Introduction
Epithelial to mesenchymal transition (EMT) is a
process by which cells undergo a morphological switch from the
epithelial polarized phenotype to the mesenchymal fibroblastoid
phenotype, induced by transforming growth factor-β (TGF-β). EMT is
characterized by loss of epithelial differentiation markers, such
as E-cadherin, and the induction of mesenchymal markers, such as
vimentin and fibronectin. EMT was shown to play a key role in
various processes during embryonic development, chronic
inflammation and fibrosis (1,2).
Moreover, EMT was noted during tumor cell invasion and metastasis
in various solid tumors, such as lung cancer (3–5).
Mesenchymal cells arising from EMT contribute to the process of
tumor cell invasion. Taken together, these findings suggest that
EMT is a crucial event for cancer cells to acquire invasive and
metastatic phenotypes.
Lung cancer is the leading cause of cancer-related
death in Japan and worldwide. Patient prognosis remains poor
despite recent improvements in chemotherapies and
molecular-targeted therapies. The identification of sensitive and
specific biomarkers that predict metastasis, prognosis and drug
sensitivity may have a clinically significant effect on lung cancer
treatment strategies (6–9). Studies have described the involvement
of EMT in various tumors (3–5). A
number of studies reported that transforming growth factor-β1
(TGF-β1)-induced EMT is correlated with carcinogenesis, metastasis
and resistance to chemotherapy in lung cancer (5,10–16).
These findings demonstrated that suppression of EMT may be used as
a target for the chemoprevention and treatment of lung cancer. The
Smad signal pathway is well known as a major transducer of TGF-β
signaling. However, the potential signaling pathway during EMT of
lung cancer requires further investigation.
This study analyzed the protein expression profiles
of lung cancer cells to clarify the signaling mechanisms of EMT
using two-dimensional difference gel electrophoresis (2D-DIGE) and
mass spectrometry. Heat shock protein 27 (HSP27) was identified as
a molecule that increased during TGF-β1-induced EMT in lung cancer
cells in a Smad-independent manner. Furthermore, the suppression of
HSP27 using specific small interfering RNA (siRNA) accelerated
TGF-β1-induced EMT in A549 cells, a lung adenocarcinoma cell
line.
Materials and methods
Cell culture
This study used well-characterized lung
adenocarcinoma cell lines (17,18).
A549 was purchased from the American Type Culture Collection;
RERF-LC-KJ and LC2-ad were obtained from the Riken Cell Bank
(Ibaraki, Japan); and PC9 was obtained from Immuno-Biological
Laboratories (Gunma, Japan). Lung cancer cell lines were maintained
in RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum.
2D-DIGE and mass spectrometry
On reaching 80–90% confluence, the cells were washed
twice with phosphate-buffered saline, scraped off into a 1.5-ml
tube and briefly centrifuged. The cell pellets were incubated for
30 min in lysis buffer containing 6 M urea, 2 M thiourea, 1% Triton
X-100 and 3%
[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate. Following
centrifugation at 15,000 rpm for 30 min, the cell proteins were
recovered from the supernatant and the protein concentration was
measured using a Protein Assay Kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The proteins were labeled with fluorescent dyes
developed for the 2D-DIGE system, as previously described (GE
Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) (19,20).
The gels were scanned at appropriate wavelengths for Cy3 and Cy5
dyes with Typhoon (GE Healthcare Bio-Sciences Corp.). The spots
were detected and quantified using DeCyder software (GE Healthcare
Bio-Sciences Corp.). Protein identification was performed by mass
spectrometry. Mass spectrometric analysis of tryptic digests was
performed using Magic 2000 (GE Healthcare Bio-Sciences Corp.) and
peptide mass mapping was performed using Mascot search.
