Introduction
Desmoplastic small round cell tumor (DSRCT) is a
rare, aggressive, malignant tumor that predominantly affects young
males at a median age of 19 years (range 7–58) and with a
male-to-female ratio ranging from 5:1 to 10:1 (1,2). DSRCT
is a member of the small round blue cell tumor family, which
includes small-cell carcinoma, Merkel cell carcinoma, synovial
sarcoma, Ewing’s sarcoma/primitive neuroectodermal tumor,
neuroblastoma, lymphoma, rhabdomyosarcoma and DSRCT (3). No standard therapy is currently
available for patients with DSRCT and the prognosis of DSRCT
remains extremely poor (2). In this
study, we report a case of long-term survival of metastatic DSRCT
treated with multimodal therapy, including multiagent chemotherapy,
radiation therapy and therapeutic endoscopy.
Case history
A 24-year-old male was admitted to our hospital with
a chief complaint of hematemesis. The patient had neither
significant medical history nor family history. Physical
examination revealed only that the patient was anemic. Laboratory
examination was as follows: hemoglobin, 11.4 g/dl (normal range
13.5–16.7 g/dl); white blood cell count, 6,200/dl (normal range
2,900–8,900/dl); platelet count, 13.2×1010/dl (normal
range, 15.9–38.9×1010/dl); C-reactive protein, 0.1 mg/dl
(normal value ≤0.2 mg/dl); aspartate aminotransferase, 52 IU/l
