Introduction
Ewing’s sarcoma (ES) is the second most common bone
tumor after osteosarcoma in children and adolescents. ES are
aggressive tumors with a tendency towards recurrence following
resection and pronounced proclivity toward early hematogenous
metastases to lungs and bone. No hereditary or congenital
syndromes, environmental or known risk factors have been associated
with the occurrence of ES. In 90% of cases, Ewing’s sarcoma family
tumor (ESFT) cells harbor the translocation t(11;22)(q24;q12), and
in the remaining 10% the variant translocation is t(21;12)(22;12)
(1,2). Although peak incidence occurs between
the ages of 10 and 20 years, patients of younger or older ages
account for almost 30% of the cases (3). Poor prognostic factors include tumor
≥8 cm, pelvic primary, presence of metastases and age >15 at the
time of diagnosis (4). Older
patients with sarcoma also have a higher risk of thromboembolism
(5).
Case report
A 52-year-old healthy female presented with a 2-week
history of pain in her right posterior thigh. The pain originated
in the patient’s right buttock and radiated to the back of the knee
without a radicular component. Motor strength, sensory function and
reflexes were normal. Musculoskeletal examination was within normal
limits without tenderness of the hips or back.
Despite the use of non-steroidal anti-inflammatory
medications and physical therapy the pain persisted. On subsequent
reassessment, a radicular component was present. Magnetic resonance
imaging (MRI) demonstrated an irregular-shaped right presacral mass
with heterogeneous short T1 inversion recovery (STIR)
hyperintensity (Fig. 1A) and T1
hypointensity (Fig. 1B). Additional
evaluation with computed tomography (CT) of the pelvis revealed a
complex mass with central necrosis and a thick enhancing wall
extending into the right S2 neural foramen with lytic bone
destruction (Fig. 1C) measuring
6×6×5 cm with compression of the right piriformis muscle
posteriorly (Fig. 1D). No
adenopathy or correlation to gynecologic structures of the pelvis
were found.
An initial core needle biopsy of the mass revealed a
small, round blue cell neoplasm, suggestive of a primitive
neuroectodermal tumor. The tumor was markedly positive for vimentin
