Introduction
TP53 tumor suppressor gene is one of the most
commonly mutated genes in human neoplasia, and approximately 80% of
these mutations are missense mutations (1,2). The
gene product, p53 protein, is a nuclear transcriptional activator
that is activated by post-translational modification, including
phosphorylation and acetylation, in response to DNA-damaging
stresses. Activated p53 is stabilized, accumulates in the nucleus
and binds to p53-responsive elements (p53REs) in the promoter
region of p53-downstream genes (3).
Transactivation of these genes, including p21WAF1,
MDM2, p53AIP1, BAX, NOXA and
PUMA, results in cell cycle arrest and apoptosis.
Most p53 mutants with a single amino acid
substitution found in human neoplasm lose the ability to bind to
p53REs, and this functional defect is thought to be one of the most
important oncogenic events caused by TP53 mutation (4). Therefore, the translocation of p53
into the nucleus is crucial for normal p53 function. Cytoplasmic
sequestration of wild-type p53 was observed in undifferentiated
neuroblastoma, breast cancer, retinoblastoma, colorectal carcinoma
and glioblastoma cells (5–7). In all these cells, wild-type p53 is
inactivated since it is retained in the cytoplasm. Although the
precise mechanism underlying the cytoplasmic sequestration remains
unclear, several molecular mechanisms have been proposed: i) a
mutation in the bipartite sequence of p53 (residues 305 and 306)
(8) or a truncated mutation of the
nuclear localization motif receptor protein importin-α (9); ii) hyperactive nuclear export by an
MDM2-dependent pathway (10); and
iii) overexpression of cytoplasmic tethering proteins, such as
mortalin (11), cullin 7 (12) and PARC (13). The mutations in the bipartite
sequence have been analyzed comprehensively, and these mutants were
shown to lose transactivation activity in a yeast functional assay
(14).
In contrast to tumor-derived loss-of-function p53
mutants, other types of p53 mutants (super p53s) have a stronger
ability to induce apoptosis than wild-type p53. Among these, a p53
mutant with a serine to phenylalanine substitution at residue 121
(S121F) has a distinct affinity to bind p53REs from wild-type p53
(15). S121F induces a more potent
apoptosis than wild-type p53 in mammalian cell lines. The
transcriptional activity of S121F for downstream genes, however, is
less efficient than that of wild-type p53 (16). In addition, different expression
profiles among super p53s have been reported (17). These results suggest that
transactivation-independent cytoplasmic activity occurs in
p53-dependent apoptosis and that S121F may be a diverged mutant
with enhanced cytoplasmic activity.
To test this hypothesis, we expressed wild-type and
S121F p53 in the nucleus or cytoplasm of p53-null SF126
glioblastoma cells using a p53 mutant with an arginine to glycine
substitution at residue 306 (R306G), and analyzed them for
induction of apoptosis and transactivation of p53-downstream genes
following the p53 induction.
Materials and methods
Construction of stable SF126 glioblastoma
cell lines
The plasmids pCR259-WTp53, pCR259-S121F and
pCR259-R306G were previously constructed (14). pCR259-S121F-R306G was constructed by
inserting a small fragment of pCR259-R306G into the
Bsu36I/EagI site of pCR259-S121F. The small
NheI/EagI fragments of the four pCR259-based plasmids
were inserted into the NheI/NotI site of pcDNA5/TON
(Invitrogen, Carlsbad, CA, USA). The resulting plasmids were
designated pcDNA5/TON-WTp53, pcDNA5/TON-S121F, pcDNA5/TON-R306G and
pcDNA5/TON-S121F-R306G, respectively. The stable SF126 cell lines
expressing tetracycline-inducible p53 were constructed according to
the protocol described in the T-Rex™ System (Invitrogen) using the
four pcDNA5/TON-based plasmids. For each category, several stable
clones were selected by hygromycin B (100 μg/ml) and two
independent stable clones were used.
Western blot analysis
The cell lines were harvested and the cells were
resuspended in lysis buffer containing 50 mM Tris-HCl, pH 8.0, 150
mM NaCl, 5 mM EDTA and 1% protease inhibitor cocktail
(Sigma-Aldrich, St. Louis, MO, USA). Cell lysates were centrifuged
for 10 min at 4°C. The supernatants were resolved by SDS-PAGE and
transferred to PVDF membranes. The membranes were incubated with
anti-p53 (FL-393; Santa Cruz Biotechnology, Santa Cruz, CA, USA)
and mouse anti-actin (Sigma-Aldrich), followed by incubation with
goat anti-rabbit Alexa Fluor 680 IgG (Invitrogen) and goat
anti-mouse IR Dye 800 CW IgG (Rockland, Gilbertsville, PA, USA).