(normal range 13–33 IU/l); alanine aminotransferase, 99 IU/l
(normal range 8–42 IU/l); alkaline phosphatase, 588 IU/l (normal
range 115–359 IU/l); γ-glutamyl transpeptidase, 380 IU/l (normal
range 9–54 IU/l); and total bilirubin, 0.7 mg/dl (normal range
0.3–1.3 mg/dl). Renal function tests were normal.
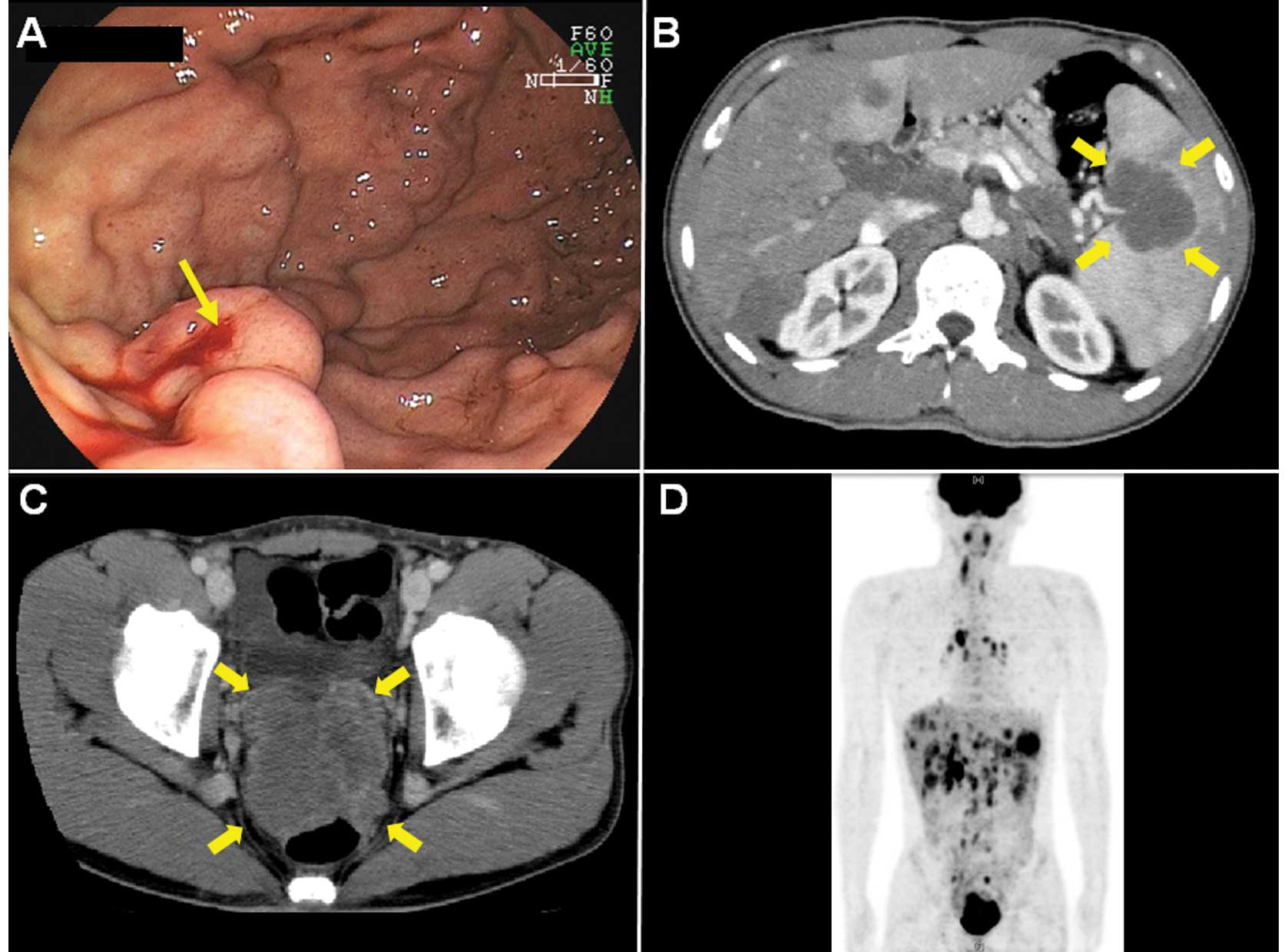
Endoscopy showed oozing bleeding from varicose veins
located on the greater curvature of the upper gastric body
(Fig. 1A). Spontaneous hemostasis
was obtained. Computed tomography (CT) demonstrated that
compression of the splenic vein by the splenic hilar tumor appeared
to cause the gastric varices (Fig.
1B). CT revealed the presence of a well-enhanced, bulky and
lobulated mass on the pelvic floor (Fig. 1C) with a splenic hilar tumor,
multiple liver and lung tumors, and marked lymph node swellings
(particularly in the hepatic portal region).
18F-fluorodeoxyglucose positron emission tomography
showed multiple accumulation of a glucose analog in the same
lesions detected using CT (Fig.
1D).
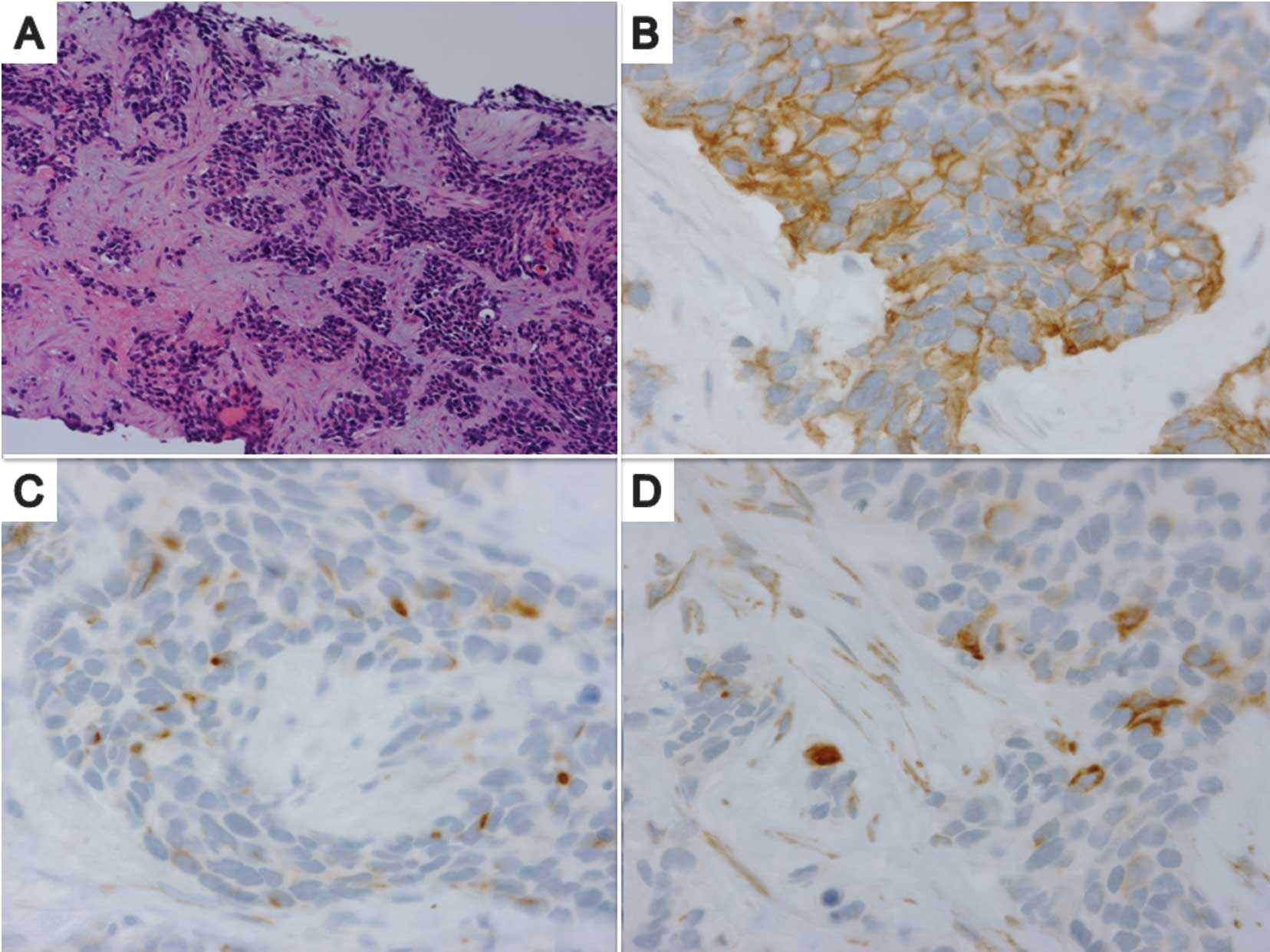
A needle biopsy specimen from the liver tumor
revealed the presence of a poorly differentiated tumor with a
variable size and shape, composed of nests of small round cells
surrounded by a prominent desmoplastic stroma (Fig. 2A). Immunohistochemically, tumor
cells coexpressed an epithelial marker (cytokeratin, Fig. 2B), a mesenchymal marker (desmin,