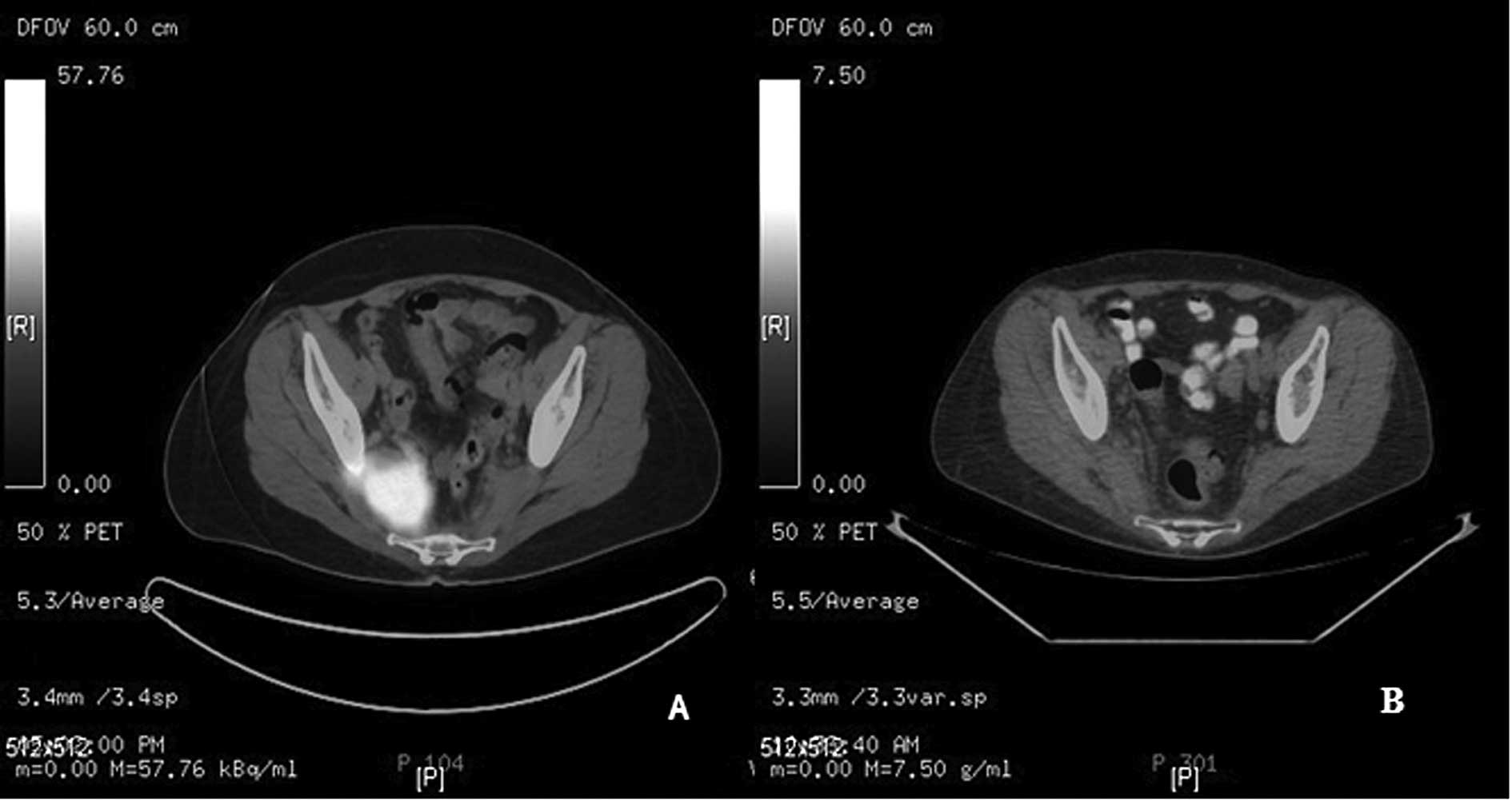
(Fig. 2A) and CD99 (Fig. 2B). Staging workup with a positron
emission tomography-computed tomography (PET-CT)image (Fig. 3A) demonstrated a large avid
fluorodeoxyglucose (FDG) uptake at the level of the lower sacrum
measuring approximately 5.8 cm. There was no other area of
abnormally increased uptake of FDG in the whole body PET-CT. An
open biopsy revealed a better preserved tumor with identical
histological features testing positive for the ES translocation
(22q12), and a diagnosis of ES stage III (T2bN0M0) was made.
The patient was treated with 17 cycles of
vincristine 2 mg/m2, doxorubicine 75 mg/m2,
cyclophosphamide 1200 mg/m2 with mesna rescue
alternating with ifosfamide 1800 mg/m2 and etoposide 100
mg/m2 (IE) as described by Grier et al (6). Re-assessment after four cycles
demonstrated a significant response. The involvement of S1 and S2
nerve roots represented a significant surgical challenge. As a
result, the patient received external beam radiation at a dose of
56 Gy followed by 13 additional cycles of adjuvant chemotherapy.
Post-treatment CT of the pelvis demonstrated a near complete
resolution of the mass with minimal residual infiltrative soft
tissue changes within the right presacral space. PET imaging
demonstrated normalization of FDG uptake within the pelvic mass
(Fig. 3B). Six weeks post-adjuvant
chemotherapy, the patient presented with acute shortness of breath.
PET-CT demonstrated an area of increased FDG uptake within the
bilateral pulmonary hila, representing interval development of
metastatic disease. A subsequent biopsy of the lung confirmed the
diagnosis of metastatic ES. Two weeks later the patient succumbed
to the disease after opting for comfort care.
Discussion
The mortality rate for ES is extremely high when
treated with surgery or radiation therapy alone for local control
of the disease (7). Over the past 5
decades, advances in chemotherapy, surgery and radiation therapy
have improved the prognosis of patients with ESFTs (8). The limited number of patients over the
age of 40 and the exclusion of these patients from the majority of
trials render these findings difficult to generalize (9).
