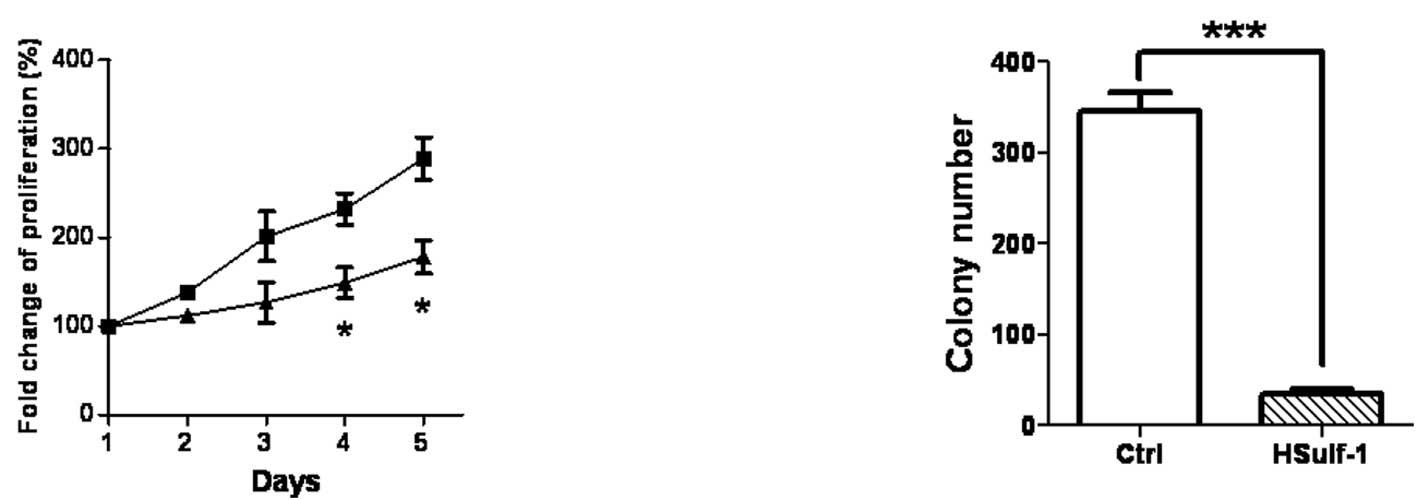Introduction
Gastric cancer is the second most malignant type of
cancer worldwide. It is often diagnosed at a late stage, and
despite the current regimen of surgical, chemo-, radio- and other
adjuvant therapies, gastric cancer has a poor prognosis with a
5-year survival rate of less than 20–25% in the USA, Europe and
China (1). This has galvanized
efforts to identify new therapeutic targets for treating this
lethal disease.
Emerging evidence revealed that gastric cancer
results from a combination of factors, including Helicobacter
pylori-induced inflammation and deregulation of the Wnt and
Hedgehog signaling pathways (2–4). The
Hedgehog (Hh) family of proteins controls multiple cellular
functions, including cell growth, survival and outcome, as well as
body patterning and organ morphogenesis (5). Deregulated Hh pathway activation plays
a role in various types of cancer including glioma, basal cell
carcinoma, medulloblastoma, lung, breast, pancreatic and gastric
cancers (6–8). Hh signaling is controlled by two
transmembrane proteins, Patched (Ptch1) and Smoothened (SMO). In
the absence of the Hh ligand, PTCH1 inhibits SMO, causing cleavage
of GLI1 to the N-terminal repressor form. Once Hh binds to PTCH1,
the inhibitory effect on SMO is released, causing active
full-length GLI1 to transport into the nucleus and activate
transcription of the Hh target genes, including GLI1, PTCH1, HHIP
and C-MYC (9–11).
Human sulfatase-1 (HSulf-1) has emerged as a
negative regulator of Hh signaling. It disrupts the distribution of
the Hh morphogen and Hh signaling transduction by desulfating cell
surface heparan sulfate proteoglycans (HSPGs) (12–14).