Fig. 2C) and the Wilms’ tumor 1
protein (Fig. 2D). Chromogranin,
cluster of differentiation antigen (CD) 99 and CD56 were negative.
From these findings, the patient was provided with a definite
diagnosis of pelvic cavity-origin DSRCT with multiple-organ
metastases (4,5).
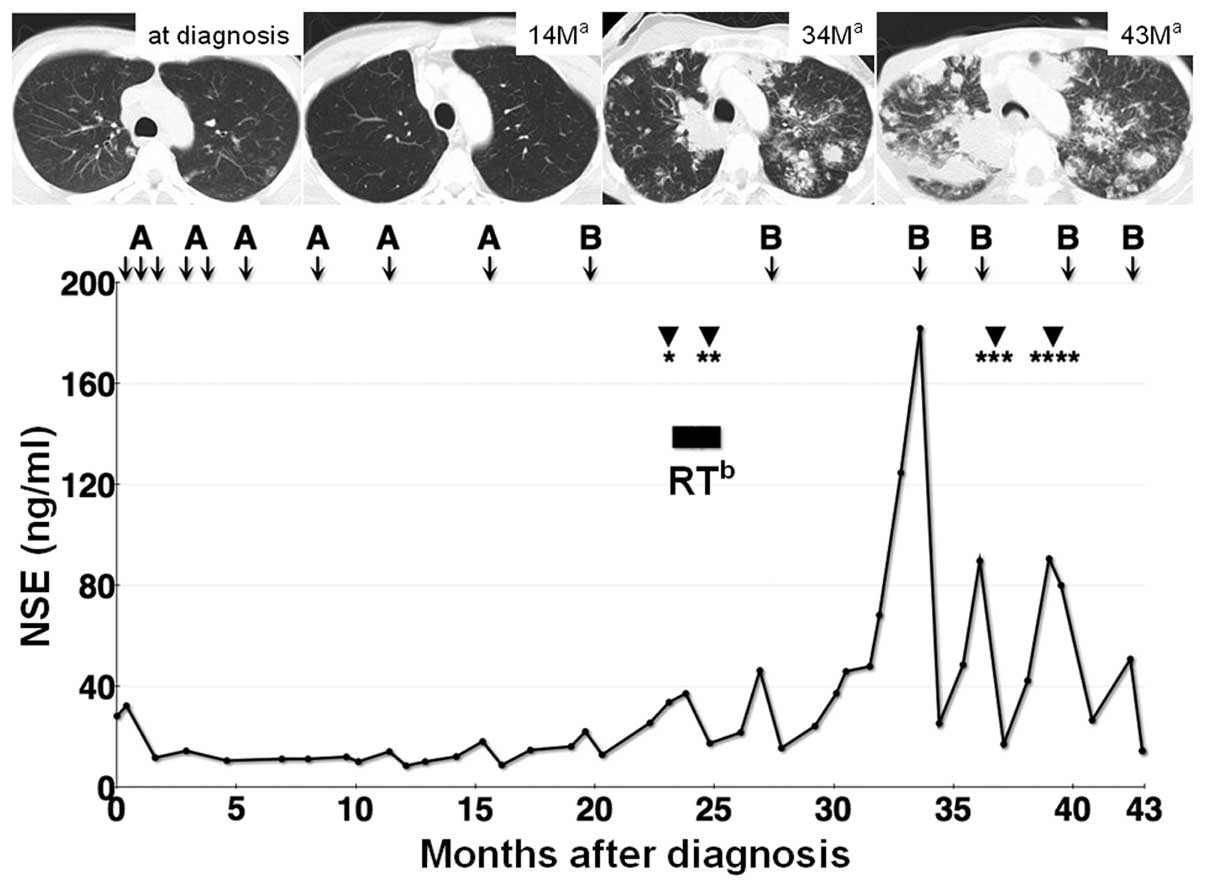
The clinical course of this case is shown in
Fig. 3. The patient was initially
treated with multiagent chemotherapy using cyclophosphamide,
pirarubicin, vincristine, ifosfamide and etoposide, according to
the Ewing’s sarcoma protocol, which is a modified protocol of the
P6 regimen using pirarubicin instead of doxorubicin (modified P6
regimen) (2,6). During each course of this
chemotherapy, the patient suffered from severe nausea and vomiting.
The patient required frequent blood transfusions and continuous use
of granulocyte colony-stimulating factor due to severe bone marrow
suppression. The multiple pulmonary metastases were almost
eradicated following four courses of the modified P6 regimen and
the patient reached 15 months of progression-free survival after
the application of this modified P6 regimen (Fig. 3). After the nine courses of
treatment, second-line chemotherapy based on the PAVEP regimen
(doxorubicin, cyclophosphamide, etoposide and cisplatin) (7) was introduced due to disease