Expression of both p53 and β-actin was visualized using an Odyssey
Infrated Imaging System (LI-COR, Lincoln, NE, USA).
Immunofluorescent analysis
Each cell line was cultured on poly-D lysine-coated
Lab-Tek Chamber Slides™ (Nalge Nunc, Rochester, NY, USA) until 70%
confluence was achieved. At 24 h after the addition of 10 ng/ml
doxycycline or phosphate-buffered solution (PBS), the cells were
fixed with acetone-methanol (1:1) and incubated for 20 min at
−20°C. After washing with PBS and blocking with 5% non-fat milk in
PBS containing 0.05% Tween-20 for 1 h, cells were incubated with
FITC-conjugated mouse anti-p53 (DO-1 FITC; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) diluted at 1:500, stained with
propidium iodide and then visualized on an LSM5 PASCAL (Carl Zeiss,
Jena, Germany).
Cell proliferation assay
Cells (4×103) were seeded and incubated
in a 96-well plate for 24 h. Doxycycline (10 ng/ml) or PBS was
added to the medium and the cells were then cultured until 72 h at
37°C. At 0, 24, 48 and 72 h after the addition of doxycycline, 10
μl of the Cell Counting Kit-8 (Dojin Laboratories, Kumamoto, Japan)
was added to each well and the cells were incubated for 2 h at
37°C. Absorbance at 490 nm was read with a microplate reader. Each
data point is derived from triplicate experiments. The absorbance
values at 24, 48 and 72 h were normalized by the value at 0 h.
Cell cycle analysis by
fluorescence-activated cell sorting
Cells (1×106) were seeded and incubated
in a 10-cm culture plate for 24 h, and then incubated in the
presence of doxycycline (10 ng/ml). After 24 h, the cells were
collected and stained with propidium iodide (50 μg/ml). The stained
cells were filtered through 50-μm nylon mesh and analyzed using a
Cytomics FC500 (Beckman Coulter, Miami, FL, USA). The subfraction
of cells in each phase of the cell cycle was calculated using
Multicycle software (Phoenix Flow Systems, San Diego, CA, USA). The
average subfraction value of two independent cell lines was
calculated.
Quantitative real-time PCR analysis
Total RNA was extracted from cells in the presence
or absence of doxycycline using an RNeasy Mini kit (Qiagen,
Gaithersburg, MD, USA). RNA (1 μg) was converted to cDNA using a
High-Capacity cDNA Reverse Transcription kit (Applied Biosystems,
Foster City, CA, USA) with random hexamers. TaqMan Gene Expression
Assay was performed on the ABI 7500 real-time PCR System (Applied
Biosystems) according to the manufacturer’s protocol. Assay ID was
as follows; GAPDH, Hs99999905_m1; ACTB (β-actin), Hs99999903_m1;
MDM2, Hs01066938_m1; p21 (CDKN1A), Hs00355782_m1; BAX,
Hs00180269_m1; NOXA (PMAIP1), Hs00560402_m1; PUMA (BBC3),
Hs00248075_m1; and P53AIP1, Hs00223141_m1. The expression level of
each p53 target gene was collected by either β-actin or GAPDH (data
not shown). A relatively induced expression was measured as a ratio
of the collected value of doxycycline presence against that of
doxycycline absence. Two independent clones were analyzed, and the
data were shown as a mean of four replicates with an error bar of
standard deviation.