HSulf-1 was cloned as the first human ortholog of the
developmentally regulated putative Quail sulfatase QSulf-1
(13) and was found to be
downregulated in a number of cancer cells including ovarian,
breast, renal tumor, hepatocellular carcinoma, myeloma and head and
neck squamous carcinoma (15–21).
HSulf-1 expression inhibited cancer cell growth, cell motility and
invasion in multiple cancer cells, and promoted stress-induced
apoptosis (15–17). Furthermore, HSulf-1 expression led
to a reduction in vascular density and increased breast cancer cell
apoptosis in xenografts, suggesting that HSulf-1 inhibits both
angiogenesis and tumorigenesis in vivo (18,19).
However, the role of HSulf-1 in gastric cancer tumorigenesis
remains to be elucidated. Findings of our previous study
demonstrated that the HSulf-1 expression is downregulated in
gastric cancer cells, and that HSulf-1 gene silencing is associated
with a high level of promoter hypermethylation (22).
In this study, we also investigated the function of
HSulf-1 in gastric cancer cell proliferation. Expression of HSulf-1
in monoclonal MKN28 gastric cancer cells suppressed cell
proliferation and colony formation. Notably, HSulf-1 inhibited
GLI1-mediated transcription and the downregulated expression of Hh
downstream target genes, including GLI1, PTCH1, C-MYC, CCND1 and
BCL2. These data strongly support that HSulf-1 may inhibit gastric
cancer cell proliferation by blocking the Hh pathway.
Materials and methods
Cell culture and establishment of
HSulf-expressing monoclonal MKN28 stable cells
The human gastric cancer cell line MKN28 was
obtained from the China Center for Type Culture Collection (Wuhan,
China). Cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin
(100 IU/ml) and streptomycin (100 μg/ml) in a humidified incubator
at 37°C with a 5% CO2 atmosphere. Cells
(1×104) were plated in 12-well plates one day prior to
transfection. Monoclonal cells stably expressing HSulf-1 or empty
vector were established following plasmid transfection and
Geneticin (G418 sulfate) selection. The stable monoclonal cell
lines were then cultured in DMEM supplemented with 10% FBS and 0.5
mg/ml G418. Cells were maintained at 37°C in a humid incubator with
5% CO2.
Quantitative real-time PCR (RT-PCR)
RNA extraction and RT-PCR were performed as
previously described (22). The
primer sequences were as follows: 18S rRNA, 5′-CAGCCA
CCCGAGATTGAGCA-3′ (forward) and 5′-TAGTAGCGACG GGCGGTGTG-3′
(reverse); HSulf-1, 5′-ACTGTACCAATCG GCCAGAG-3′ (forward) and
5′-CCTCCTTGAATGGGTGA AGA-3′ (reverse). Semi-quantitative RT-PCR was
used to examine the expression levels of Hh downstream target
genes. The primers used are shown in Table I. The procedure was performed as
described in a previous study (22).
 | Table IPrimers for semi-quantitative RT-PCR
analysis. |
Table I
Primers for semi-quantitative RT-PCR
analysis.
