Introduction
Telomeres are disposable DNA sequences that preserve
chromosomal integrity during mitosis. Human telomerase is a
ribonucleoprotein comprising human telomerase RNA (hTR) and related
proteins, which prevents telomere degradation, loss, rearrangement
or end-to-end fusion (1). One of
these related proteins, human telomerase reverse transcriptase
(hTERT), uses hTR as a template to continuously synthesize
telomeric DNA sequences at the ends of chromosomes. Although
telomerase activity in normal cells is only detected in cells with
proliferative potential, such as germ line and hematopoietic cells,
as well as activated lymphocytes (2), the majority of malignant carcinoma
cells exhibit telomerase activity. For example, telomerase activity
is detected in up to 80% of non-small cell lung carcinoma cells
(3,4), suggesting that the inhibition of
telomerase activity in tumor cells blocks telomeric repair, leading
to a gradual reduction in telomere length during each round of
replication and subsequent cell senescence and death. Therefore,
telomerase has received much attention in the investigation into
cancer treatment strategies. Studies (5,6) have
confirmed that telomerase inhibition by approaches including the
use of exogenous antisense oligonucleotides are capable of
inhibiting intracellular telomerase activity and therefore blocking
cell growth and inducing apoptosis.
In the present study, 95D giant-cell lung carcinoma
cells, which exhibit a high hTERT expression, were used for RNAi
experiments, wherein siRNAs specifically targeting hTERT mRNA were
transfected into cells and the effects of hTERT reduction on tumor
cell growth and proliferation were analyzed. This study provides
experimental evidence for the application of RNAi technology to the
treatment of lung cancer and also provides new data on the role of
telomerase in lung cancer.
Materials and methods
Materials
The following cells were used in this study: L78 and
NCI-H520 human squamous cell lung cancer cells, A549 and LTEP-α-2
human lung adenocarcinoma cells, NCI-H460 large-cell lung cancer
cells, 95D giant-cell lung carcinoma cells, 3T3 mouse embryonic
fibroblasts and 293T human embryonic kidney cells. Cells were
provided by and stored in the National Key Laboratory of
Respiratory Diseases, Guangzhou, China. The PCR primers for the
amplification of the hTERT and β-actin (internal control) genes
were designed using Primer 5 software, and the specificity was
confirmed by GenBank Blast analysis. The primers were commercially
synthesized (Invitrogen, Shanghai, China) and diluted to 10
μM using RNase-free water prior to use. The primer sequences
and lengths of the amplified fragments are shown in Table I.
 | Table IThe primer sequences and lengths of
the amplified hTERT and β-actin PCR fragments. |
Table I
The primer sequences and lengths of
the amplified hTERT and β-actin PCR fragments.
| Gene | | Primer sequences | Lengths (bp) |
|---|
| hTERT | Upstream primers |
5′-GCGTTTGGTGGATGATTTCT-3′ | 131bp |
| Downstream
primers |
5′-CAGGGCCTCGTCTTCTACAG-3′ | |
| β-actin | Upstream primers |
5′-TTCCTGGGCATGGAGTCCT-3′ | 187bp |
| Downstream
primers |
5′-TGATCTTCATTGTGCTGGGTG-3′ | |
Detection of hTERT mRNA levels
For each cell line, vials of cells were retrieved
from liquid nitrogen, placed in a 37°C water bath and agitated
frequently to rapidly thaw the cells. The cells were then
transferred to a centrifuge tube with 10 volumes of complete cell
culture medium and pelleted by low-speed centrifugation. The
supernatant was then removed and the cells were washed once with
culture medium. The cells were diluted in fresh culture medium,
seeded into flasks and incubated at 37°C. The following day, the
medium was changed and the cells were continually cultured.
To passage the cells, the culture medium was
aspirated from the flask, 1 ml trypsin was added and the flask was
gently swirled to cover all of the cells with trypsin. The cells
were incubated with trypsin for 2 to 5 min, during which time the
cells were observed by microscopy. Once the cytoplasms had shrunk
and the spaces between cells were enlarged, an equal volume of
culture medium with 10% fetal bovine serum was added immediately to
stop trypsinization. The cells were pipetted up and down repeatedly
until all of the cells were dislodged and suspended. The cells were
then seeded into new flasks and fresh medium containing serum was
added.
To freeze cells for storage, uncontaminated cells in
the exponential phase of growth were selected and fed with fresh
medium one day prior to freezing. The cells were then trypsinized
and collected as previously described and then pelleted by
centrifugation at 1000 rpm for 10 min. The supernatant was removed
gently and the cells were resuspended in cell freezing medium. The
suspension was transferred to 2-ml cryovials and gradually frozen
by first cooling at 4°C for 10 min, then freezing at −20°C for 2 h
and finally transferring the cells to −70°C for storage.
Extraction of total RNA from lung cancer
cells
The culture medium from healthy lung cancer cells
(1×106) was aspirated, and the cells were washed with
PBS and then directly lysed by adding 1 ml TRIzol reagent to the
culture flask. After pipetting several times, the cells were
incubated at room temperature for 5 min, transferred into 1-ml
microfuge tubes, inverted 10 times and held at room temperature for
2 min. For every 1 ml TRIzol, 0.2 ml chloroform was added to the
microfuge tube, and the cells were mixed by vortexing for 15 sec to
completely mix the solutions. The tube was held at room temperature
for 5 min, and the solution was separated into three layers by
centrifugation at 12,000 × g for 15 min at 4°C.
After centrifugation, the top aqueous phase
containing the RNA was transferred into a new 1.5-ml microfuge tube
and 0.5 ml isopropanol was added (a 0.5:1 ratio of
isopropanol:TRIzol). The tube was inverted 5 times to mix the
solutions and then held at room temperature for 10 min. The mixture
was then separated by centrifugation at 12,000 × g for 10 min at
4°C. The supernatant was carefully removed, leaving the pellet
intact. Pre-chilled 75% ethanol (1 ml; prepared by diluting in
RNase-free water) was added to each tube. The tube was gently
agitated and then separated by centrifugation at 7,500 × g for 5
min at 4°C. The supernatant was discarded, and as much of the
residual ethanol was removed as possible. The microfuge tube was
placed in a vacuum dryer for approximately 2 min, or until the RNA
pellet became transparent. Depending on the size of the RNA pellet,
20–40 μl RNase-free water was added to the tube to dissolve
the RNA pellet; an RNase inhibitor was also added at a ratio of
1:30. The purity of the isolated RNA was then assessed by
spectrometry; the optical density
(OD)260/OD280 of the samples was between 1.8
and 2.0, indicating little protein contamination and a high purity
that was ideal for our experiments. The integrity of the RNA was
also examined by 2% agarose gel electrophoresis.