Results
Cytoplasmic sequestration of p53 by R306G
mutation
To examine the cytoplasmic activity of wild-type p53
and the S121F mutant, we constructed a series of stable SF126
glioblastoma cell lines. These cells expressed wild-type p53,
S121F, R306G or S121F-R306G double mutants in the presence of
doxycycline (Fig. 1). To examine
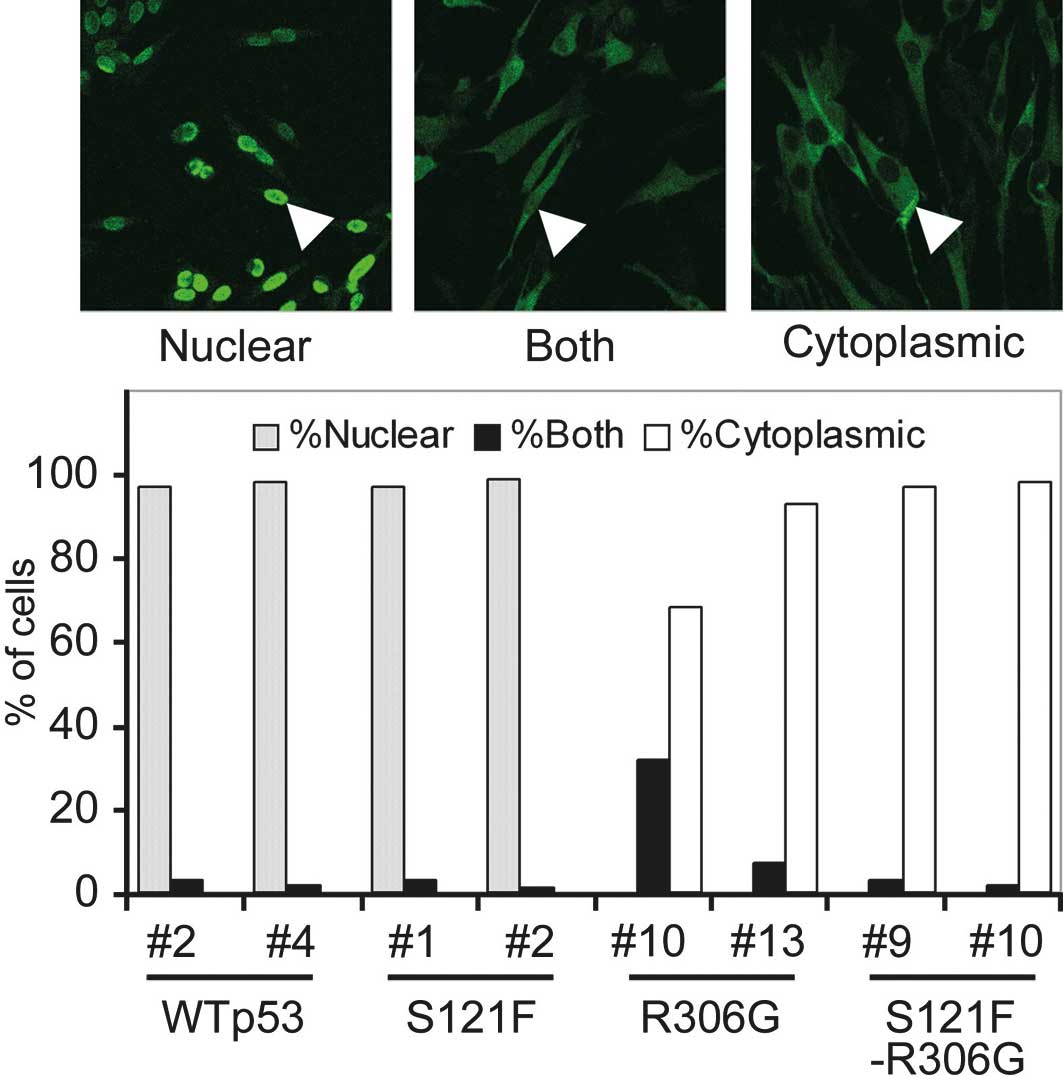
the cellular localization of doxycycline-induced p53, an
immunofluorescent analysis was performed (Figs. 1 and 2). Both wild-type p53 and S121F localized
mostly to the nucleus. In the presence of the R306G mutation in the
same p53 molecule, wild-type p53 and S121F were exclusively
sequestered from the nucleus to the cytoplasm. To quantify the
degree of sequestration of p53, 100 cells of each cell clone were
scored as having nuclear, cytoplasmic or both nuclear and
cytoplasmic patterns (Fig. 2). Both
wild-type and S121F thoroughly localized in the nucleus (>95% of
cells), and no cell showed a cytoplasmic pattern. By contrast, both
wild-type and S121F with R306G localized in the cytoplasm (>95%)
or exhibited cytoplasmic and nuclear patterns. None of the cells
expressing p53 exhibited any nuclear pattern. These results
indicate that R306G sequestered p53 from the nucleus to the
cytoplasm. This result is reasonable since R306G is a mutation in
the bipartite sequence of p53 (residues 305 and 306) as described
above.