| Gene name | Primer sequences
(5′-3′) | Amplicon size
(bp) |
|---|
| GLI1 |
5′-TACTCACGCCTCGAAAACCT-3′
(forward)
5′-GTCTGCTTTCCTCCCTGATG-3′ (reverse) | 352 |
| PTCH1 |
5′-ATGCTGGCGGGATCTGAGTTCGACT-3′
(forward)
5′-GGGTGTGGGCAGGCGGTTCAAG-3′ (reverse) | 174 |
| PTCH2 |
5′-GATGTGCTCTGCTGCTTCTCCA-3′
(forward)
5′-CTGCCTTCTGCCTTGTCTCCTC-3′ (reverse) | 283 |
| HHIP |
5′-GCCTCGCATTCCATCCCAATTAC-3′
(forward)
5′-ACCCATCCATTTCTTCCATATCATCC-3′ (reverse) | 297 |
| C-MYC |
5′-TTCGGGTAGTGGAAAACCAG-3′
(forward)
5′-CAGCAGCTCGAATTTCTTCC-3′ (reverse) | 203 |
| MYCN |
5′-CATCCACCAGCAGCACAACTAT-3′
(forward)
5′-CTCAAGCTCTTAGCCTTTGGGG-3′ (reverse) | 138 |
| CCND1 |
5′-AGCTCCTGTGCTGCGAAGTGGAAAC-3′
(forward)
5′-AGTGTTCAATGAAATCGTGCGGGGT-3′ (reverse) | 480 |
| FOXF1 |
5′-GCCGTATCTGCACCAGAACA-3′
(forward)
5′-CGTTGAAAGAGAAGACAAACTCCTT-3′ (reverse) | 116 |
| FOXM1 |
5′-GGAGCAGCGACAGGTTAAGG-3′
(forward)
5′-GTCGTGCAGGGAAAGGTTGT-3′ (reverse) | 244 |
| BCL2 |
5′-TTTGAGTTCGGTGGGGTCAT-3′
(forward)
5′-TGACTTCACTTGTGGCCCAG-3′ (reverse) | 275 |
| 18S rRNA |
5′-CAGCCACCCGAGATTGAGCA-3′
(forward)
5′-TAGTAGCGACGGGCGGTGTG-3′ (reverse) | 254 |
Immunoblotting
For total cell lysates, the cell pellet was
resuspended in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1%
Triton X-100, 0.5% sodium deoxycholate and 0.1% SDS), mixed with
protease inhibitors and vortexed for 5 sec. Following incubation on
ice for 15 min, the suspension was centrifuged at 14,000 rpm at 4°C
for 5 min and the supernatant was collected for immunoblotting.
Total cell lysates (20 μg) were subjected to SDS-PAGE and
transferred to a PVDF membrane (GE Healthcare, Chalfont, UK). The
membrane was blocked by 5% milk for 2 h and incubated with primary
antibodies overnight at 4°C, followed by horseradish
peroxidase-conjugated secondary antibody (Pierce, Rockford, IL,
USA) and Super Signal West Pico reagent (Pierce). The primary
antibodies included anti-HSulf-1 (1:250; Abnova Corp., Walnut, CA,
USA) and anti-β-actin (1:5000; Sigma Aldrich, St. Louis, MO,
USA).
Cell proliferation assay
Cell growth rates were determined by a CellTiter
96® AQueous non-radioactive cell proliferation assay kit
(Promega, Madison, WI, USA). Briefly, the stable cell lines were
plated into 96-well tissue culture plates at 5×102
cells/well. After the cells were cultured for a number of days as
indicated, MTS solutions were added to the medium and incubated for
1.5 h at 37°C. The absorbance at 490 nm was measured using a
microplate reader model 680 (Bio-Rad, Hercules, CA, USA) and the
normalized absorbance was plotted against culture time to determine
the number of viable cells.
Colony formation assay
The stable cells expressing empty vector or HSulf-1
were seeded in triplicate in 100-mm dishes at 1×103
cells/dish. Fresh medium containing 0.5 mg/ml G418 was changed
every 3–4 days. After being cultured for 2–3 weeks, cells were
fixed with 4% paraformaldehyde for 10 min, washed with PBS several
times, stained with 0.1% crystal violet for 20 min and
photographed. Clones with more than 50 cells were counted.
Luciferase reporter assay
MKN28 cells at a density of 5×103
cells/well were seeded into 24-well plates prior to transfection.
8xGli-BS or 8xmutGli-BS luciferase reporter plasmid was
co-transfected with 20 ng TK-Renilla (pRL-TK) (Promega) internal
control plasmid and DNA expression vectors as indicated. After
cells were cultured for 48 h, the luciferase activity was measured
by the Dual-Glo Luciferase Assay System (Promega). The ratio
between firefly luciferase activity (8xGli or Mut Gli) and renilla
activity was calculated to assess the Gli transcriptional
activity.
Statistical analysis
Data are expressed as the standard error of the mean
(SEM) unless specifically indicated otherwise. The student’s t-test
was used for statistical analysis of the data. P<0.05 was
considered to be statistically significant.