The RNA concentration was calculated based on the
OD260 value using the formula: concentration of RNA =
OD260 value × dilution factor × 40 μg/ml. Based
on this calculation, total RNA was further diluted to 1000
ng/μl using RNase-free water for convenience during the
reverse transcription reaction, which was performed immediately.
The remaining RNA was stored at −80°C.
Reverse transcription
For each sample, 1 μg total RNA was used for
the reverse transcription reaction (reaction mixture are shown in
Table II) using a PCR thermocycler
(Biometra, T-personal 48, Germany) and the PrimeScript RT reagent
kit (Takara, Japan). The reverse transcription reaction was
performed at 37°C for 15 min, and the reaction was terminated by
heating to 85°C for 5 sec.
| Table IIReverse transcription reaction
mixtures. |
Table II
Reverse transcription reaction
mixtures.
| Reagents | Dose (μl) | Final
concentration |
|---|
| 5X PrimeScript™
Buffer | 4.0 | 1X |
| PrimeScript™ RT
Enzyme Mix I | 1.0 | |
| Oligo dT primer | 1.0 | 25pmol/l |
| Random 6 mers | 1.0 | 50pmol/l |
| RNase-free
dH2O | 12.0 | |
| Total RNA (1000
ng/μl) | 1.0 | 50
ng/μl |
Optimization of PCR conditions
Different annealing temperatures and PCR cycle
parameters were tested to optimize the PCR conditions, which were
critical for our experiments. The PCR conditions used for
optimization were: initial DNA denaturation at 95°C for 15 min and
then 40 cycles of denaturation at 94°C for 60 sec, annealing at
58°C for 60 sec and elongation at 72°C for 60 sec, followed by a
final elongation step at 72°C for 10 min. The annealing temperature
was the parameter that best reflected the specificity of the target
gene. During annealing temperature optimization, four annealing
temperatures, 55°C, 56°C, 57°C and 58°C, were used for the regular
PCR experiment based on the theoretical annealing temperature
calculated by the Primer 5 software (reaction mixture shown in
Table III). For optimization of
the cycle number for target gene amplification, PCR experiments
were performed with 20–40 cycles, differing by increments of 4. The
PCR products were analyzed by agarose gel electrophoresis to detect
the specific amplified products.
| Table IIIPCR reaction mixtures. |
Table III
PCR reaction mixtures.
| Reagents | Dose
(μl) | Final
concentration |
|---|
| Premix Ex Taq™
(2X) | 10.0 | 1X |
| Upstream primers
(10 μM) | 0.4 | 0.2
μmol/l |
| Downstream primers
(10 μM) | 0.4 | 0.2
μmol/l |
| cDNA solution | 1.0 | |
|
ddH2O | 8.2 | |
Analysis of regular PCR products
Each PCR product (2 μl) was separated by
electrophoresis using a 1.0% agarose gel containing ethidium
bromide (0.5 μg/ml) for 40 min at 100 V. The DL2000 DNA
marker (ShineGene, Shanghai, China) was used as the molecular
standard, and the gel was observed under UV light.
Detection of the relative levels of hTERT
mRNA
For each sample, 1 μl cDNA was used for
RT-PCR using the SYBR-Green I method (the reaction mixture is shown
in Table IV) to detect the
relative expression levels of hTERT mRNA. The reaction conditions
were optimized according to the steps described previously in this
section. The reaction conditions used to detect hTERT levels were:
initial DNA denaturation at 95°C for 15 min and then 40 cycles of
denaturation at 94°C for 15 sec, annealing at 58°C for 30 sec and
elongation at 72°C for 15 sec.
| Table IVQuantitative real-time RT-PCR
reaction mixtures. |
Table IV
Quantitative real-time RT-PCR
reaction mixtures.
| Reagents | Dose
(μl) | Final
concentration |
|---|
| Premix Ex Taq™
(2X) | 10.0 | 1X |
| Upstream primers
(10 μM) | 0.4 | 0.2
μmol/l |
| Downstream primers
(10 μM) | 0.4 | 0.2
μmol/l |
| cDNA solution | 1.0 | |
| RNase Free
dH2O | 8.2 | |
The Ct values for the hTERT and β-actin genes in
each sample were determined. After normalization to the internal
control gene, the data were analyzed by using the 2-ΔΔCt
method (5), in which ΔCt =
Ct(hTERT) - Ct(β-actin) and ΔΔCt = ΔCtsample group -
ΔCtcontrol group. The relative level of hTERT mRNA was
2-ΔΔCt.
Transfection of hTERT siRNAs and the
analysis of the effects of hTERT knockdown: design and synthesis of
siRNAs
Based on the hTERT gene sequence in Genbank
(NM_003219, NM_198253, NM_198254 and NM_198255) and following the
rules of siRNA design, three pairs of 21-bp siRNAs were engineered.
These siRNAs were then synthesized by Ambion (Austin, TX, USA):
siRNA-1 sense strand: 5′-GGA ACACCAAGAAGUUCAU-TT-3′ (1521–1539) and
anti-sense strand: 5′-AUGAACUUCUUGGUGUUCC-TT-3′; siRNA-2 sense
strand: 5′-CGCUCUUUUUCUACCGGAA-TT-3′ (1769–1787) and anti-sense
strand: 5′-UUCCGGUAGAAAAAGA GCC-TT-3′; siRNA-3 sense strand:
5′-GGUCUUUCUUUUA UGUCAC-TT-3′ (1728–1746) and anti-sense strand:
5′-GUGA CAUAAAAGAAAGACC-TT-3′.
The Basic Local Alignment Search Tool (http://www.ncbi.nlm.nih.gov/BLAST/) was used to
confirm that the sequences were not homologous to other genes. In
addition, non-specific negative control siRNAs were also designed
and synthesized. siRNAs were labeled with red fluorescent protein
for the visual detection of transfection efficiency.
Transfection of hTERT siRNAs into lung
cancer cells
siRNAs were delivered into the target cells using a
reverse transfection method. Different concentrations of siRNA were
transfected using the cationic liposome transfection reagent siPORT
NeoFX (Ambion) into the target cells as per the manufacturer's
instructions using 50 nmol/well siRNA in 12-well plates as follows:
lung cancer cells in the exponential phase of growth were
trypsinized and suspended in antibiotic-free OPTI-MEM (Invitrogen,
Carlsbad, CA, USA). The cell density was adjusted to
1×105/ml and stored in a culture incubator at 37°C with
5% CO2 until use.
In 47 μl OPTI-MEM, 3 μl siPORT NeoFX
was added and then incubated at room temperature for 10 min. In
47.5 μl OPTI-MEM, 2.5 μl 20 μM siRNA was
added, and the above two solutions were mixed to form transfection
complexes; held at room temperature for 20 min.