progression. To reduce adverse events, we modified the PAVEP
regimen by using pirarubicin instead of doxorubicin and shortening
the period of the regimen from five to two days (modified PAVEP
regimen). The P6, modified P6, PAVEP and modified PAVEP regimens
are shown in Table I.
 | Table IChemotherapy regimens reported
previously and used in this case. |
Table I
Chemotherapy regimens reported
previously and used in this case.
| Dose | Day |
|---|
| P6 (6) |
| Courses 1, 2, 3 and
6 |
|
Cyclophosphamide | 2.1
g/m2 | 1–2 |
| Doxorubicin | 25
mg/m2 | 1–3 |
| Vincristine | 0.67
mg/m2 | 1–3 |
| Courses 4, 5 and
7 |
| Ifosfamide | 1.8
g/m2 | 1–5 |
| Etoposide | 100
mg/m2 | 1–5 |
| PAVEP (7) |
| Doxorubicin | 40
mg/m2 | 1 |
|
Cyclophosphamide | 300
mg/m2 | 1–3 |
| Etoposide | 75
mg/m2 | 1–3 |
| Cisplatin | 100
mg/m2 | 4 |
| Modified P6 |
| Courses 1, 3, 5 and
7 |
|
Cyclophosphamide | 2
g/m2 | 1–2 |
| Pirarubicin | 20
mg/m2 | 1–3 |
| Vincristine | 2
mg/m2 | 1 |
| Courses 2, 4 and
6 |
| Ifosfamide | 2.5
g/m2 | 1–5 |
| Etoposide | 100
mg/m2 | 1–5 |
| Modified PAVEP |
| Pirarubicin | 40
mg/m2 | 2 |
|
Cyclophosphamide | 450
mg/m2 | 1–2 |
| Etoposide | 110
mg/m2 | 1–2 |
| Cisplatin | 100
mg/m2 | 1 |
Obstructive jaundice caused by portal
lymphadenopathy developed 23 months following diagnosis. Endoscopic
biliary drainage using a plastic stent was successfully performed.