Results
Establishment of a monoclonal
MKN28-HSulf-1 stable cell line
To investigate the role of HSulf-1 in gastric
cancer, we first established a gastric cell line that was able to
stably express HSulf-1. As previously reported, endogenous HSulf-1
was downregulated in a number of gastric cancer cell lines
including MKN28 (22). We therefore
transfected pcDNA3.1/Myc-His(-)HSulf-1 or an empty vector plasmid,
pcDNA3.1/Myc-His(-), into this cell line. Following G418 selection
the monoclonal MKN28-HSulf-1 cell line was established, which
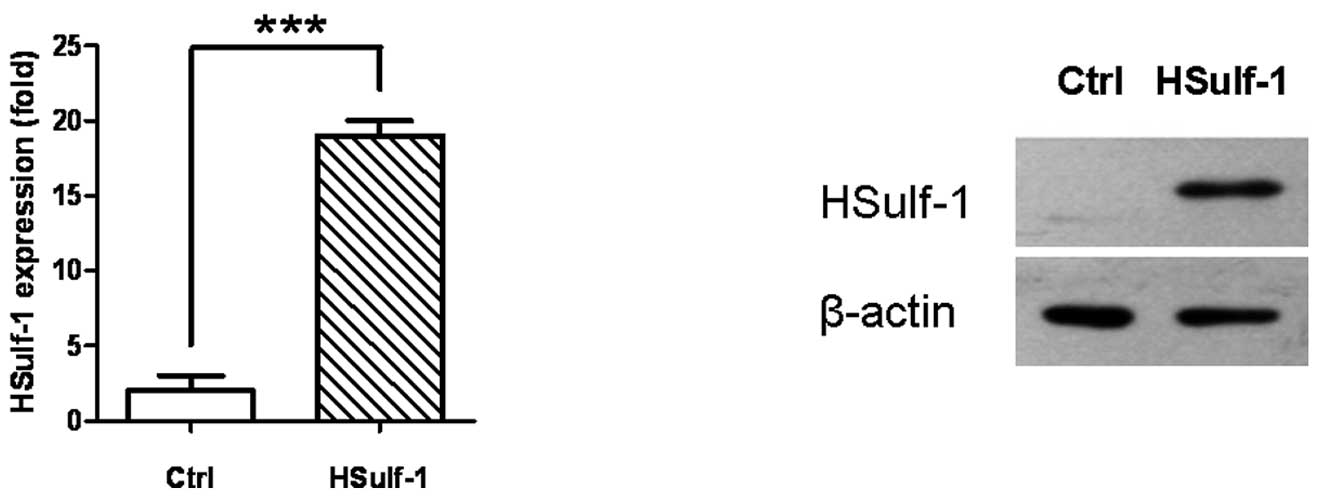
expressed a significantly higher amount of HSulf-1 mRNA and protein
than the empty vector control (Ctrl) as verified by both
quantitative real-time RT-PCR (Fig.
1A) and immunoblotting (Fig.
1B). The results shown in Fig.
1 are representative of at least three independent experiments
with similar results.
HSulf-1 expression inhibits gastric
cancer cell proliferation
To determine the effect of HSulf-1 expression on
gastric cancer cell proliferation, a MTS assay and a clone
formation assay were utilized. Compared with control cells,
HSulf-1-expressing monoclonal MKN28 cells showed a significantly
reduced growth rate starting from day 4 (Fig. 2A). HSulf-1-expressing MKN28 cells
also formed many fewer colonies, as shown in the colony formation
assay (Fig. 2B). These data
indicated that HSulf-1 expression may significantly suppress the
growth and proliferation of MKN28 gastric cancer cells, consistent
with previous studies showing that HSulf-1 inhibits the growth of a
number of tumors, including ovarian, breast, myeloma and
hepatocellular carcinoma cells (15–21).
Data shown in Fig. 2 are
representative of three independent experiments with similar
results.