In each well of the 12-well plate, 900 μl
cell suspension was added, followed by 100 μl transfection
mixture. The plate was gently agitated and then incubated for 6 h,
after which the medium was replaced. After 48 h, red fluorescent
protein expression was observed in the cells using an inverted
fluorescent microscope to estimate the transfection efficacy.
Detection of hTERT mRNA levels
Total RNA was extracted from the transfected lung
cancer cells after 48 h, and the hTERT expression levels were
measured by real-time PCR using the 2-ΔΔCt method described
previously. The siRNA-mediated suppression rate of hTERT mRNA was
calculated as follows: suppression rate (%) = (1 – the relative
level of hTERT mRNA in the siRNA-transfected group ÷ the relative
level of hTERT mRNA in the negative control group) × 100% (6).
Analysis of cell apoptosis
The 95D cells were transfected with 100 nmol/l
siRNA, and the cells were harvested after 48 h using EDTA-free
trypsin. The cells were then washed twice with PBS and pelleted by
centrifugation (2000 rpm for 5 min per wash); 1–5×105
cells were harvested. Cells were resuspended in 500 μl
binding buffer, and 5 μl annexin V-EGFP was added, followed
by 5 μl propidium iodide (PI). The solutions were mixed well
and held in the dark at room temperature for 5 to 15 min. The cells
were then analyzed by flow cytometry; the green fluorescence of
annexin V-EGFP was detected using the FITC channel (FL1) and the PI
red fluorescence was detected through the PI channel (FL2 or
FL3).
The MTT assay was used to detect the inhibition of
cell proliferation by hTERT siRNA in lung cancer cells. Cells in
the exponential phase of growth were transfected in 96-well plates,
where each well contained 200 μl cell culture. At 12, 24, 48
and 72 h after transfection, 20 μl MTT (5 mg/ml) was added
to each well and the plate was incubated at 37°C with 5%
CO2 for 4 h. The culture medium was then removed and 150
μl DMSO was added in each well. The plate was agitated for
10 min and then read by a plate reader for the absorbance (A) at
570 nm; the experiment was repeated three times. The inhibition
rate of cell proliferation = (1 - the average A of the experimental
group ÷ the average A of the control group) × 100%.
Statistical analysis
SPSS12.0 software was used for the statistics
analysis. The experimental data are shown as the mean ± standard
deviation. Where the data followed a normal distribution with equal
variance, one-way analysis of variance (ANOVA) was used to compare
the means of multiple samples, and the LSD-t test was used for the
pair-wise comparison of the means of multiple samples. For the
comparison of the means of two independent samples, the paired
t-test was used. P<0.05 was considered to be statistically
significant.
Results
Observation of the growth of lung cancer
cells
By microscopic observation, lung cancer cells grew
well and adhered to the bottom of the culture flask. The cells
exhibited irregular polygonal spindle or oval shapes, the
cytoplasms were transparent and the nuclei indistinct. Cells
manifesting these morphological characteristics were collected for
total RNA extraction, RT-PCR and quantification.
Detection of hTERT levels in lung cancer
cells
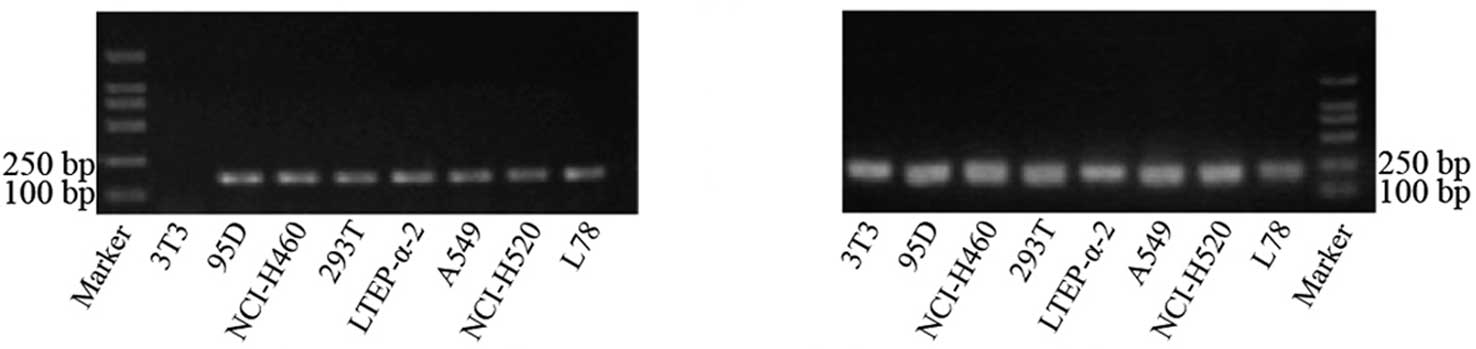
The annealing temperature for the amplification of
the hTERT and β-actin genes was 58°C, since our results showed that
non-specific amplification could be avoided at this temperature. In
determining the optimum number of cycles, the intensity of the
target gene band increased at an exponential rate after 30 to 40
amplification cycles, demonstrating that the amplification products
increased exponentially. Cycle numbers greater than 40 caused
enzyme saturation and a plateau effect; therefore, the optimized
PCR protocol used 40 cycles. The electrophoresis results of the PCR
products of hTERT and β-actin are shown in Fig. 1.
Detection of the relative levels of hTERT
mRNA in lung cancer cell lines
The levels of hTERT expression were detected in
multiple lung cancer cell lines, including L78 and NCI-H520 human
squamous cell lung cancer cells, A549 and LTEP-α-2 human lung
adenocarcinoma cells, NCI-H460 large-cell lung cancer cells and 95D
giant-cell lung carcinoma cells. The 3T3 mouse embryonic fibroblast
cell line does not express hTERT and was therefore used as the
negative control, and 293T human kidney cells, which are known to
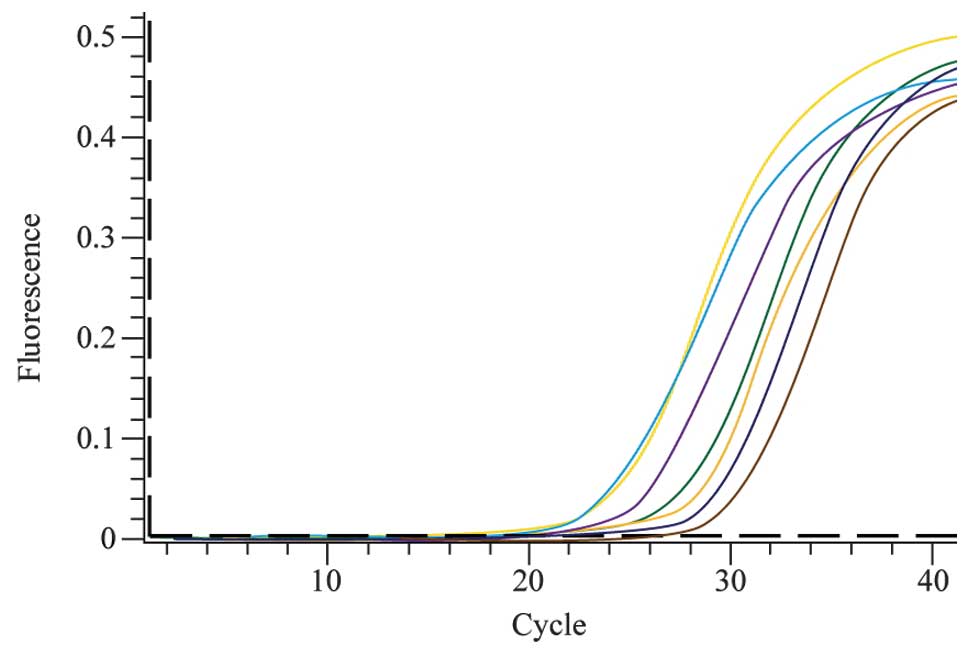
express hTERT, were used as the positive control. The real-time
quantitative PCR amplification curves of the target gene are shown
in Fig. 2.