However, stent obstruction occurred two months after the initial
placement of the plastic stent. Subsequently, we removed the stent
and inserted a metal stent, which was followed by irradiation of
the hepatic portal region using a total dose of 42.5 Gy, at 1.8 Gy
per fraction. The patient had massive hematemesis 37 months
following diagnosis caused by the active bleeding from a known
varicose vein and endoscopic hemostasis using metal clips was
obtained successfully. Furthermore, metal stent obstruction caused
by tumor ingrowth occurred 39 months following diagnosis, which was
relieved by inserting a plastic stent into the metal stent placed
previously. After 15 courses of chemotherapy, radiation therapy and
four instances of endoscopic therapy (Fig. 3), the patient experienced sudden
respiratory arrest caused by bronchial obstruction by the
parabronchial multiple lung metastatic lesions. Despite intensive
care, including intubation and one course of chemotherapy, the
patient succumbed to the disease 43 months after diagnosis.
Discussion
DSRCT was first described in 1989 by Gerald and
Rosai (8). DSRCT is a rare
aggressive tumor with few long-term survivors and the prognosis of
patients with DSRCT has not improved substantially since the first
description of the disease. The rarity of this tumor has prevented
the development of standard therapy for DSRCT. DSRCT has been
reported as being associated with a characteristic reciprocal
translocation [t(11; 22)(p13; q12)], which fuses the Ewing’s
sarcoma gene on chromosome 22 to the Wilms’ tumor gene on
chromosome 11 (9). This
translocation is reflected by the immunohistological expression of
the Wilms’ tumor 1 protein in the tumor (10). The neuron-specific enolase (NSE) and
CA125 proteins are tumor markers that are elevated in DSRCT
patients and correlate specifically with response to treatment
(11). The serum NSE levels in our
case correlated well with clinical response, as shown in Fig. 3. The prolonged (15 months)
progression-free survival obtained following multiagent
chemotherapy suggests that DSRCT is a chemosensitive disease, as
mentioned in previous studies (1,12).
Efforts have been made to establish treatments aimed
at controlling this disease. One of them was the combination of
aggressive surgery (to resect visible disease), radiation therapy
to the tumor bed and myeloablative multiagent chemotherapy
(6). A recent report from the
Memorial Sloan-Kettering Cancer Center experience (2) showed that overall survival in 66
patients was 44% at three years and 15% at five years using a
combination of the P6 regimen, surgical debulking and radiotherapy
to a dose of 30 Gy. However, more than half of these patients had
no distant metastasis. Currently, there is no standard therapy for
patients with DSRCT, particularly for inoperable/metastatic DSRCT
cases, and there are few reports of metastatic DSRCT treatment
(13). To the best of our
knowledge, this is the first case of metastatic DSRCT in a patient
who lived for more than three years without surgical resection
(14). Multi-institutional
randomized control trials for DSRCT are not available due to the
rarity of the disease. Another attempt at developing treatments for
this disease was the use of a novel molecularly targeted therapy
and a new chemical agent (15). We
examined the expression of c-kit, androgen receptor, CD20 and
epidermal growth factor receptor using a biopsy sample (16,17).
However, we found that none of the markers were expressed in tumor
cells; therefore, we could not use any molecularly targeted agent
in this case.
In this study, we used pirarubicin instead of
doxorubicin in the P6 regimen administered to this patient, as
pirarubicin may be relatively superior to doxorubicin regarding
side effects (18). Moreover, the
use of pirarubicin in a multiagent regimen for DSRCT was previously
reported (6,7). Although our study shows only one case
of treatment for metastatic DSRCT, the two modified regimens
selected for our case may represent a treatment modality for DSRCT
patients that has the potential advantages of decreased toxicity
and improved completion rate of the chemotherapy regimen.
In conclusion, DSRCT is an aggressive but
chemosensitive disease and continuous chemotherapy using an
appropriate regimen with possible supportive care is essential for
the long-term survival of these patients. This case report may
present a treatment option for this rare disease.
Acknowledgements
We are grateful to the patient and his family for
their permission to report our experience with his case.
References
|
1
|
Gerald WL, Miller HK, Battifora H,
Miettinen M, Silva EG and Rosai J: Intra-abdominal desmoplastic
small round-cell tumor. Report of 19 cases of a distinctive type of
high-grade polyphenotypic malignancy affecting young individuals.