HSulf-1 is capable of inhibiting the Hh
signaling pathway
HSulf-1 has emerged as a negative regulator of Hh
signaling through the disruption of Hh signaling transduction by
desulfating HSPG (17–19). Therefore, we investigated the effect
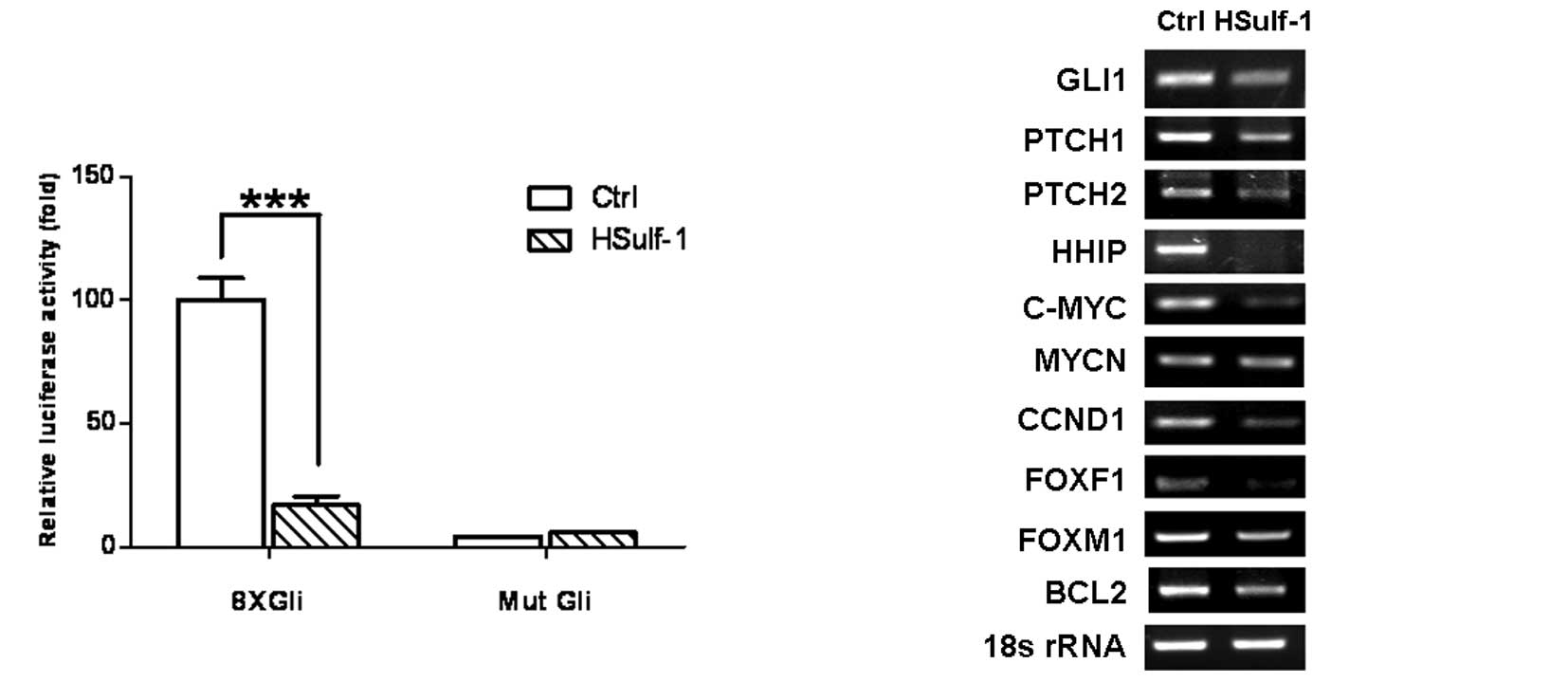
of HSulf-1 on Hh signaling. Hh pathway activation was evaluated by
a Gli luciferase reporter assay and semi-quantitative RT-PCR for Hh
target genes. 8xGli luciferase reporter activity was significantly
elevated in MKN28 cells as compared to mutant Gli reporter (Mut
Gli)-transfected cells, indicating that the Hh pathway is activated
in MKN28 cells (Fig. 3A). Notably,
the introduction of HSulf-1 significantly inhibited 8xGli reporter
activity (Fig. 3A, HSulf-1 vs.