Inhibition of cell proliferation by
cytoplasmically localized wild-type p53 and S121F
To examine the effect of cytoplasmically localized
wild-type p53 and the S121F mutant on cell proliferation, we
cultured two independent cell lines for each category (wild-type,
S121F, R306G, S121F-R306G and null p53) and estimated the viable
cells at 24, 48 and 72 h after p53 induction by doxycycline (data
not shown). The results at 48 h are shown in Fig. 3A. Although the cytoplasmic
sequestration of wild-type p53 considerably disturbed the
inhibitory effect against cell proliferation by wild-type p53, some
inhibitory effects remained. The cytoplasmic sequestration of S121F
did not disrupt the strong inhibitory effect on cell proliferation.
These results indicate a cytoplasmic function of p53 on cell
proliferation in both wild-type p53 and S121F.
Induction of apoptosis by cytoplasmically
localized wild-type p53 and S121F
To evaluate the ability of wild-type p53 and the
S121F mutant to induce apoptosis, each cell line was cultured and
analyzed for a percentage of cell-cycle phase by
fluorescence-activated cell sorting 24 h following the p53
induction by doxycycline (Fig. 3B and
C). As shown in Fig. 3B,
compared to the null p53 control, wild-type p53 clearly arrested
cells at the G1 (49.9–59.4%) and G2 + M (12.3–32.1%) phases of the
cell cycle and subsequently reduced the S-phase fraction
(33.1–2.3%). The sub-G1 fraction (apoptosis fraction) was only
slightly increased (4.7–6.2%) at 24 h, whereas at 48 h a
substantial increase was observed (1.7–16.1%; data not shown). The
cytoplasmic sequestration of p53 did not affect the cell cycle (G1,
60.5%; G2 + M, 31.6%), but slightly affected both the S phase
(5.9%) and the sub-G1 phase (2%). S121F markedly increased the
sub-G1 fraction (44.5%) without an increase in the G1 (33.1%) and
G2 + M (8.3%) fractions, indicating strong apoptotic induction
without cell cycle arrest. The cytoplasmic sequestration of S121F
affected the sub-G1 (26.7%), G1 (41.2%) and G2 + M (19.6%)
fractions only partially, indicating that a strong induction of
apoptosis of S121F was retained despite the cytoplasmic
sequestration. These results were consistent with the results of
the cell proliferation analysis and again indicated a cytoplasmic
function of p53 on both cell cycle arrest and apoptosis.
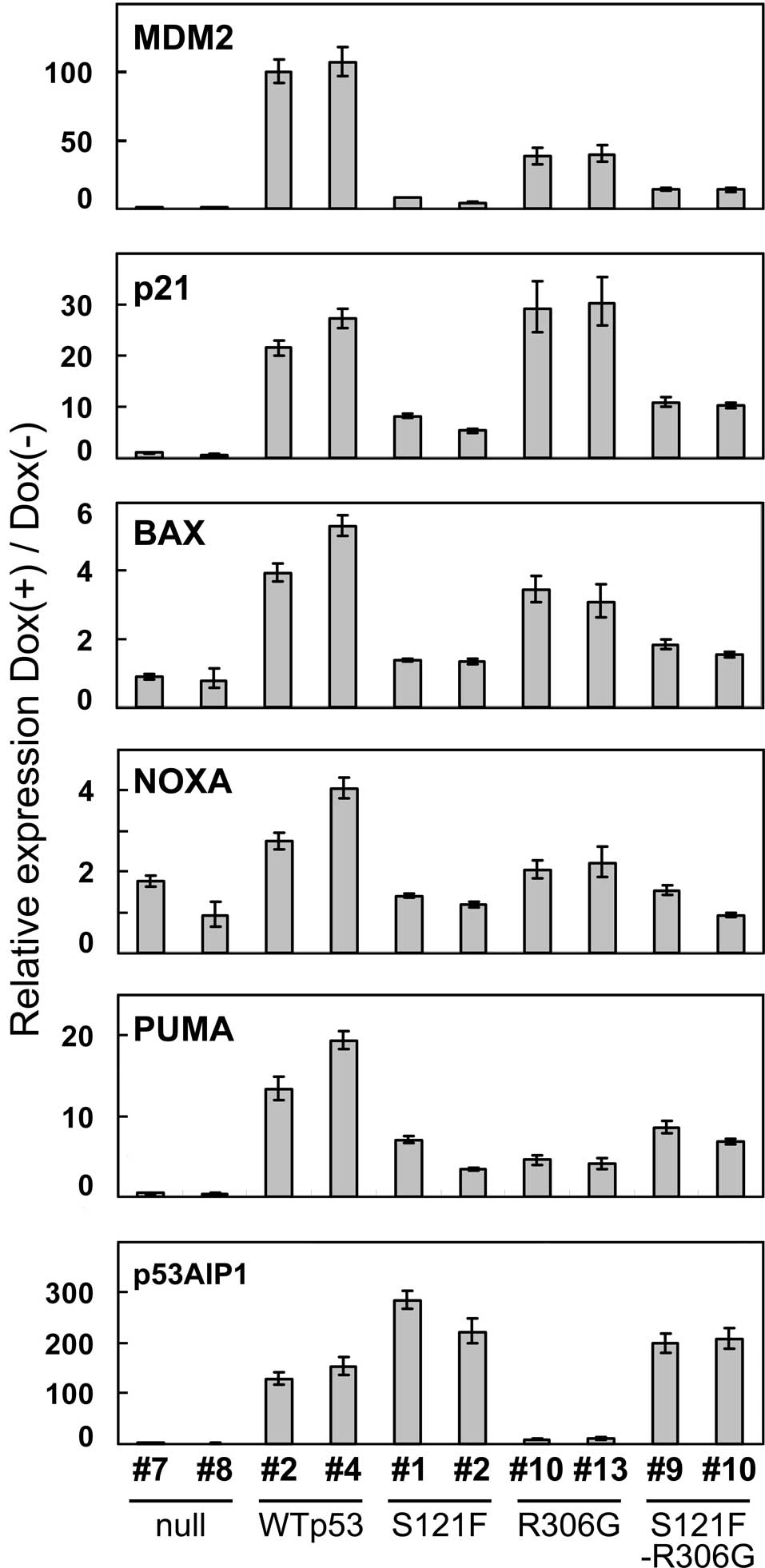
Transactivation of p53 target genes by