Antibodies and Western blot analysis
The cells were lysed in buffer (pH 7.6) containing
50 mM Tris-HCl, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1%
Nonidet P-40 and 0.5% sodium-deoxycholate. The lysates were
maintained on ice for 30 min and then centrifuged at 13,000 × g for
30 min. The supernatant was collected, and 10 μg of protein were
separated by gel electrophoresis on 12% gels and transferred to
nitrocellulose membranes by immunoblotting using a
chemiluminescence system (GE Healthcare Bio-Sciences Corp.). The
antibodies for detecting HSP27, Smad2, Smad3, phospho-Smad2
(p-Smad2), phospho-Smad3 (p-Smad3) and β-actin were purchased from
Cell Signaling Technology (Beverley, MA, USA).
Small interfering RNA (siRNA)
transfection
Transfections were performed at ~50% cell
confluency. Briefly, 1.0×105 A549 cells per well (6
wells) were seeded in OPTI-MEM I (Gibco 3198) without supplementary
antibiotics. To prepare siRNA, 4 μl of Lipofectamine™ 2000
(Invitrogen, Carlsbad, CA, USA), which is a transfection reagent of
siRNA, were mixed with 196 μl of OPTI-MEM I (Solution I). In
addition, 100 pmol siRNA solution was diluted with 200 μl of
OPTI-MEM I and incubated for 5 min at room temperature (Solution
II). Solutions I and II were then mixed and incubated for 20 min at
room temperature. siRNA and this reagent complex were added to the
A549 cells in OPTI-MEM I (2 ml/well). The final concentration of
siRNA was 50 nM. The A549 cells were incubated for 48 h after
transfection. siGENOME SMART pool Human SMAD2 no. M003561
(Dharmacon Inc., Lafayette, CO, USA) was used as siSmad2, siGENOME
SMART pool Human SMAD3 no. M020067 (Dharmacon) as siSmad3 and
siGENOME SMART pool Human HSPB1 no. M005269 (Dharmacon) as siHSP27,
with siGENOME SMART pool non-targeting siRNA no. D001206
(Dharmacon) as the control.
Results
Identification of the proteins whose
expression was affected by EMT of A549 cells
TGF-β1 is known to stimulate the EMT of A549 lung
cancer cells (1–4). In this study, A549 cells treated with
5 ng/ml of TGF-β1 for 72 h were designated as A549/TGF-β1. We
observed that while the parent A549 cells exhibited a classic
epithelial morphology, A549/TGF-β1, by contrast, appeared less
uniformly epithelial (Fig. 1A).
2D-DIGE was used to compare the protein expression
profiles of A549 cells to those of A549/TGF-β1, in order to
identify the protein populations whose expression was associated
with EMT in lung cancer. Fig. 1B
shows a representative Cy5 image unique to A549/TGF-β cells. More
than 2,000 spots were noted using 2D-DIGE. Computer-assisted
quantitative analysis identified 32 protein spots with increased
intensity and 21 protein spots with decreased intensity when
compared to A549 cells (p<0.05) (Fig. 1B). Mass spectrometry successfully
identified 53 proteins. Of these, 14 had a >2.0-fold change in
the level of expression in A549/TGF-β1 cells compared to A549 cells
(Table I). The most significant
increase in expression levels in A549/TGF-β1 cells compared to A549
cells was observed with HSP27 protein, which increased by 9.2-fold.
HSP27 protein expression levels were also higher in RERF-LC-KJ and
LC2-ad cells, which also showed the EMT phenomenon, following
treatment of the respective parent cells with TGF-β1. By contrast,
HSP27 expression levels in PC9 cells were not affected by exposure
to TGF-β1 (Fig. 1C). PC9 cells did
not exhibit signs of EMT following stimulation with TGF-β1 (data
not shown).
 | Table IA list of the identified proteins, the
expression of which was affected by EMT of A549 cells. |
Table I
A list of the identified proteins, the
expression of which was affected by EMT of A549 cells.