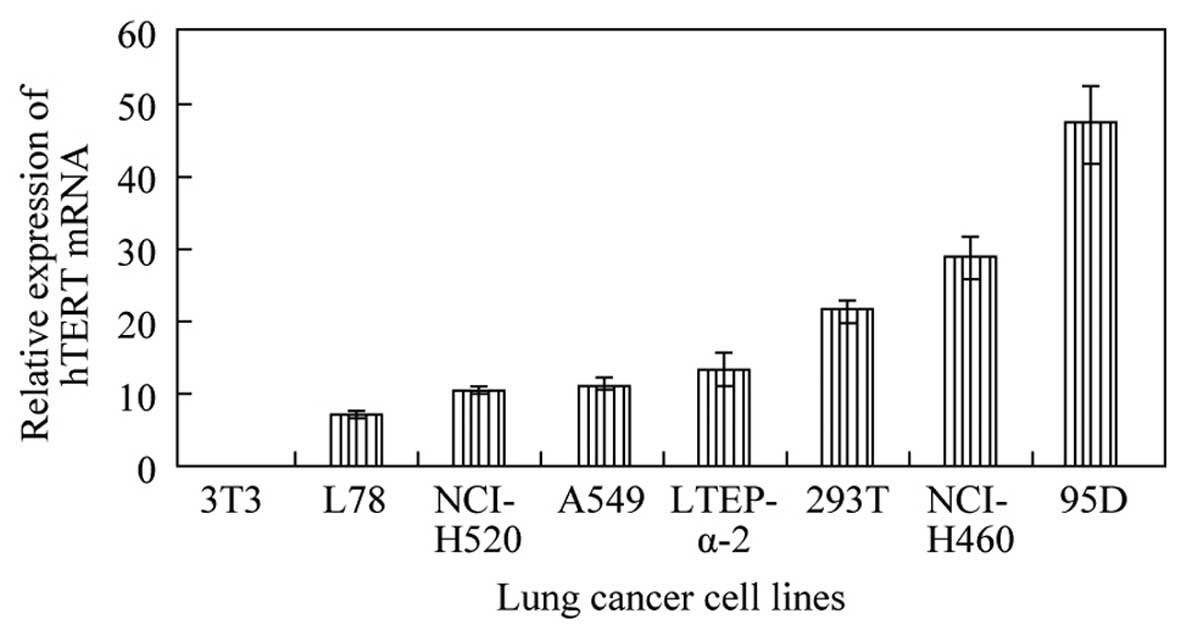
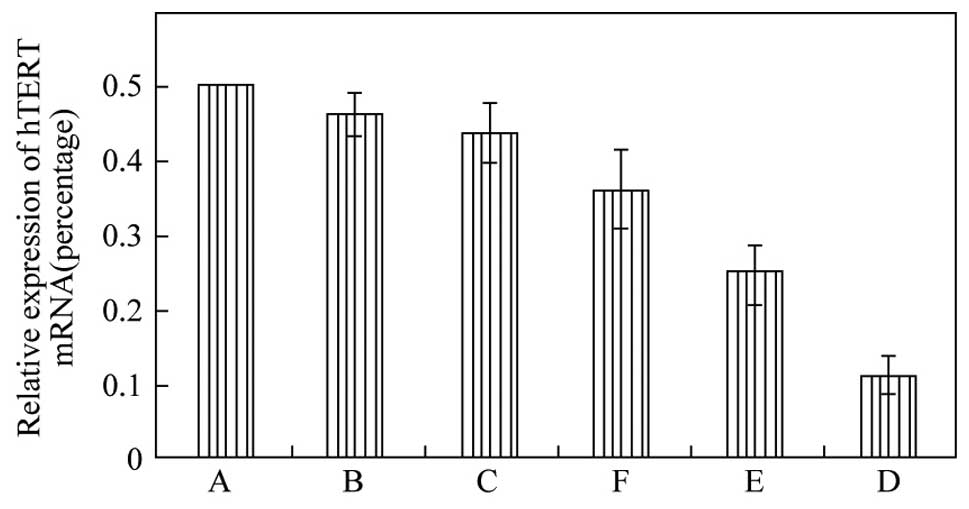
After statistical calculation, 95D human giant-cell
lung carcinoma cells showed the highest relative level of hTERT
mRNA, followed by NCI-H460 large-cell lung cancer cells (Fig. 3). The levels of hTERT mRNA in these
two cell lines exceeded those in the squamous cancer and
adenocarcinoma cell lines tested. Based on these data, the 95D cell
line was selected for the RNAi experiments.
Detection of the effects of hTERT siRNAs
on hTERT mRNA levels
To knock down hTERT levels, 95D cells were
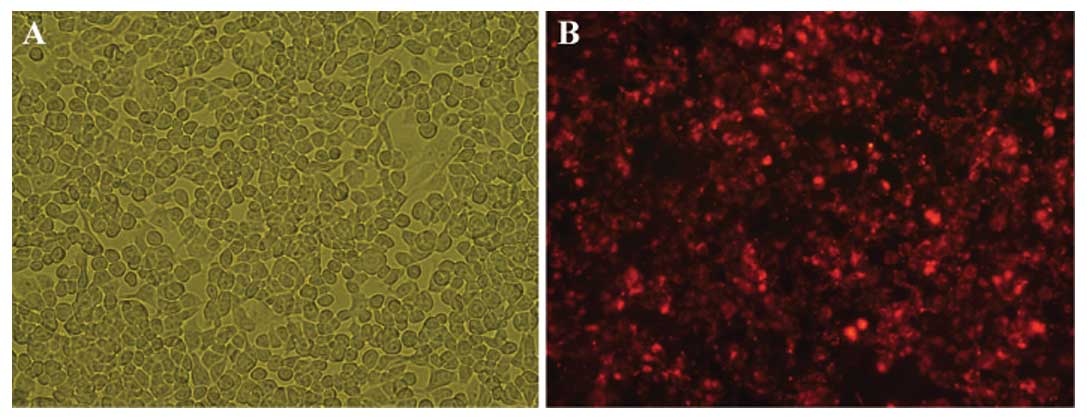
transfected with hTERT or control siRNAs. After 24 h, the majority
of control siRNA-transfected cells remained adherent to the culture
flask and showed no change in cell morphology (Fig. 4A), indicating that the transfection
caused little cytotoxicity. By fluorescent microscopy, transfected
95D cells fluoresced red, whereas non-transfected cells were not
detected (Fig. 4B). Subsequently,
the reverse siRNA transfection efficacy was estimated to be greater
than 90%, which is ideal for optimal target gene knockdown.