cytoplasmically localized wild-type p53 and S121F
To examine the effect of cytoplasmic sequestration
on transcriptional activation by wild-type p53 and S121F,
transcripts of the p53-downstream genes, MDM2,
p21WAF1, BAX, NOXA, PUMA and
p53AIP1, were quantitated by real-time quantitative PCR
analysis at 24 h after p53 induction (Fig. 4). Of the six genes, all except
p53AIP1 were less efficiently transactivated by S121F than
by wild-type p53. The results showing a lower ability of S121F than
wild-type p53 on transactivation were mostly consistent with our
previous findings, with the exception of the result of
p53AIP1 (17), which is
consistent with the previous hypothesis that S121F may cause a
transactivation-independent apoptotic pathway. Notably, the
cytoplasmic sequestration of wild-type p53 did not completely
inactivate transactivation, but it reduced the level of
transactivation in 5 of the 6 target genes (wiht the exception of
p21WAF1). Of note, the cytoplasmic sequestration of S121F
did not change the expression profile of the target genes. The
ability of cytoplasmically sequestered S121F on transactivation was
also confirmed when the expression level of each p53 target gene
was collected by GAPDH (data not shown).
Discussion
The cytoplasmic sequestration of p53 did not
completely inactivate p53 function, suggesting the cytoplasmic
function of p53. This finding may be the reason that mutations on
the bipartite sequence in human tumors are extremely rare. In this
context, Goldman et al showed that in a neuroblastoma cell
line expressing cytoplasmically sequestered wild-type p53, p53
target genes (p21WAF1 and MDM2) were up-regulated
following cell irradiation (18).
These results also suggest that wild-type p53 retains some
functional activity when it is sequestered in the cytoplasm,
although p53 homologues, such as p63 and p73, may have been
involved in the result. By contrast, our experimental system was a
p53-specific inducible system; therefore, involvement of p53
homologue activation is unlikely.
Our previous knowledge of p53-dependent apoptosis
was that after genotoxic stress, activated p53 transactivated its
downstream genes in a sequence-specific manner in the cell nucleus
and induced apoptosis in cells through the direct or indirect
induction of the downstream protein(s); however, a
transactivation-independent mechanism for p53-dependent apoptosis
has been reported by several laboratories (19,20).