| Spot no. | Fold change | NCBI ID no./accession
no.a | Protein
identifieda | MWb | Total scorec | No. of
hit-peptides |
|---|
| 1975 | 9.2 | gi|662841 | Heat shock protein
27 | 22427 | 336 | 17 |
| 1694 | 4.6 | gi|63252900 | Tropomyosin 1 α chain
isoform 4 | 32856 | 769 | 22 |
| 1662 | 3.0 | gi|48735337 | Prolyl 4-hydroxylase,
β polypeptide | 57480 | 555 | 18 |
| 129 | 2.9 | gi|4504763 | Integrin α-V isoform
1 precursor | 117062 | 143 | 5 |
| 321 | 2.6 | gi|2202753 | Programmed cell death
6 interacting protein isoform 1 | 96590 | 519 | 9 |
| 917 | 2.4 | gi|12804537 | WD repeat domain
77 | 37442 | 273 | 5 |
| 122 | 2.3 | gil303599 | Calpastatin | 76780 | 161 | 3 |
| 2199 | 2.3 | gi|5031635 | Cofilin 1
(non-muscle) | 18719 | 218 | 10 |
| 170 | 2.3 | gi|19743823 | Integrin β 1 isoform
1A precursor | 88357 | 301 | 11 |
| 1952 | 2.1 | gi|5138999 | NADH-ubiquinone
reductase | 30401 | 585 | 14 |
| 124 | 2.1 | gi|21361331 | Carbamoyl-phosphate
synthetase 1 isoform b precursor | 165975 | 472 | 9 |
| 1737 | −4.9 | gi|38051823 | Plasminogen | 93263 | 40 | 1 |
| 1177 | −2.6 | gi|1168056 | Ornithine
aminotransferase, OAT | 48847 | 508 | 12 |
| 1056 | −2.3 | gi|182439 | Fibrinogen γ
chain | 50077 | 385 | 12 |
To confirm the increased expression of HSP27 protein
observed by 2D-DIGE, Western blot analysis was performed using
HSP27 antibody. Western blot analysis showed that the expression of
HSP27 was increased in A549/TGF-β1 cells compared to A549 cells
(Fig. 1D). Therefore, HSP27 protein
was used in further functional studies of EMT.
Knockdown of HSP27 in A549 and
A549/TGF-β1 cells and the effect on EMT
To investigate the effect of HSP27 on TGF-β1-induced
EMT, HSP27 protein was knocked down in A549 cells. Control or
specific HSP27 siRNA was transfected into A549 cells for 24 h, and
the A549 and A549/TGF-β1 cells were examined for signs of
morphological change after 48 h. Notably, silencing of HSP27 in
A549/TGF-β1 cells enhanced spindle integration, resulting in an
additive effect with TGF-β1-induced EMT (Fig. 2A). The expression of EMT markers was
evaluated to confirm the occurrence of EMT by Western blot
analysis. The expression levels of EMT markers in A549 and
A549/TGF-β1 cells treated with HSP27 siRNA were examined.
A549/TGF-β1 cells treated with HSP27 siRNA exhibited a reduced
E-cadherin expression and increased N-cadherin expression when
compared to A549/TGF-β1 cells treated with control siRNA (Fig. 2B). These observations suggest that
the inhibition of HSP27 protein, which accelerates the EMT process,
is mediated by TGF-β1.