Detection of hTERT mRNA levels in cells
after transfection of hTERT siRNAs
The level of hTERT mRNA was significantly reduced
(P<0.01) 48 h after transfection in 95D cells transfected with
either siRNA-1 or siRNA-2 compared to cells transfected with either
the liposome alone or with the negative control siRNA. The level of
hTERT mRNA in cells transfected with siRNA-3 was also significantly
decreased (P<0.05). Further calculations showed that the hTERT
suppression rate in cells transfected with siRNA-1 and siRNA-2 was
77.33±5.13 and 50.67±8.02%, respectively, whereas the suppression
rate was only 27.67±10.26% in cells transfected with siRNA-3
(Fig. 5). Due to the highest hTERT
mRNA suppression rate, siRNA-1 was selected for the subsequent
experiments.
The siRNA dose-dependent changes in hTERT
expression in lung cancer cells
Cells were transfected with three different
concentrations of siRNA-1 (50, 80 or 100 nmol/l) or negative
control siRNAs, and the extent of hTERT expression knockdown was
determined. No significant difference was detected in the levels of
hTERT mRNA between the 50 nmol/l and negative control groups
(P>0.05), while the 80- and 100-nmol/l groups showed significant
reductions in hTERT mRNA levels compared to the control group
(P<0.01). The difference in hTERT knockdown between the 80- and
100-nmol/l groups was not significantly different (P>0.05;
Table V)
| Table VhTERT expression levels in lung
cancer cells transfected with hTERT siRNAs of different
concentrations. |
Table V
hTERT expression levels in lung
cancer cells transfected with hTERT siRNAs of different
concentrations.
| Groups | Relative expression
of hTERT mRNA |
|---|
| Negative
control | 0.87±0.08 |
| 50 nmol/l
siRNA | 0.75±0.06 |
| 80 nmol/l
siRNA | 0.35±0.11a |
| 100 nmol/l
siRNA | 0.23±0.05a |
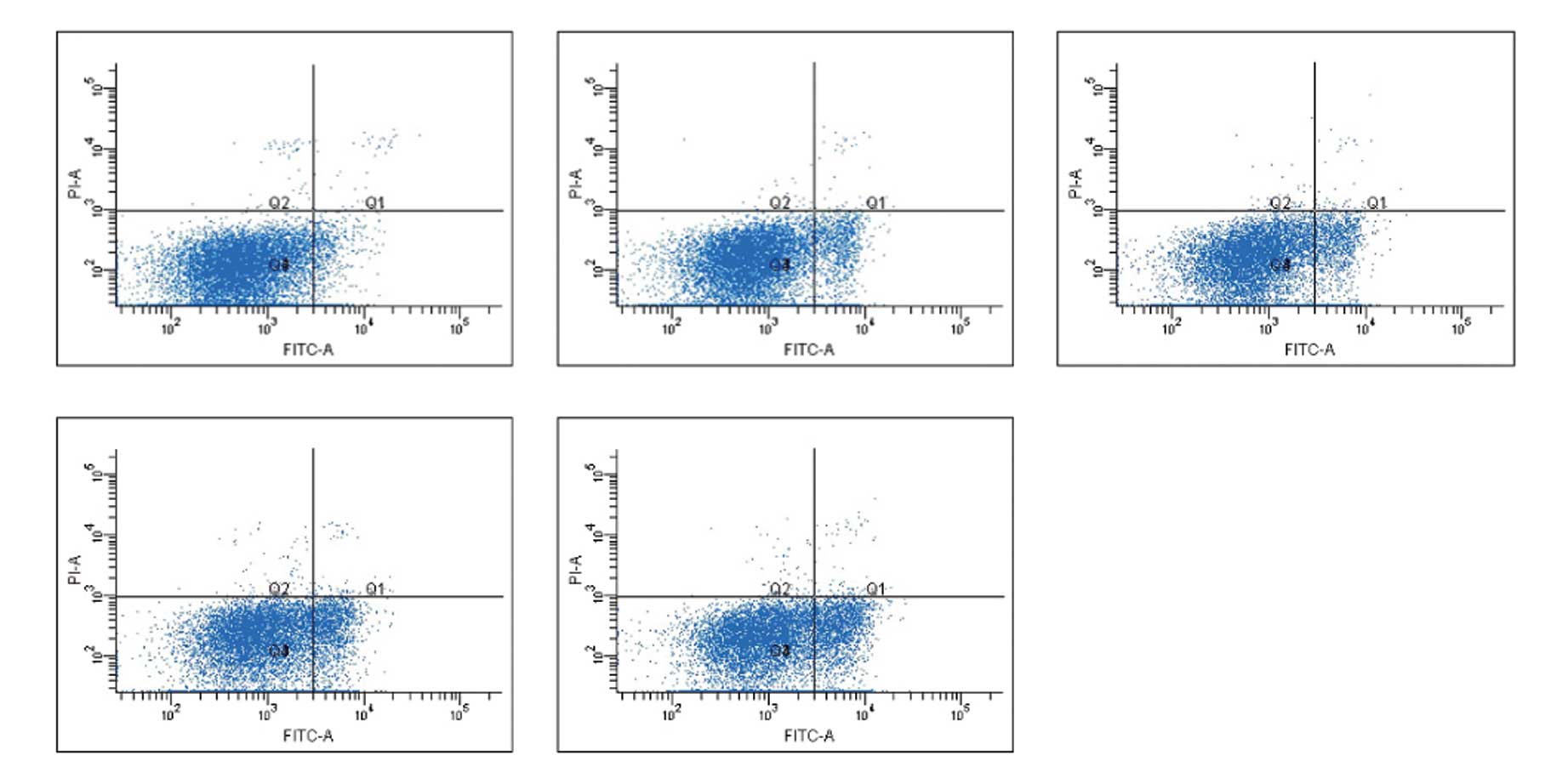
Detection of the apoptotic fraction in
hTERT siRNA-expressing lung cancer cells
The effect of hTERT knockdown on cancer cell
apoptosis was analyzed by flow cytometry. As shown in Fig. 6, the right lower quadrant (FITC+,
PI−) shows early apoptotic cells, whereas the right upper quadrant
(FITC+, PI+) shows late apoptotic cells. The apoptotic fraction was
determined by adding the early apoptotic fraction to the late
apoptotic fraction; apoptotic fractions of cells in different
groups are shown in Table III.
