the aetiology of folate deficiency in
ulcerative colitis. Gut. 22:456–461. 1981.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Ditonno I, Novielli D, Celiberto F, Rizzi
S, Rendina M, Ierardi E, Di Leo A and Losurdo G: Molecular pathways
of carcinogenesis in familial adenomatous polyposis. Int J Mol Sci.
24(5687)2023.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Mao R, Krautscheid P, Graham RP, Ganguly
A, Shankar S, Ferber M and Hegde M: ACMG Laboratory Quality
Assurance Committee. Genetic testing for inherited colorectal
cancer and polyposis, 2021 revision: A technical standard of the
American College of Medical Genetics and Genomics (ACMG). Genet
Med. 23:1807–1817. 2021.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Bhattacharya P and McHugh TW: Lynch
Syndrome. In: StatPearls [Internet]. StatPearls Publishing,
Treasure Island, FL, 2023.
|
|
116
|
Cohen PR and Kurzrock R: Germline testing
of mismatch repair genes is needed in the initial evaluation of
patients with muir-torre syndrome-associated cutaneous sebaceous
neoplasms: A case series. Cureus. 15(e33975)2023.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Park SY, Kim KS and Min HJ: Peutz-Jeghers
syndrome and sinonasal diseases: A case report and literature
review. Ear Nose Throat J: Sep 19, 2022 (Epub ahead of print).
|
|
118
|
Garofola C, Jamal Z and Gross GP: Cowden
Disease. In: StatPearls [Internet]. StatPearls Publishing, Treasure
Island, FL, 2023.
|
|
119
|
Dal Buono A, Gaiani F, Poliani L and Laghi
L: Juvenile polyposis syndrome: An overview. Best Pract Res Clin
Gastroenterol. 58-59(101799)2022.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Soares de Lima Y, Arnau-Collell C, Muñoz
J, Herrera-Pariente C, Moreira L, Ocaña T, Díaz-Gay M,
Franch-Expósito S, Cuatrecasas M, Carballal S, et al: Germline
mutations in WNK2 could be associated with serrated polyposis
syndrome. J Med Genet. 60:557–567. 2023.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Ahmed M: Colon cancer: A clinician's
perspective in 2019. Gastroenterology Res. 13:1–10. 2020.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Burnett-Hartman AN, Lee JK, Demb J and
Gupta S: An update on the epidemiology, molecular characterization,
diagnosis, and screening strategies for early-onset colorectal
cancer. Gastroenterology. 160:1041–1049. 2021.PubMed/NCBI View Article : Google Scholar
|