Correlation between HSP27 and the Smad
signaling pathway
The Smad signaling pathway contributes to
TGF-β1-induced EMT. Additionally, this signaling pathway modulated
TGF-β1-induced EMT in A549 cells. A549/TGF-β1 cells treated with
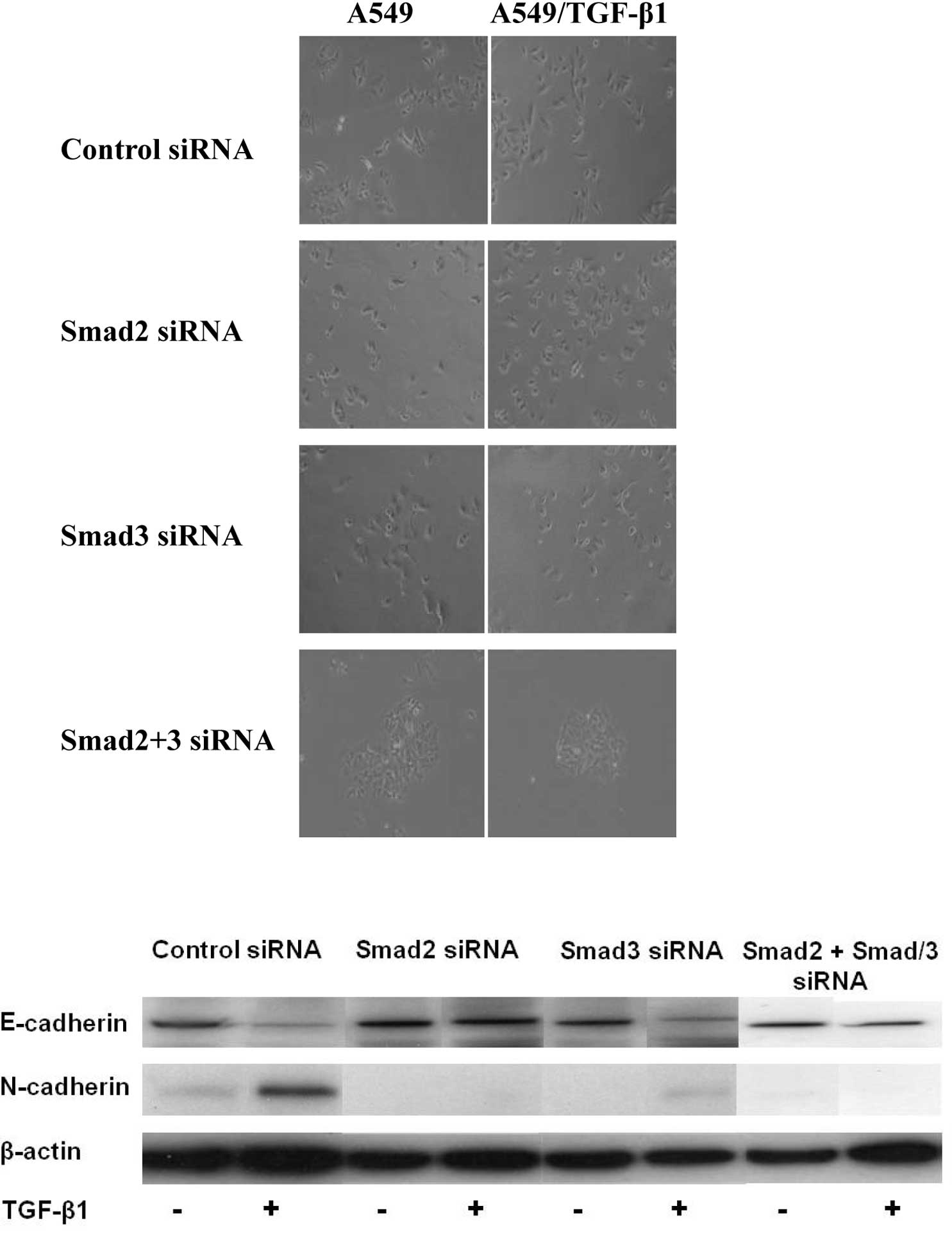
siRNAs of Smad2, Smad3 or Smad2/3 suppressed the TGF-β1-induced EMT
morphologically (Fig. 3A). The
expression levels of EMT markers were examined in A549 cells
following transfection of siRNA of Smad2 or Smad3. Cells depleted
of Smad2 or Smad3 still expressed E-cadherin and suppressed
N-cadherin expression, resulting in the suppression of
TGF-β1-induced EMT in A549/TGF-β1 cells (Fig. 3B). To further investigate the role
of HSP27 on TGF-β1-induced EMT, we evaluated the correlation
between HSP27 and the Smad signaling pathway. HSP27 protein levels
increased by exposure following the transfection of Smad2 plus
Smad3 siRNAs in A549/TGF-β1 cells (Fig.