Ctrl), suggesting that HSulf-1 downregulates Hh signaling.
Furthermore, HSulf-1 significantly downregulated the expression of
Hh pathway target genes, including GLI1, PTCH1, PTCH2, HHIP, C-MYC,
CCND1, FOXF1, FOXM1 and BCL2 (Fig.
3B). Taken together, these results markedly support that the
expression of HSulf-1 is capable of downregulating Hh signaling,
correlating with its growth inhibition in gastric cancer cells. The
data shown in Fig. 3 represent
three independent experiments with similar results.
Discussion
In this study, HSulf-1 was found to play a
significant role in suppressing the growth and proliferation of
gastric cancer in MKN28 cells. Furthermore, it was determined that
HSulf-1 expression in gastric cancer cells may significantly
inhibit the Hh signaling pathway, providing new mechanistic insight
into HSulf-1-mediated growth inhibition in gastric cancer.
Emerging evidence revealed that HSulf-1 may function
as a novel tumor suppressor in various types of cancer (15–21).
It was recently reported that HSulf-1 is downregulated in several
types of tumors including ovarian, breast, hepatocellular
carcinoma, renal tumor cells and head and neck squamous carcinoma
cells (15–21). Re-expression of HSulf-1 in ovarian,
hepatocellular carcinoma or head and neck squamous carcinoma cells
retarded cell proliferation and motility, and enhanced
stress-induced apoptosis (15–17,20).
More significantly, HSulf-1 expression in breast, myeloma or
hepatocelluar carcinoma cell-derived xenografts blocked
angiogenesis and tumor invasion in vivo (18,19).
Notably, HSulf-1 is upregulated in primary pancreatic
adenocarcinoma, and overexpression of HSulf-1 reduced growth
capacity but increased invasiveness in pancreatic cancer (23,24).
These studies suggested that HSulf-1 may play various roles in
various types of cancer. Whether or not HSulf-1 plays a role in
gastric cancer tumorigenicity remains to be elucidated. Our
previous study suggested that HSulf-1 expression is downregulated
in gastric cancer cells and that the gene silencing of HSulf-1 is
associated with promoter hypermethylation (22). In the present study, we investigated
whether HSulf-1 regulates the proliferation and growth of gastric
cancer cells. HSulf-1 expression in MKN28 gastric cancer cells was
found to markedly suppress cell proliferation and growth (Fig. 2A-B), consistent with previous
studies, which revealed that HSulf-1 inhibits the proliferation of
multiple types of cancer cells (15–21).
As a newly identified member of the endosulfatase
family, HSulf-1 selectively desulfates cell surface HSPGs (12–14).
Sulfated HSPGs play a pivotal role in mediating Wnt and Hh ligand
distribution, stability and ligand-receptor binding (12,14,25).
Therefore, desulfation by HSulf-1 re-expression may interfere with
Wnt and Hh signaling (14,25). Sulfated HSPGs also serve as
co-receptors for multiple growth factors and cytokines. Thus,
desulfation by HSulf-1 leads to the abrogation of several receptor
tyrosine kinase signaling pathways, particularly heparin-binding
growth factors including fibroblast growth factor 2, vascular
endothelial growth factor, hepatocyte growth factor, PDGF and
heparin-binding epidermal growth factor (15–18).
Since aberrant Hh activation has been shown to play a role in the
tumorigenesis of multiple cancers, including gastric cancer
(6–8), and Hh signaling regulates cell growth
and survival, we explored the hypothesis that HSulf-1 regulates the
activity of Hh signaling in gastric cancer cells. Notably, the
activated Gli transcription activity in MKN28 gastric cancer cells
was eliminated by HSulf-1 expression (Fig. 3A), indicating that HSulf-1 may
abrogate Hh signaling activity in gastric cancer. Furthermore, we
examined the effect of HSulf-1 on the expression of Hh pathway
target genes and observed the significant downregulation of GLI1,
PTCH1, PTCH2, HHIP, C-MYC, CCND1, FOXF1, FOXM1 and BCL2 (Fig. 3B). As Hh target genes, GLI1, PTCH1/2
and HHIP are responsible for the fine-tuning of Hh signaling via
positive and negative feedback loops (10,11,26–28).