Compared with cells transfected with liposome alone, the apoptotic
fractions in cells transfected with 50, 80 and 100 nmol/l hTERT
siRNA-1 were significantly increased (P<0.01). Compared with
cells transfected with the negative control siRNAs, the apoptotic
fraction in the 50-nmol/l group was not significantly different
(P>0.05), while the apoptotic fractions in the 80- and
100-nmol/l groups were significantly increased (P<0.01). The
apoptotic fraction was also significantly different between cells
transfected with transfection reagent alone and cells transfected
with the negative control siRNA (P<0.05; Table VI), suggesting an inherent
apoptotic effect of siRNA transfection on the target cells.
| Table VIThe effect of hTERT siRNA on cell
apoptosis in 95D cells. |
Table VI
The effect of hTERT siRNA on cell
apoptosis in 95D cells.
| Groups | Apoptosis rate
(%) |
|---|
| siPORT NeoFX
control | 4.33±1.12 |
| Negative
control | 9.40±1.83b |
| 50 nmol/l
siRNA | 12.40±1.51a |
| 80 nmol/l
siRNA | 18.36±1.40a,c |
| 100 nmol/l
siRNA | 22.87±3.37a,c |
The effect of hTERT expression knockdown
on lung cancer cell proliferation
MTT assays were performed to determine the
proliferative effects of hTERT expression knockdown on lung cancer
cells, and the results are shown in Fig. 3. At 12 h post-hTERT siRNA
transfection, no significant inhibition of 95D cell proliferation
was detected when compared to the negative control (P>0.05), but
a significant inhibitory effect was detected at 24 h
post-transfection and was enhanced at 48 h; the reduction in
proliferation was attenuated at 72 h post-transfection. The
inhibition rate in the hTERT siRNA-transfected cells was
significantly different from that in the negative control cells at
24, 48 and 72 h post-transfection (P<0.05) in that the
inhibitory effect of hTERT siRNA on cell proliferation was
significantly higher at 48 h post-transfection when compared to
that at 12, 24 and 72 h post-transfection (P<0.01).
Discussion
The single copy hTERT gene is 41878 bp in length and
is located on chromosome 5p15.33; the length of its coding region
is 4018 bp. We designed specific primers based on the hTERT
sequence and examined hTERT mRNA expression in a variety of lung
cancer cells by real-time RT-PCR. The results showed that the hTERT
mRNA expression was relatively high in a variety of lung cancer
cells, whereas the highest level of hTERT expression was detected
in the highly metastatic 95D human giant-cell lung carcinoma cell
line, followed by NCI-H460 cells, which are also large-cell lung
carcinoma cells. Compared to squamous cell carcinoma and
adenocarcinoma, the incidence of large-cell lung cancer is not
high, but it is an occult cancer that develops rapidly, has poor
treatment efficacy and prognosis and has a high degree of
malignancy. The World Health Organization classifies giant-cell and
clear-cell carcinomas as two subtypes of large-cell lung cancer,
and the pathological morphology, classification and development of
the two subtypes are relatively complex (7). Most cells found in large-cell lung
cancer are of neuroendocrine origin; therefore, this cancer is also
known as large-cell neuroendocrine carcinoma (LCNEC). LCNEC,
together with typical and atypical carcinoid and small-cell lung
carcinoma, constitutes bronchopulmonary neuroendocrine tumors
(8). These tumor cells, derived
from the neuroendocrine cells in bronchial lung epithelia, have a
characteristic microscopic morphology and biological behavior and
have been a focus of cancer research in recent years.
Currently, there are numerous immunomarkers used to
detect bronchopulmonary neuroendocrine tumors, most commonly
neuron-specific enolase, synaptophysin and chromogranin A, although
each has a different sensitivity and specificity (9). Therefore, searching for the best panel
of immunomarkers to detect the differentiation of neuroendocrine
tumors is crucial. Less than 10% of typical and atypical carcinoid
tumors manifest telomerase activity, while 90% of both small-cell
lung cancers and LCNEC show active telomerase (3). Telomerase-positive tumor cells often
have high proliferation indices and a high degree of malignancy
(10). In this study, the cell line
with the highest relative expression of hTERT mRNA was a highly
metastatic large-cell lung cancer line of immortalized cells with a
high proliferation rate that was able to be passaged for a number
of generations. Considering the biological characteristics of
LCNEC, hTERT may be a significant marker in the detection of
malignant lung cancer, particularly the marker for bronchopulmonary
tumors with a high degree of malignancy and the characteristics of
neuroendocrine tumors. It is obvious that hTERT is a tumor marker
with high sensitivity and specificity. The detection of hTERT in
combination with the histopathological examination of lung tumors
may provide an early reference in diagnosing lung cancer and its
potential for recurrence.
Since hTERT expression regulates telomerase activity
in normal and tumor cells, hTERT is a key factor in determining
telomerase activity in the cell and may be a target for tumor
eradication. Therefore, siRNAs specifically targeting hTERT may
down-regulate telomerase activity, induce apoptosis of cancer cells
and inhibit cancer cell proliferation by reducing hTERT expression.
Kosciolek et al (11)
studied hTERT in various human cancer cells using RNAi and found
that siRNAs efficiently and specifically inhibited the level of
hTERT mRNA and, in turn, reduced telomerase activity by greater
than 70%. Other reports have confirmed that siRNAs inhibit hTERT
expression in liver and breast cancer and leukemia cells, and
inhibit the proliferation of cancer cells (12). In this study, we used specific
siRNAs targeting hTERT and found that the decrease in hTERT
expression negatively affected lung cancer cell growth and
apoptosis.
The proper and rational design and synthesis of
siRNAs is critical to the success of RNAi experiments. In addition
to the conventional theoretical factors such as G + C content,
fragment length and the avoidance of homology with other coding
sequences, attention should be paid to four principles. First,
siRNA-binding sites should be located further than 50 to 100 bp
downstream of the start codon in the target sequence. Additionally,
there should not be a large amount of repeat sequences. The
sequences in the 5′ and 3′ untranslated regions and the region near
the start codon should be avoided since these regions are rich in
regulatory protein-binding sites. These non-coding region-binding
proteins or translation initiation complexes may affect RNA-induced
silencing complex formation and the ability of the complex to bind
mRNA, which weakens the effect of RNAi. Second, beginning from the
AUG start codon in the mRNA sequence, ‘AA’ repeat sequences should
be identified, since the 19 nucleotides at the 3′ end of this
repeat sequence are potential effective siRNA targeting sites.