Am J Surg Pathol. 15:499–513. 1991. View Article : Google Scholar
|
|
2
|
Lal DR, Su WT, Wolden SL, Loh KC, Modak S
and LaQuaglia MP: Results of multimodal treatment for desmoplastic
small round cell tumors. J Pediatr Surg. 40:251–255. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gerald WL, Ladanyi M, de Alava E,
Cuatrecasas M, Kushner BH, LaQuaglia MP and Rosai J: Clinical,
pathologic, and molecular spectrum of tumors associated with
t(11;22)(p13;q12): desmoplastic small round-cell tumor and its
variants. J Clin Oncol. 16:3028–3036. 1998.PubMed/NCBI
|
|
4
|
Chang F: Desmoplastic small round cell
tumors: cytologic, histologic, and immunohistochemical features.
Arch Pathol Lab Med. 130:728–732. 2006.PubMed/NCBI
|
|
5
|
Zhang PJ, Goldblum JR, Pawel BR, Fisher C,
Pasha TL and Barr FG: Immunophenotype of desmoplastic small round
cell tumors as detected in cases with EWS-WT1 gene fusion product.
Mod Pathol. 16:229–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kushner BH, LaQuaglia MP, Wollner N, et
al: Desmoplastic small round-cell tumor: prolonged progression-free
survival with aggressive multimodality therapy. J Clin Oncol.
14:1526–1531. 1996.PubMed/NCBI
|
|
7
|
Farhat F, Culine S, Lhommé C, et al:
Desmoplastic small round cell tumors: results of a four-drug
chemotherapy regimen in five adult patients. Cancer. 77:1363–1366.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gerald WL and Rosai J: Case 2.
Desmoplastic small cell tumor with divergent differentiation.
Pediatr Pathol. 9:177–183. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ladanyi M and Gerald W: Fusion of the EWS
and WT1 genes in the desmoplastic small round cell tumor. Cancer
Res. 54:2837–2840. 1994.PubMed/NCBI
|
|
10
|
Hill DA, Pfeifer JD, Marley EF, Dehner LP,
Humphrey PA, Zhu X and Swanson PE: WT1 staining reliably
differentiates desmoplastic small round cell tumor from Ewing
sarcoma/primitive neuroectodermal tumor. An immunohistochemical and
molecular diagnostic study. Am J Clin Pathol. 114:345–353.
2000.
|
|
11
|
Fizazi K, Farhat F, Theodore C, Rixe O, Le
Cesne A, Comoy E and Le Chevalier T: Ca125 and neuron-specific
enolase (NSE) as tumour markers for intra-abdominal desmoplastic
small round-cell tumours. Br J Cancer. 75:76–78. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Al Balushi Z, Bulduc S, Mulleur C and
Lallier M: Desmoplastic small round cell tumor in children: a new
therapeutic approach. J Pediatr Surg. 44:949–952. 2009.PubMed/NCBI
|
|
13
|
Mrabti H, Kaikani W, Ahbeddou N, Abahssain
H, El Khannoussi B, Amrani M and Errihani H: Metastatic
desmoplastic small round cell tumor controlled by an
anthracycline-based regimen: review of the role of chemotherapy. J
Gastrointest Cancer. 8–February;2011.[Epub ahead of print].
|
|
14
|
Miwa S, Kitamura S, Shirai T, et al:
Desmoplastic small round cell tumour successfully treated with
caffeine-assisted chemotherapy: a case report and review of the
literature. Anticancer Res. 30:3769–3774. 2010.PubMed/NCBI
|
|
15
|
Thijs AM, van der Graaf WT and van Herpen
CM: Temsirolimus for metastatic desmoplastic small round cell
tumor. Pediatr Blood Cancer. 55:1431–1432. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fine RL, Shah SS, Moulton TA, et al:
Androgen and c-Kit receptors in desmoplastic small round cell
tumors resistant to chemotherapy: novel targets for therapy. Cancer
Chemother Pharmacol. 59:429–437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sankhala KK and Chawla SP: Review:
desmoplastic small round cell tumor: current treatment approach and
role of targeted therapy. Clin Adv Hematol Oncol. 7:476–478.
2009.PubMed/NCBI
|
|
18
|
Zhai L, Guo C, Cao Y, et al: Long-term
results of pirarubicin versus doxorubicin in combination
chemotherapy for aggressive non-Hodgkin’s lymphoma: single center,
15-year experience. Int J Hematol. 91:78–86. 2010.
|