3C). Furthermore, expression of Smad2, Smad3, p-Smad2 and
p-Smad3 was unchanged by the HSP27-specific siRNA transduction
(Fig. 3D). The results suggest that
HSP27 was up-regulated by TGF-β1 exposure in a Smad-independent
manner and contributed to EMT in lung cancer cells.
Discussion
This is the first study to report that HSP27 is
involved in TGF-β1-induced EMT in A549 lung cancer cells and is
independently activated by the Smad signaling pathway. A number of
studies have demonstrated that EMT change is involved in
carcinogenesis and drug resistance in lung cancer (5,10–15).
For example, a significant association was found between EMT and
K-Ras dependency in K-Ras-addicted lung cancer cells (5). In lung adenocarcinoma cells, thyroid
transcription factor, a master regulator of lung morphogenesis,
inhibited the process of EMT in response to TGF-β (10). As for drug sensitivity, the
induction of EMT contributed to the acquired resistance of
epidermal growth factor receptor tyrosine kinase inhibitor in lung
cancer (11–13). Thus, the inhibition of EMT is a
novel potential target for the chemoprevention and treatment of
lung cancer.
TGF-β1 is a significant mediator of EMT and is
involved in epithelial to mesenchymal interactions during lung
carcinogenesis. The Smad pathway is a major transducer of TGF-β
signaling (21). Smad2 and Smad3
are phosphorylated by the TGF-β type I receptor and form complexes
with Smad4. These complexes accumulate in the nucleus of the cell,
regulating the transcription of target genes and playing a crucial
role in the control of cell proliferation, differentiation,
apoptosis and cell migration. Apart from Smad-mediated
transcription, TGF-β type I and II receptors also allow
Smad-independent TGF-β responses. TGF-β receptors activate
alternative signaling effectors, such as mitogen-activated protein
kinase, phosphatidylinositol-3-kinase and Rho-like GTPases in
response to TGF-β (21). The
association between the direct activation of the Smads and other
signaling pathways often defines cellular responses to TGF-β.
However, the question remains as to which signaling pathway is
involved in EMT in lung cancer.
HSP27 is one of the protein chaperones that
transport and stabilize proteins within cells. HSP27 modulates
thermo-tolerance, regulation of cell development and
differentiation, as well as chaperone activity (22–24).
HSP27 can also inhibit apoptosis by binding to cytochrome c and
preventing its interaction with Apaf-1 and pro-caspase 9 (22). In addition, HSP27 has been shown to
interact with TGF-β (25). Numerous
human tumor cells express high levels of HSP27, suggesting that
HSP27 plays a role in carcinogenesis (26). In non-small cell lung cancer (NSCLC)
patients, high expression levels of HSP27 are thought to inversely
correlate with metastasis, poor prognosis and resistance to
chemotherapy (27–29). Therefore, HSP27 is a potential
diagnostic and prognostic marker in NSCLC patients. The present
study showed that HSP27 was significantly up-regulated by TGF-β1
stimulation in lung cancer cells. Furthermore, the inhibition of
HSP27 enhanced TGF-β1-induced EMT in a Smad-independent manner.
HSP27 may be involved in the TGF-β signaling pathway, although not
via an EMT negative feedback loop.
In conclusion, we found that HSP27 was up-regulated
by TGF-β1-induced EMT in a Smad-independent manner in lung cancer
cells. TGF-β1, which induces EMT in lung cancers, simultaneously
induced a molecule that negatively modulated EMT. Our data suggest
that HSP27 provides an effective clinical strategy in lung cancer
patients whose tumors are dependent on TGF-β1-induced EMT. Further
investigation of the association between HSP27 and TGF-β signaling
in lung cancer through the regulation of EMT is crucial.