Tumors of the pelvis have a poorer prognosis when
compared with other sites. Whether this is related to the challenge
of achieving local control or the proximity to critical deep
structures remains to be elucidated. Current treatment
recommendations are based on the available literature that is
limited by selection bias, small study size, non-standard radiation
therapy technique and lack of randomized trials comparing the two
management approaches. The role of surgery in treating ES is
controversial; however, certain studies suggest that resection with
chemotherapy and irradiation positively impacts patient survival
(10). Radiotherapy is usually
applied at doses of 40–45 Gy for microscopic residues and 50–60 Gy
for macroscopic disease (11).
Treatment of adult patients follows the same principles. However,
tolerability of therapies in adults are taken into account when
transferring treatment protocols conceived for patients under the
age of 30 years (11).
Although most cases of ES present as localized
disease, overt metastases are capable of developing rapidly.
Microscopic metastatic disease has been postulated to be present at
the time of presentation. However, its spread is held in check by
as yet unidentified factors secreted by the primary tumor. When the
primary tumor is removed or irradiated, the loss of the putative
suppressive factors may permit the metastases to grow. The use of
chemotherapy in conjunction with surgery or radiation therapy to
treat presumed metastatic disease has substantially improved
survival (12).
Beyond specific clinical trials, patients with
metastatic disease receive similar therapy to that administered for
localized disease, with appropriate local treatment of metastases,
usually radiotherapy. Certain studies have suggested benefit from
intensive chemotherapy followed by autologous stem cell rescue, but
randomized trials have not yet been performed and the benefit of
stem cell transplant remains unproven. Patients with recurrent
disease fare poorly, with 5-year survival rates of less than 20%
(11). Current studies show that,
following achieving remission in patients with non-metastatic ES,
30–40% of these patients are likely to develop recurrence of local
or metastatic disease (13). The
majority of these studies report a time range of 2–10 years between
commencing treatment and development of recurrence (14). Patients relapsing later than 2 years
from initial diagnosis have more favorable outcomes (11).
This case report raises the importance of early
metastasis in a treatment-responsive patient. According to current
guidelines, initial work-up for staging in a non-metastatic ES is
followed by reassessment of treatment response after 3–6 treatment
cycles using focal PET-CT. In treatment responsive ES, local
therapy is followed by additional chemotherapy. A surveillance
follow-up imaging every 2–3 months for the first three years is
recommended for localized, non-metastatic ES. The current case has
demonstrated complex issues with localized pelvic ES in an older
patient who initially responded well to chemo-radiation therapy,
with complete resolution of the tumor. Despite adequate control of
the local disease, multimodal therapy did not appear to affect
metastasis. Although sarcomas are notorious for metastasis into
lungs and bone, the utility of PET-CT and MRI for detecting
subclinical recurrence or metastases has not been established
during the treatment period. Only the primary site is evaluated by
imaging for treatment response 10–12 weeks in to therapy.
In patients with lung metastases, the resection of
residual metastases after chemotherapy, and whole lung irradiation,
may grant a survival advantage (11). Most intensive therapies with
additional agents have failed to markedly increase long-term
survival in patients with metastatic disease. The impact of
chemotherapy on metastasis of ES patients over the age of 30
remains to be elucidated. A recent intergroup study suggested that
the addition of IE to traditional regimens may confer a local
control benefit (15). Similar
effects, with the addition of IE on survival benefit, have also
been reported by another randomized trial among patients with
non-metastatic pelvic ES (6).
At present, patients should be offered participation
in a clinical trial when available. Barring trial participation,
multimodality therapy, as described above is recommended.
References
|
1
|
Grier HE: The Ewing family of tumors.
Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr Clin
North Am. 44:991–1004. 1997.
|
|
2
|
McManus AP, Gusterson BA, Pinkerton CR and
Shipley JM: The molecular pathology of small round-cell tumours –
relevance to diagnosis, prognosis, and classification. J Pathol.
