Introduction
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is
characterized by congenital aplasia of the uterus and the upper two
thirds of the vagina in females demonstrating normal development of
secondary sexual characteristics and a normal 46, XX karyotype.
MRKH may be isolated but it is more frequently associated with
renal, vertebral and, to a lesser extent, auditory and cardiac
defects. The first indication of MRKH syndrome is a primary
amenorrhea in young females that otherwise present with the normal
development of secondary sexual characteristics and normal external
genitalia, with normal and functional ovaries (1).
The supernumerary ovary is an ectopic ovary that is
not connected to the utero-ovarian, broad or infundibulopelvic
ligaments. This is one of the rarest gynecological abnormalities
(2). A supernumerary ovary in MRKH
syndrome is extremely rare. A search of Google Scholar, Medscape
and PubMed revealed only one case with a benign tumor in the
literature (3). Therefore, we
describe the first case of cancer of the supernumerary ovary.
Written informed patient consent was obtained from the patient.
Case report
Patient
A 31-year-old nullipara was admitted to the Korea
University Anam Hospital, Korea University College of Medicine
(Seoul, Korea) with a 2-week history of intermittent pain in the
lower abdomen and back. On ultrasound examination, an 11.8×8.3-cm
cauliflower-like mass was noted on the left side of the pelvic
cavity. A computed tomography (CT) scan revealed an ill-defined,
irregular soft tissue mass with extensive calcification in the
pelvic cavity.
The patient had been admitted to our hospital 4
years previously with chief complaints of primary infertility and
amenorrhea. On admission, the patient was observed to be
age-appropriate with normal development of the breasts, pubic hair
and external genitalia. The karyotype was normal 46, XX. A
diagnostic laparoscopy revealed an absent upper vagina and two
irregularly shaped uterine bodies positioned near each of the
infundibulopelvic ligaments. Both ovaries were normal in
appearance. A hypoplastic and non-functioning left kidney was
diagnosed, as the left kidney was not visualized in the renal scan.
Additionally, a scoliotic change in the thoracic spine was observed
by a chest X-ray. After two months, the patient underwent an
Abbé-McIndoe procedure.
At the time of the current admission, the laboratory
results were within the normal limits, with the exception of an
elevated CA 125 level of 1,870.0 IU/ml. During surgery, a 25-cm
pelvic mass was observed to occupy the left pelvic cavity. Multiple
peritoneal seeding, omental invasion and rectal wall adhesions were
detected. Following adhesiolysis, the bilateral ovaries and
salpinges were demonstrated to be slightly enlarged, but normal in
their contour and position. The uterus had enlarged since the
previous surgery. A pelvic exenteration, omentectomy, sigmoid colon
resection en-bloc, bilateral pelvic lymph node dissection and right
external iliac node sampling were performed with a peritoneal
washing cytology.
Both ovaries exhibited normal histological features,
but with numerous carcinoma cell implants on their surface.
Furthermore, the pelvic mass itself contained areas of well-defined
ovarian tissue with cystic follicles. This latter finding and the
immunohistochemistry results revealed that a serous tumor had
developed from a third ovary, as opposed to the mesothelium.
The patient received post-operative chemotherapy.
Sixteen months post-surgery, the patient was stable and did not
demonstrate signs of recurrence.