Third, the 3′ end should be designed to contain dTdT or dTdG
overhangs to increase the stability of the siRNA molecules and to
improve their resistance to degradation by ribozymes, thereby
prolonging the knockdown effect. The fourth principle is that
negative and positive control siRNAs should be included. Negative
control siRNA sequences should have an identical or a similar
composition as the selected siRNA, but show no degradative effect
on the target mRNA (13,14). The purpose of designing a positive
control is to optimize transfection and detection conditions.
Housekeeping genes are favorable positive controls in most cells.
We used GAPDH-specific siRNAs as the positive control in the
preliminary experiments. After GAPDH siRNAs were transfected into
the target cells, GAPDH mRNA expression was significantly
suppressed by over 85% compared to non-transfected cells; these
data aided in determining the appropriate transfection conditions
and experimental methods for the hTERT RNAi experiments.
Specific siRNAs only interfere with hTERT mRNA
expression after entering the cells, and highly efficient siRNA
transfection into the lung cancer cells was therefore of particlar
significance to these experiments (15). Methods of transfecting siRNAs
include calcium phosphate precipitation, electroporation,
microinjection, DEAE-dextran and polybrene polymer complex methods
and cationic liposomal delivery methods. Although most of these
methods are characterized by low transfection efficacy, high
cytotoxicity, poor reproducibility and a narrow range of
applications, the cationic liposomal method has been widely used
due to the ease of use, widespread application, high transfection
efficacy and non-immunogenicity. Although certain studies have
achieved very high transfection efficiencies by using retroviral or
adenoviral vector-mediated siRNA delivery (16,17),
the biological safety of these methods requires further study. In
our experiments, siRNAs were transiently transfected using siPORT
NeoFX, which is an inert liposomal reagent that can be effectively
used for adherent cell transfection without obvious cytotoxicity.
We also used the reverse transfection method (18), in which, instead of the conventional
transfection where cells are plated in advance and the transfection
reagent and siRNAs are added once the cells grow to a certain
confluence, the cells in suspension are plated into wells that
already contain the siRNA complexes and are allowed to adhere and
expand. We used fluorescence microscopy to monitor the cell plating
and transfection efficacy, which for 95D lung cancer cells was
greater than 90%. Thus, compared with the conventional transfection
method, the reverse transfection method is time-efficient and
increases the transfection efficacy, which may be due to the larger
area of the non-adherent cells to which the siRNA transfection
complexes bind. No significant change in cell morphology following
transfection was observed, suggesting a lack of obvious
cytotoxicity. Therefore, the reverse transfection method may be
suitable for high-throughput siRNA transfection.
After hTERT siRNAs were effectively transfected into
95D lung cancer cells, real-time RT-PCR was employed to measure the
hTERT mRNA levels to confirm RNAi-induced hTERT knockdown.
According to previous in vitro studies in mammalian cells,
RNAi exhibited a time-dependent effect following the transfection
of chemically synthesized siRNAs (19). This effect appeared at 12 h
post-transfection, reached maximal knockdown at 48 h and gradually
decreased after 72 h. Based on our results, we selected 48 h
post-transfection, at which point the most significant hTERT
knockdown was measured as the detection time point in our
experiments. The results showed that siRNA-1 and siRNA-2
significantly suppressed hTERT mRNA levels at 48 h
post-transfection, with suppression rates of 77.33±5.13 and
50.67±8.02%, respectively, whereas siRNA-3 showed a relatively low
suppression rate of 27.67±10.26%. Based on these data, siRNA-1 was
selected for our RNAi experiments.
In addition to the conventional blank control group
in our experiments, the negative control and the liposome alone
control groups were also included in our experiments and were
critical in determining the specificity of RNAi. The negative
control group used non-specific siRNAs and was included to exclude
any effects caused by non-specific siRNAs on gene silencing. The
liposome alone control group was used to identify potential effects
of the transfection reagent on the experimental results. The
results showed that, compared to the negative control and the
liposome alone control groups, hTERT siRNA transfection groups
using 80 and 100 nmol/l significantly reduced hTERT mRNA
expression, whereas the transfection group with 50 nmol/l hTERT
siRNA was not statistically different from the two control groups.
These data demonstrate a concentration-dependent effect of hTERT
siRNA on hTERT mRNA expression. Therefore, the siRNA-mediated hTERT
suppression can be enhanced with increased concentrations of
siRNA.
RNAi targeting hTERT expression inhibited the growth
of lung cancer cells and induced cell apoptosis. Data from MTT
assays showed significant growth inhibition at 24 h
post-transfection; this inhibition reached maximum levels at 48 h
and then began to attenuate, which is consistent with the
time-dependent effect of siRNA on hTERT mRNA levels. These data are
consistent with the hypothesis that the siRNA-dependent
down-regulation of hTERT expression inhibits telomerase activity,
promotes the progressive shortening of telomeres and thus
effectively inhibits tumor cell growth. These data further suggest
that hTERT is the rate-limiting factor of telomerase activity and
is a favorable target of RNAi.
Flow cytometry revealed that lung cancer cells
undergo apoptosis and necrosis following the suppression of hTERT
mRNA expression (20). At 48 h
post-transfection, apoptosis was detected in lung cancer cells and
the apoptotic fraction was up to 22.87±3.37%, although the increase
in the apoptotic fraction was not as significant as that of the
suppression rate of hTERT mRNA. The reason for this difference may
be that the change in telomerase activity may not directly affect
apoptosis, which is jointly regulated by a complex pathway composed
of a variety of pro- and anti-apoptotic genes jointly. In addition,
some investigators have identified a so-called telomere extension
bypass in cancer cells, which is the alternative pathway of
telomere extension. Although the exact mechanisms remain to be
elucidated, this bypass may maintain telomeric integrity through
DNA recombination to inhibit cell apoptosis and promote cell
immortalization (21). Therefore,
the clinical application of hTERT siRNA for the treatment of lung
cancer may require other confirmed apoptosis-inducing agents.
This study used chemically synthesized siRNAs to
perform in vitro RNAi experiments using lung cancer cells.
Chemical synthesis technology is established, easy to operate and
has wide applications; however, the chemically synthesized siRNAs
are susceptible to RNase degradation and therefore have shorter
durations for RNAi of no longer than 7 days (22). Our ultimate aim in using RNAi is to
effectively treat lung cancer, which requires the continuous action
of siRNAs in vivo. Methods used to express siRNAs in
vivo include using siRNA expression cassettes (SECs) and siRNA
expression vectors. SECs contain the siRNA expression template
obtained by PCR, and the PCR product can be transfected directly
into cells for expression without first being cloned into a vector
(23). However, since it is
difficult to transfect PCR products into cells and the PCR products
do not contain drug resistance genes, stable cell transfection
cannot be conducted, as the in vivo biological function is
transient and is not suitable for long-term suppression (24,25).