178:116–121. 1996.
|
|
3
|
Stiller CA, Bielack SS, Jundt G and
Steliarova-Foucher E: Bone tumours in European children and
adolescents, 1978–1997 report from the Automated Childhood Cancer
Information System project. Eur J Cancer. 42:2124–2135. 2006.
|
|
4
|
Leavey PJ, Mascarenhas L, Marina N, Chen
Z, Krailo M, Miser J, Brown K, Tarbell N, Bernstein ML, Granowetter
L, Gebhardt M and Grier HE; Children’s Oncology Group. Prognostic
factors for patients with Ewing sarcoma (EWS) at first recurrence
following multi-modality therapy: a report from the Children’s
Oncology Group. Pediatr Blood Cancer. 51:334–338. 2008.PubMed/NCBI
|
|
5
|
Athale U, Cox S, Siciliano S and Chan AK:
Thromboembolism in children with sarcoma. Pediatr Blood Cancer.
49:171–176. 2007. View Article : Google Scholar
|
|
6
|
Grier HE, Krailo MD, Tarbell NJ, Link MP,
Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers
PA, Donaldson SS, Moore S, Rausen AR, Vietti TJ and Miser JS:
Addition of ifosfamide and etoposide to standard chemotherapy for
Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl
J Med. 348:694–701. 2003.
|
|
7
|
Nesbit ME: Ewing’s sarcoma. CA Cancer J
Clin. 26:174–180. 1976.
|
|
8
|
Indelicato DJ, Keole SR, Shahlaee AH, Shi
W, Morris CG, Gibbs CP Jr, Scarborough MT and Marcus RB Jr: Impact
of local management on long-term outcomes in Ewing tumors of the
pelvis and sacral bones: the University of Florida experience. Int
J Radiat Oncol Biol Phys. 72:41–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bacci G, Balladelli A, Forni C, Ferrari S,
Longhi A, Benassi MS, Briccoli A, Serra M and Picci P: Adjuvant and
neoadjuvant chemotherapy for Ewing sarcoma family tumors in
patients aged between 40 and 60: report of 35 cases and comparison
of results with 586 younger patients treated with the same
protocols in the same years. Cancer. 109:780–786. 2007.
|
|
10
|
Sciubba DM, Petteys RJ, Garces-Ambrossi
GL, Noggle JC, McGirt MJ, Wolinsky JP, Witham TF and Gokaslan ZL:
Diagnosis and management of sacral tumors. J Neurosurg Spine.
10:244–256. 2009. View Article : Google Scholar
|
|
11
|
Paulussen M, Bielack S, Jurgens H and
Casali PG; ESMO Guidelines Working Group. Ewing’s sarcoma of the
bone: ESMO clinical recommendations for diagnosis, treatment and
follow-up. Ann Oncol. 20(Suppl 4): 140–142. 2009.
|
|
12
|
Skubitz KM and D’Adamo DR: Sarcoma. Mayo
Clin Proc. 82:1409–1432. 2007. View Article : Google Scholar
|
|
13
|
Bacci G, Ferrari S, Longhi A, Donati D, De
Paolis M, Forni C, Versari M, Setola E, Briccoli A and Barbieri E:
Therapy and survival after recurrence of Ewing’s tumors: the
Rizzoli experience in 195 patients treated with adjuvant and
neoadjuvant chemotherapy from 1979 to 1997. Ann Oncol.
14:1654–1659. 2003.
|
|
14
|
Hanna SA, David LA, Gikas PD, Tindall AJ,
Cannon SR and Briggs TW: Very late local recurrence of Ewing’s
sarcoma – can you ever say ‘cured’? A report of two cases and
literature review. Ann R Coll Surg Engl. 90:W12–W15. 2008.
|
|
15
|
Yock TI, Krailo M, Fryer CJ, Donaldson SS,
Miser JS, Chen Z, Bernstein M, Laurie F, Gebhardt MZ, Grier HE and
Tarbell NJ; Children’s Oncology Group. Local control in pelvic
Ewing sarcoma: analysis from INT-0091 – a report from the
Children’s Oncology Group. J Clin Oncol. 24:3838–3843.
2006.PubMed/NCBI
|