Pathological findings
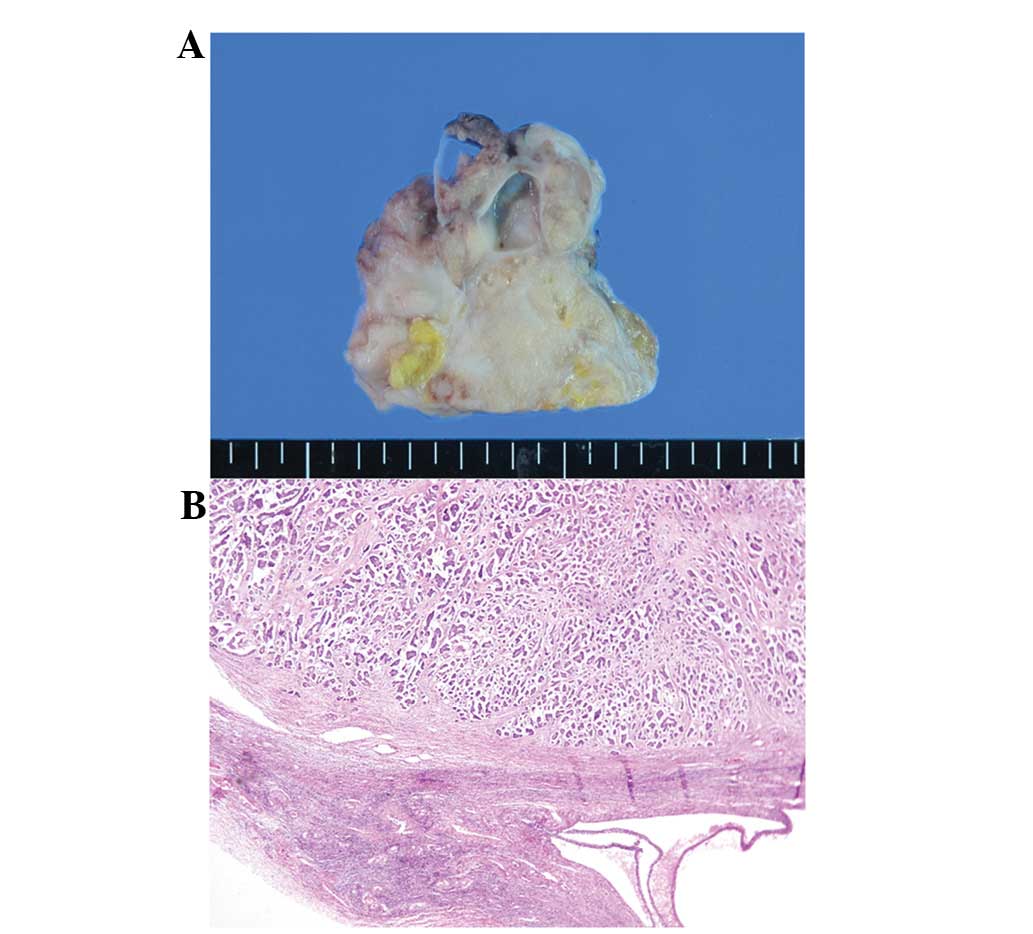
The en-bloc resected pelvic mass consisted of an
ill-defined tumor mass, a segmentallyresected rectum and duplicate
uterine bodies with attached bilateral adnexae (Fig. 1). On dissection, the right uterus
measured 4.7×2.0×1.5 cm, the attached ovary measured 8.×1.8×0.5 cm,
and the salpinx measured 16 cm in length and 0.5 cm in diameter.
The left uterus measured 4.7×3.7×2.0 cm, the attached ovary
measured 7.0×2.3×1.5 cm, and the salpinx measured 7.2 cm in length
and 0.5 cm in diameter. The serosal surface of the uterus, both
ovaries and the mesosalpinx had multiple fungating nodules; the
largest of which was 1.3 cm in diameter. Both uteri had separate
uterine cavities and blind ends. The endometrial cavities of the
right and left uteri measured 1.5 and 1.1 cm, respectively. The
poorly demarcated yellow-gray tumor mass, measuring 8.0×7.0×5.0 cm,
invaded the uterine and rectal walls. The lump of omental tissue
measured 38.0× 5.4×2.0 cm and had multiple granular mass-like
lesions.
Histologically, the main tumor in the peritoneum was
a serous papillary carcinoma, characterized by the thin or complex
papillary growth of cuboidal-to-columnar serous-type cells.