Acknowledgements
We thank Dr Koichi Hagiwara for the helpful
discussion.
References
|
1
|
Grünert S, Jechlinger M and Beug H:
Diverse cellular and molecular mechanisms contribute to epithelial
plasticity and metastasis. Nat Rev Mol Cell Biol. 4:657–665.
2003.PubMed/NCBI
|
|
2
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh A, Greninger P, Rhodes D, Koopman L,
Violette S, Bardeesy N and Settleman J: A gene expression signature
associated with ‘K-Ras addiction’ reveals regulators of EMT and
tumor cell survival. Cancer Cell. 15:489–500. 2009.
|
|
6
|
Beer DG, Kardia SL, Huang CC, et al:
Gene-expression profiles predict survival of patients with lung
adenocarcinoma. Nat Med. 8:816–824. 2002.PubMed/NCBI
|
|
7
|
Potti A, Mukherjee S, Petersen R, Dressman
HK, Bild A, Koontz J, Kratzke R, Watson MA, Kelley M, Ginsburg GS,
West M, Harpole DH Jr and Nevins JR: A genomic strategy to refine
prognosis in early-stage non-small-cell lung cancer. New Eng J Med.
355:570–580. 2006. View Article : Google Scholar
|
|
8
|
Seike M, Yanaihara N, Bowman ED, Zanetti
KA, Budhu A, Kumamoto K, Mechanic LE, Matsumoto S, Yokota J,
Shibata T, Sugimura H, Gemma A, Kudoh S, Wang XW and Harris CC: Use
of a cytokine gene expression signature in lung adenocarcinoma and
the surrounding tissue as a prognostic classifier. J Natl Cancer
Inst. 99:1257–1269. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seike M, Goto A, Okano T, Bowman ED,
Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H, Gemma
A, Kudoh S, Croce CM and Harris CC: MiR-21 is an EGFR regulated
anti-apoptotic factor in lung cancer in never-smokers. Proc Natl
Acad Sci USA. 106:12085–12090. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saito RA, Watabe T, Horiguchi K, Kohyama
T, Saitoh M, Nagase T and Miyazono K: Thyroid transcription
factor-1 inhibits transforming growth factor-beta-mediated
epithelial-to-mesenchymal transition in lung adenocarcinoma cells.
Cancer Res. 69:2783–2791. 2009. View Article : Google Scholar
|
|
11
|
Thomson S, Buck E, Petti F, Griffin G,
Brown E, Ramnarine N, Iwata KK, Gibson N and Haley JD: Epithelial
to mesenchymal transition derived from repeated exposure to
gefitinib determines the sensitivity to EGFR inhibitors in A549, a
non-small cell lung cancer cell line. Cancer Res. 65:9455–9462.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yauch RL, Januario T, Eberhard DA, Cavet
G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S,
Modrusan Z, Lin CY, O’Neill V and Amler LC: Epithelial versus
mesenchymal phenotype determines in vitro sensitivity and predicts
clinical activity of erlotinib in lung cancer patients. Clin Cancer
Res. 11:8686–8698. 2005. View Article : Google Scholar
|
|
13
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soltermann A, Tischler V, Arbogast S,
Braun J, Probst-Hensch N, Weder W, Moch H and Kristiansen G:
Prognostic significance of epithelial-mesenchymal and
mesenchymal-epithelial transition protein expression in non-small
cell lung cancer. Clin Cancer Res. 14:7430–7437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bremnes RM, Veve R, Gabrielson E, Hirsch
FR, Baron A, Bemis L, Gemmill RM, Drabkin HA and Franklin WA:
Hirsch tissue microarray analysis used to evaluate biology and
prognostic significance of the E-cadherin pathway in non-small-cell
lung cancer. J Clin Oncol. 20:2417–2428. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deeb G, Wang J, Ramnath N, Slocum HK,
Wiseman S, Beck A and Tan D: Altered E-cadherin and epidermal
growth factor receptor expressions are associated with patient
survival in lung cancer: a study utilizing high-density tissue
microarray and immunohistochemistry. Mod Pathol. 17:430–439. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gemma A, Takenaka K, Hosoya Y, Matuda K,
Seike M, Kurimoto F, Ono Y, Uematsu K, Takeda Y, Hibino S,
Yoshimura A, Shibuya M and Kudoh S: Altered expression of several
genes in highly metastatic subpopulations of a human pulmonary
adenocarcinoma cell line. Eur J Cancer. 37:1554–1561. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gemma A, Li C, Sugiyama Y, Matsuda K,
Minegishi Y, Noro R, Nara M, Seike M, Yoshimura A, Ogawa N, Uesaka
H, Shionoya A, Kawakami A, Kosaihira S and Kudoh S: Anticancer drug
clustering in lung cancer based on gene expression profiles and
sensitivity database. BMC Cancer. 6:1742006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seike M, Kondo T, Mori Y, Gemma A, Kudoh
S, Sakamoto M, Yamada T and Hirohashi S: Proteomic analysis of
intestinal epithelial cells expressing stabilized beta catenin.