The Hh pathway induces cell proliferation through upregulation of
C-MYC, cell cycle regulator CCND1 and FOXM1, and promotes survival
of cells via BCL2 (28–31). Collectively, the data confirmed that
inhibition of Hh signaling may be one of the major mechanisms
mediating HSulf-1-induced growth inhibition in gastric cancer
cells. However, the results do not exclude the possibility that
HSulf-1 may mediate its growth inhibition via multiple mechanisms
including modulation of the Wnt and heparin-binding growth factor
signaling pathways.
In conclusion, we demonstrated that HSulf-1 may
function as a tumor suppressor in gastric cancer, as it
significantly retarded cell growth and downregulated the Hh
signaling pathway in gastric cancer cells. Since HSulf-1
potentiates the effects of histone deacetylase (HDAC) inhibitors
(32), and its epigenetic silencing
in ovarian cancer is implicated in chemoresistance (33), strategies targeting the epigenetic
reactivation of HSulf-1 in combination with HDAC inhibitors may
prove to be useful therapeutic modalities in treating gastric
cancer (34).
Acknowledgements
This study was partially supported by funds from the
National Key Basic Research Project (No. 2007CB914401), the
National Key Basic Research and Development (973) Program of China
(No. 06CB503905) and the China Natural Science Foundation (No.
30770475).
References
|
1
|
Hartgrink HH, Jansen EP, van Grieken NC
and van de Velde CJ: Gastric cancer. Lancet. 374:477–490. 2009.
View Article : Google Scholar
|
|
2
|
Ding SZ, Goldberg JB and Hatakeyama M:
Helicobacter pylori infection, oncogenic pathways and
epigenetic mechanisms in gastric carcinogenesis. Future Oncol.
6:851–862. 2010. View Article : Google Scholar
|
|
3
|
Merchant JL, Saqui-Salces M and El-Zaatari
M: Hedgehog signaling in gastric physiology and cancer. Prog Mol
Biol Transl Sci. 96:133–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martin J, Donnelly JM, Houghton J and
Zavros Y: The role of sonic hedgehog reemergence during gastric
cancer. Dig Dis Sci. 55:1516–1524. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Varjosalo M and Taipale J: Hedgehog:
functions and mechanisms. Genes Dev. 22:2454–2472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang L, Xie G, Fan Q and Xie J: Activation
of the hedgehog-signaling pathway in human cancer and the clinical
implications. Oncogene. 29:469–481. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scales SJ and de Sauvage FJ: Mechanisms of
Hedgehog pathway activation in cancer and implications for therapy.
Trends Pharmacol Sci. 30:303–312. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma X, Chen K, Huang S, et al: Frequent
activation of the hedgehog pathway in advanced gastric
adenocarcinomas. Carcinogenesis. 26:1698–1705. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hooper JE and Scott MP: Communicating with
Hedgehogs. Nat Rev Mol Cell Biol. 6:306–317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huangfu D and Anderson KV: Signaling from
Smo to Ci/Gli: conservation and divergence of Hedgehog pathways
from Drosophila to vertebrates. Development. 133:3–14. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Osterlund T and Kogerman P: Hedgehog
signalling: how to get from Smo to Ci and Gli. Trends Cell Biol.
16:176–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin X: Functions of heparan sulfate
proteoglycans in cell signaling during development. Development.
131:6009–6021. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morimoto-Tomita M, Uchimura K, Werb Z,
Hemmerich S and Rosen SD: Cloning and characterization of two
extracellular heparin-degrading endosulfatases in mice and humans.