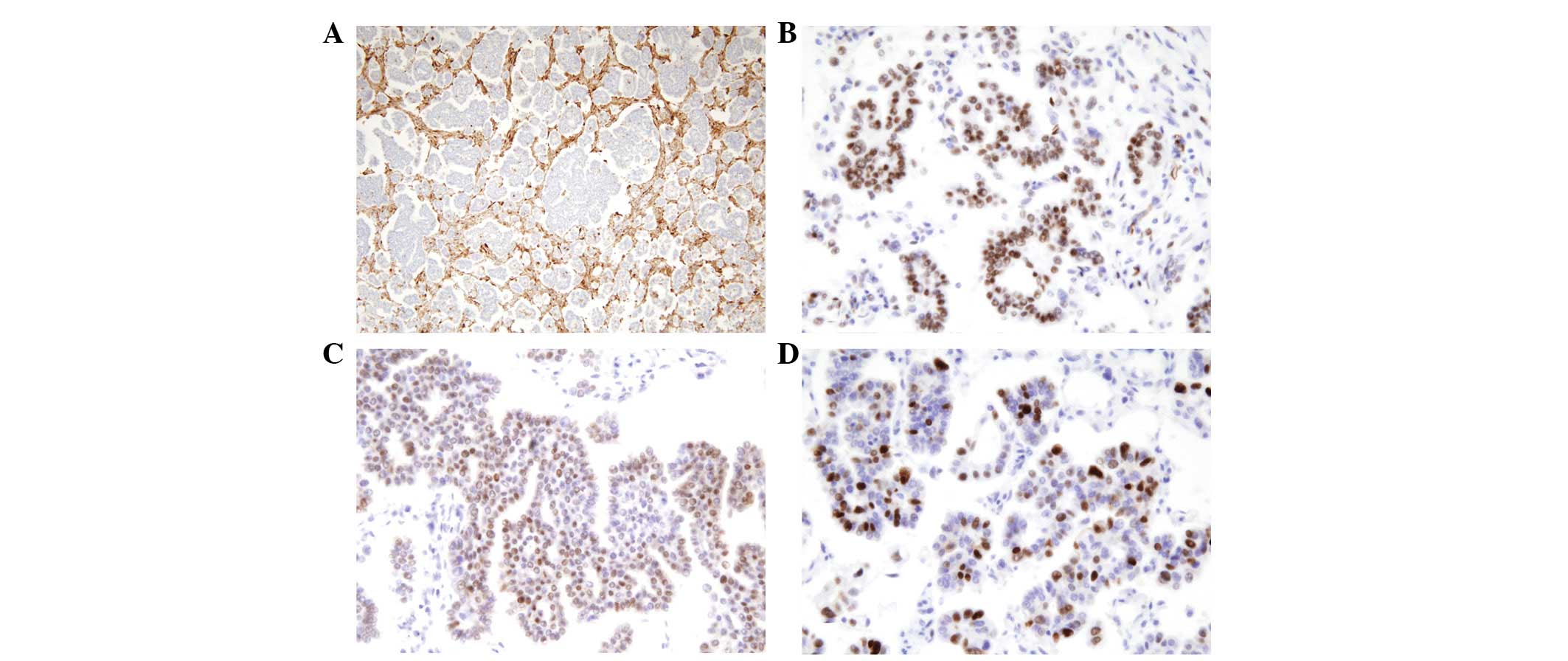
Numerous psammoma bodies were noted (Figs. 1B and C). The tumor cells stained
negative for mesothelial cell markers (D2-40 and calretinin), but
positive for the Wilms' tumor-1 (WT-1) antibody, the estrogen
receptor (ER) and the progesterone receptor (PR) in the
immunohistochemical stains. Ki-67 labeling was detected in 60–70%
of tumor cells, while p53 was expressed in numerous tumor cells
(Fig. 2A–D). The well-defined
ovarian tissue containing cystic follicles and corpora albicantia
demonstrated tumor involvement in multiple sections of the pelvic
mass (Fig. 3A). Both ovaries
exhibited normal histological features, but had multiple deposits
of serous papillary carcinoma on the surface and superficial cortex
(Fig. 3B). The mesosalpinx also
contained tumor deposits.
Both uteri had the proliferative phase of the
endometrium, and adenomyosis was present in the right-side of the
uterus.
Discussion
MRKH syndrome, first described by Mayer and further
studied by Rokitansky, Küster and Hauser, is one spectrum of
Müllerian anomalies originally characterized by the congenital
absence of a uterus and vagina in genotypic and phenotypic females
with a normal endocrine status (4,5). MRKH
syndrome affects 1 in 4,000 live female births, and is the second
most common cause of primary amenorrhea (6).
In isolated MRKH syndrome (type I), the Fallopian
tubes and ovaries are usually present, as well as variable degrees
of uterine or vaginal aplasia. Type II MRKH syndrome includes other
non-Müllerian anomalies, such as urinary tract and cervicothoracic
disorders, and hearing defects (7).
The evaluation of renal defects is mandatory, as anomalies such as
the hypoplastic and non-functioning left kidney in the study by
Capero and Gallego are common (6).
The patient in the present case also showed a scoliotic change in
the thoracic spine; however, a supernumerary ovary is an extremely
unusual accompanying anomaly with type II MRKH syndrome.
During surgery, the pelvic mass was suggested to be
a primary peritoneal carcinoma, as the ovaries had been defined to
be morphologically normal. The primary peritoneal serous carcinoma
was histologically indistinguishable from a primary ovarian serous
tumor. We propose that malignant mesothelioma arising from the
peritoneum ought to be distinguished from primary peritoneal
Müllerian neoplasms. Immunohistochemical staining for mesothelial
markers (D2-40 and calretinin) excluded the mesothelial origin. The
expression of ER and PR, as well as WT-1, favored a serous tumor of
Müllerian origin.