Cancer Res. 63:4641–4647. 2003.PubMed/NCBI
|
|
20
|
Okano T, Kondo T, Fujii K, Nishimura T,
Takano T, Ohe Y, Tsuta K, Matsuno Y, Gemma A, Kato H, Kudoh S and
Hirohashi S: Proteomic signature corresponding to the response to
gefitinib (Iressa, ZD1839), an epidermal growth factor receptor
tyrosine kinase inhibitor in lung adenocarcinoma. Clin Cancer Res.
13:799–805. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bruey JM, Ducasse C, Bonniaud P, Ravagnan
L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G,
Solary E and Garrido C: Hsp27 negatively regulates cell death by
interacting with cytochrome c. Nat Cell Biol. 2:645–652. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jia Y, Ransom RF, Shibanuma M, Liu C,
Welsh MJ and Smoyer WE: Identification and characterization of
hic-5/ARA55 as an hsp27 binding protein. J Biol Chem.
276:39911–39918. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhuang H, Jiang W, Cheng W, Qian K, Dong
W, Cao L, Huang Q, Li S, Dou F, Chiu JF, Fang XX, Lu M and Hua ZC:
Down-regulation of HSP27 sensitizes TRAIL-resistant tumor cell to
TRAIL-induced apoptosis. Lung Cancer. 68:27–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu L, Chen S and Bergan RC: MAPKAPK2 and
HSP27 are downstream effectors of p38 MAP kinase-mediated matrix
metalloproteinase type 2 activation and cell invasion in human
prostate cancer. Oncogene. 25:2987–2998. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jäättelä M: Escaping cell death: survival
proteins in cancer. Exp Cell Res. 248:30–43. 2009.PubMed/NCBI
|
|
27
|
Yao H, Zhang Z, Xiao Z, Chen Y, Li C,
Zhang P, Li M, Liu Y, Guan Y, Yu Y and Chen Z: Identification of
metastasis associated proteins in human lung squamous carcinoma
using two-dimensional difference gel electrophoresis and laser
capture microdissection. Lung Cancer. 65:41–48. 2009. View Article : Google Scholar
|
|
28
|
Malusecka E, Krzyzowska-Gruca S,
Gawrychowski J, Fiszer-Kierzkowska A, Kolosza Z and Krawczyk Z:
Stress proteins HSP27 and HSP70i predict survival in non-small cell
lung carcinoma. Anticancer Res. 28:501–506. 2008.PubMed/NCBI
|
|
29
|
Berrieman HK, Cawkwell L, O’Kane SL, Smith
L and Lind MJ: Hsp27 may allow prediction of the response to
single-agent vinorelbine chemotherapy in non-small cell lung
cancer. Oncol Rep. 15:283–286. 2006.PubMed/NCBI
|