J Biol Chem. 277:49175–49185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bernfield M, Gotte M, Park PW, et al:
Functions of cell surface heparan sulfate proteoglycans. Annu Rev
Biochem. 68:729–777. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lai J, Chien J, Staub J, et al: Loss of
HSulf-1 up-regulates heparin-binding growth factor signaling in
cancer. J Biol Chem. 278:23107–23117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lai JP, Chien JR, Moser DR, et al: hSulf1
Sulfatase promotes apoptosis of hepatocellular cancer cells by
decreasing heparin-binding growth factor signaling.
Gastroenterology. 126:231–248. 2004. View Article : Google Scholar
|
|
17
|
Lai JP, Sandhu DS, Shire AM and Roberts
LR: The tumor suppressor function of human sulfatase 1 (SULF1) in
carcinogenesis. J Gastrointest Cancer. 39:149–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Narita K, Staub J, Chien J, et al: HSulf-1
inhibits angiogenesis and tumorigenesis in vivo. Cancer Res.
66:6025–6032. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai Y, Yang Y, MacLeod V, et al: HSulf-1
and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo.
J Biol Chem. 280:40066–40073. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lai JP, Chien J, Strome SE, et al: HSulf-1
modulates HGF-mediated tumor cell invasion and signaling in head
and neck squamous carcinoma. Oncogene. 23:1439–1447. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lai JP, Sandhu DS, Moser CD, et al:
Additive effect of apicidin and doxorubicin in sulfatase 1
expressing hepatocellular carcinoma in vitro and in vivo. J
Hepatol. 50:1112–1121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Z, Fan JQ, Li J, et al: Promoter
hypermethylation correlates with the Hsulf-1 silencing in human
breast and gastric cancer. Int J Cancer. 124:739–744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J, Kleeff J, Abiatari I, et al:
Enhanced levels of Hsulf-1 interfere with heparin-binding growth
factor signaling in pancreatic cancer. Mol Cancer. 4:142005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abiatari I, Kleeff J, Li J, Felix K,
Buchler MW and Friess H: Hsulf-1 regulates growth and invasion of
pancreatic cancer cells. J Clin Pathol. 59:1052–1058. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dhoot GK, Gustafsson MK, Ai X, Sun W,
Standiford DM and Emerson CP Jr: Regulation of Wnt signaling and
embryo patterning by an extracellular sulfatase. Science.
293:1663–1666. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chuang PT, Kawcak T and McMahon AP:
Feedback control of mammalian Hedgehog signaling by the
Hedgehog-binding protein, Hip1, modulates Fgf signaling during
branching morphogenesis of the lung. Genes Dev. 17:342–347. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chuang PT and McMahon AP: Vertebrate
Hedgehog signalling modulated by induction of a Hedgehog-binding
protein. Nature. 397:617–621. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Katoh Y and Katoh M: Hedgehog target
genes: mechanisms of carcinogenesis induced by aberrant hedgehog
signaling activation. Curr Mol Med. 9:873–886. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rao G, Pedone CA, Coffin CM, Holland EC
and Fults DW: c-Myc enhances sonic hedgehog-induced medulloblastoma
formation from nestin-expressing neural progenitors in mice.
Neoplasia. 5:198–204. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bigelow RL, Chari NS, Unden AB, et al:
Transcriptional regulation of bcl-2 mediated by the sonic hedgehog
signaling pathway through gli-1. J Biol Chem. 279:1197–1205. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lai JP, Yu C, Moser CD, et al: SULF1
inhibits tumor growth and potentiates the effects of histone
deacetylase inhibitors in hepatocellular carcinoma.
Gastroenterology. 130:2130–2144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Staub J, Chien J, Pan Y, et al: Epigenetic
silencing of HSulf-1 in ovarian cancer:implications in
chemoresistance. Oncogene. 26:4969–4978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lai JP, Thompson JR, Sandhu DS and Roberts
LR: Heparin-degrading sulfatases in hepatocellular carcinoma: roles
in pathogenesis and therapy targets. Future Oncol. 4:803–814. 2008.
View Article : Google Scholar : PubMed/NCBI
|