The distinction between a supernumerary and an
accessory ovary is not always clear. In an extensive review of the
literature in 1959, Wharton described the supernumerary ovary
(8). The term ‘supernumerary ovary’
is used to include rare cases of ectopy in which the third ovary is
entirely separate from the ovary which is situated normally. The
term ‘accessory ovary’ includes cases in which excess ovarian
tissue is situated near the normally placed ovary, and may have a
connection with it, appearing to have been developed from it
(9).
There are two proposed mechanisms for the formation
of a supernumerary ovary (10).
Pearl and Plotz proposed the arrested gonocyte migration theory.
Gonocytes may be arrested as they pass retroperitoneally through
the dorsal mesentery (9).
Alternatively, the transplantation theory of the germinal ridge
following incorporation of the gonocyte was proposed by Printz
et al(11). Supernumerary
ovaries may be situated in the pelvis, the retroperitoneum, the
para-aortic area or the colonic mesentery. Supernumerary ovaries
may be multiple and functional, associated with ovarian neoplasms
or located with other congenital malformations of the genitourinary
system (12). The mass may present
as either painful or asymptomatic, and is often found incidentally
during surgery or autopsy.
From these mechanisms, we suggest that the
supernumerary ovary tends to be situated in the posterior
peritoneum, where it is hard to detect the normal-sized ovary
without expectation, compared with the anterior peritoneum. This
explains why the third ovary was not identified in the first
diagnostic scope operation in the present case.
In summary, we report the first case of cancer of
the super-numerary ovary in a patient with MRKH syndrome. Although
both ovaries were confirmed to be normal in the present patient
with MRKH syndrome, an ovarian neoplasm should be considered in
pelvic mass diagnosis.
References
|
1.
|
Morcel K, Camborieux L; Programme de
Recherches sur les Aplasies Müllériennes; Guerrier D:
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J Rare
Dis. 2:132007.
|
|
2.
|
Barik S, Dhaliwal LK, Gopalan S and
Rajwanshi A: Adenocarcinoma of the supernumerary ovary. Int J
Gynaecol Obstet. 34:75–77. 1991. View Article : Google Scholar
|
|
3.
|
Rodriguez E, Pombo F, Alvarez C and Arnal
F: Tumor in ectopic omental ovary in Mayer-Rokitansky-Kuster-Hauser
syndrome: CT findings. J Comput Assist Tomogr. 22:758–759. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Fletcher HM, Campbell-Simpson K, Walcott D
and Harriott J: Müllerian remnant leiomyomas in women with
Mayer-Rokitansky-Küster-Hauser syndrome. Obstet Gynecol.
119:483–485. 2012.
|
|
5.
|
Sultan C, Biason-Lauber A and Philibert P:
Mayer-Rokitansky-Kuster-Hauser syndrome: recent clinical and
genetic findings. Gynecol Endocrinol. 25:8–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Capraro VJ and Gallego MB: Vaginal
agenesis. Am J Obstet Gynecol. 124:98–107. 1976.PubMed/NCBI
|
|
7.
|
El Khamlichi A, Allali N and Dafiri R:
Typical form of Mayer-Rokitansky-Kuster-Hauser syndrome and ectopic
kidney. A rare association. Gynecol Obstet Fertil. 39:e40–e43.
2011.(In French).
|
|
8.
|
Wharton LR: Two cases of supernumerary
ovary and one of accessory ovary, with an analysis of previously
reported cases. Am J Obstet Gynecol. 78:1101–1119. 1959.PubMed/NCBI
|
|
9.
|
Pearl M and Plotz EJ: Supernumerary ovary.
Report of a case Obstet Gynecol. 21:253–256. 1963.PubMed/NCBI
|
|
10.
|
Cruikshank S: Supernumerary ovary:
embryology. Int J Gynaecol Obstet. 34:175–178. 1991. View Article : Google Scholar
|
|
11.
|
Printz JL, Choate JW, Townes PL and Harper
RC: The embryology of supernumerary ovaries. Obstet Gynecol.
41:246–252. 1973.PubMed/NCBI
|
|
12.
|
Zhigang Z and Wenlu S: An intrarenal
supernumerary ovary concurrent with a completely duplicated pelvis
and ureter. Int Urogynecol J Pelvic Floor Dysfunct. 18:1243–1246.
2007. View Article : Google Scholar : PubMed/NCBI
|