The aim of using siRNA expression vectors is to generate
intracellular siRNAs through a plasmid or viral vector to achieve
long-term suppression of target gene expression. The antibiotic
selection marker on the siRNA expression vector aids in the rapid
selection of siRNA-positive clones and may be the ideal approach
for a wide array of in vivo RNAi research applications and
treatment strategies.
In conclusion, hTERT is a key regulator of
telomerase activity and is potentially a tumor marker for lung
cancer detection. hTERT was highly expressed in multiple lung
cancer cell lines of different pathological types, and the highest
relative expression levels were found in large-cell lung carcinoma
cells. The significant reduction in hTERT mRNA by RNAi induced
apoptosis and inhibited the proliferation of 95D lung cancer cells,
suggesting that telomerase activity is closely associated with
tumor cell survival and proliferation. These data further suggest
that telomerase is a key target for RNAi-based lung cancer
strategies.
References
|
1
|
Raynaud CM, Sabatier L, Philipot O,
Olaussen KA and Soria JC: Telomere length, telomeric proteins and
genomic instability during the multistep carcinogenic process. Crit
Rev Oncol Hematol. 66:99–117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Djojosubroto MW, Choi YS, Lee HW and
Rudolph KL: Telomeres and telomerase in aging, regeneration and
cancer. Mol Cells. 15:164–175. 2003.PubMed/NCBI
|
|
3
|
Taga S, Osaki T, Ohgami A, Imoto H and
Yasumoto K: Prognostic impact of telomerase activity in non-small
cell lung cancers. Ann Surg. 230:715–720. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hiyama K, Hiyama E, Ishioka S, Yamakido M,
Inai K, Gazdar AF, Piatyszek MA and Shay JW: Telomerase activity in
small-cell and non-small cell lung cancers. J Natl Cancer Inst.
87:895–902. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
6
|
Xia Y, Lin RX, Zheng SJ, Yang Y, Bo XC,
Zhu DY and Wang SQ: Effective siRNA targets screening for human
telomerase reverse transcriptase. World J Gastroenterol.
11:2497–2501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
The World Health Organization histological
typing of lung tumours. Second edition. Am J Clin Pathol. 77. pp.
123–136. 1982
|
|
8
|
Schleusener JT, Tazelaar HD, Jung SH, Cha
SS, Cera PJ, Myers JL, Creagan ET, Goldberg RM and Marschke RF Jr:
Neuroendocrine differentiation is an independent prognostic factor
in chemotherapy-treated nonsmall cell lung carcinoma. Cancer.
77:1284–1291. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Graziano SL, Tatum AH, Newman NB, Oler A,
Kohman LJ, Veit LJ, Gamble GP, Coleman MJ, Barmada S and O'Lear S:
The prognostic significance of neuroendocrine markers and
carcinoembryonic antigen in patients with resected stage I and II
non-small cell lung cancer. Cancer Res. 54:2908–2913.
1994.PubMed/NCBI
|
|
10
|
Zaffaroni N, De Polo D, Villa R, Della
Porta C, Collini P, Fabbri A, Pilotti S and Daidone MG:
Differential expression of telomerase activity in neuroendocrine
lung tumours: correlation with gene product immunophenotyping. J
Pathol. 201:127–133. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kosciolek BA, Kalantidis K, Tabler M and
Rowley PT: Inhibition of telomerase activity in human cancer cells
by RNA interference. Mol Cancer Ther. 2:209–216. 2003.PubMed/NCBI
|
|
12
|
Zhou X and Zhang PH: Stable inhibition of
hTERT gene by siRNA in hepatocarcinoma cells. J Fourth Mi1 Med
Univ. 26:2233–2236. 2005.
|
|
13
|
Reynolds A, Leake D, Boese Q, Scaringe S,
Marshall WS and Khvorova A: Rational siRNA design for RNA
interference. Nat Biotechnol. 22:326–330. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ui-Tei K, Naito Y, Takahashi F, Haraguchi
T, Ohki-Hamazaki H, Juni A, Ueda R and Saigo K: Guidelines for the
selection of highly effective siRNA sequences for mammalian and
chick RNA interference. Nucleic Acids Res. 32:936–948. 2004.
View Article : Google Scholar
|
|
15
|
Pham JW, Pellino JL, Lee YS, Carthew RW
and Sontheimer EJ: A Diecr-2- dependent 80s complex cleaves
targeted mRNAs during RNAi in Drosophila. Cell. 117:83–94. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Godfrey A, Anderson J, Papanastasiou A,
Takeuchi Y and Boshoff C: Inhibiting primary effusion lymphoma by
lentiviral vectors encoding short hairpin RNA. Blood.
105:2510–2518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andersson MG, Haasnoot PC, Xu N, Berenjian
S, Berkhout B and Akusjärvi G: Suppression of RNA interference by
adenovirus virus-associated RNA. J Virol. 79:9556–9565. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Erfle H, Neumann B, Liebel U, Rogers P,
Held M, Walter T, Ellenberg J and Pepperkok R: Reverse transfection
on cell arrays for high content screening microscopy. Nat Protoc.
2:392–399. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He GP, Zhang SZ, Wang YC, Xiao CY, Ma YX,
Xu WM, Ding L, Tao DC, Sun Y and Chen YJ: Time and dose effect of
RNA interference mediated by short hairpin RNA. Progress in
Biochemistry and Biophysics. 32:258–267. 2005.
|
|
20
|
Shammas MA, Koley H, Batchu RB, Bertheau
RC, Protopopov A, Munshi NC and Goyal RK: Telomerase inhibition by
siRNA causes senescence and apoptosis in Barrett’s adenocarcinoma
cells: mechanism and therapeutic potential. Mol Cancer.
4:242005.PubMed/NCBI
|
|
21
|
Collins K and Mitchell JR: Telomerase in
the human organism. Oncogene. 21:564–579. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martinez J, Patkaniowska A, Urlaub H,
Lührmann R and Tuschl T: Single-stranded antisense siRNAs guide
target RNA cleavage in RNAi. Cell. 110:563–574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castanotto D, Li HT and Rossi IJ:
Functional siRNA expression form transfected PCR products. RNA.
8:1454–1460. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu N, Gu HJ and Li Q: Effects of
antidiabetic drug metformin on the migration and invasion abilities
of human pulmonary adenocarcinoma A549 cell line in vitro. J Thorac
Dis. 2:76–78. 2010.PubMed/NCBI
|
|
25
|
Shao WL, Wang DY and He JX: The role of
gene expression profiling in early-stage non-small cell lung
cancer. J Thorac Dis. 2:89–99. 2010.PubMed/NCBI
|