In addition, we previously indicated a lack of correlation between
p53-dependent transactivation activity and the ability to induce
apoptosis, and speculated that a transactivation-independent
mechanism may exist (17). We
excluded the nuclear function of p53, including the
sequence-specific transactivation function, by introducing R306G, a
mutation in the bipartite sequence at residues 305 and 306. A
conditional expression system of cytoplasmically sequestered p53
was constructed and we found that cytoplasmically sequestered p53
retains its ability to arrest cell proliferation (wild-type p53)
and induce apoptosis (S121F). These results strongly support a
cytoplasmic apoptotic function of p53. Notably, however,
cytoplasmically sequestered p53 transactivated downstream genes.
Therefore, we did not clarify whether cytoplasmic p53-dependent
apoptosis depends on either a direct or an indirect transactivation
mechanism or is independent of transactivation.
Additional experiments are required to evaluate
which mechanism is crucial for p53-dependent apoptosis and to
clarify the mechanism underlying super p53 (S121F)-dependent
apoptosis.
Acknowledgements
This study was supported by the Ministry of
Education, Culture, Sports, Science and Technology (C. Ishioka and
S. Kato), the Comprehensive Research and Education Center for
Planning of Drug Development (CRECENDO) of Tohoku University 21st.
Century COE Program (K. Yasuda and C. Ishioka), and the Gonryo
Medical Foundation (C. Ishioka).
References
|
1
|
Hollstein M, Rice K, Greenblatt MS, et al:
Database of p53 gene somatic mutations in human tumors and cell
lines. Nucleic Acids Res. 22:3551–3555. 1994.PubMed/NCBI
|
|
2
|
Soussi T: p53 alterations in human cancer:
more questions than answers. Oncogene. 26:2145–2156. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harris SL and Levine AJ: The p53 pathway:
positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soussi T and Lozano G: p53 mutation
heterogeneity in cancer. Biochem Biophys Res Commun. 331:834–842.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moll UM, LaQuaglia M, Benard J and Riou G:
Wild-type p53 protein undergoes cytoplasmic sequestration in
undifferentiated neuroblastomas but not in differentiated tumors.
Proc Natl Acad Sci USA. 92:4407–4411. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moll UM, Riou G and Levine AJ: Two
distinct mechanisms alter p53 in breast cancer: mutation and
nuclear exclusion. Proc Natl Acad Sci USA. 89:7262–7266. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nagpal J, Jamoona A, Gulati ND, et al:
Revisiting the role of p53 in primary and secondary glioblastomas.
Anticancer Res. 26:4633–4639. 2006.PubMed/NCBI
|
|
8
|
Liang SH and Clarke MF: A bipartite
nuclear localization signal is required for p53 nuclear import
regulated by a carboxyl-terminal domain. J Biol Chem.
274:32699–32703. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim IS, Kim DH, Han SM, et al: Truncated
form of importin alpha identified in breast cancer cell inhibits
nuclear import of p53. J Biol Chem. 275:23139–23145. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rodriguez-Lopez AM, Xenaki D, Eden TO,
Hickman JA and Chresta CM: MDM2 mediated nuclear exclusion of p53
attenuates etoposide-induced apoptosis in neuroblastoma cells. Mol
Pharmacol. 59:135–143. 2001.PubMed/NCBI
|
|
11
|
Kaul SC, Deocaris CC and Wadhwa R: Three
faces of mortalin: a housekeeper, guardian and killer. Exp
Gerontol. 42:263–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Andrews P, He YJ and Xiong Y: Cytoplasmic
localized ubiquitin ligase cullin 7 binds to p53 and promotes cell
growth by antagonizing p53 function. Oncogene. 25:4534–4548. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nikolaev AY, Li M, Puskas N, Qin J and Gu
W: Parc: a cytoplasmic anchor for p53. Cell. 112:29–40. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kato S, Han SY, Liu W, et al:
Understanding the function-structure and function-mutation
relationships of p53 tumor suppressor protein by high-resolution
missense mutation analysis. Proc Natl Acad Sci USA. 100:8424–8429.
2003. View Article : Google Scholar
|
|
15
|
Freeman J, Schmidt S, Scharer E and Iggo
R: Mutation of conserved domain II alters the sequence specificity
of DNA binding by the p53 protein. EMBO J. 13:5393–5400.
1994.PubMed/NCBI
|
|
16
|
Saller E, Tom E, Brunori M, et al:
Increased apoptosis induction by 121F mutant p53. EMBO J.
18:4424–4437. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kakudo Y, Shibata H, Otsuka K, Kato S and
Ishioka C: Lack of correlation between p53-dependent
transcriptional activity and the ability to induce apoptosis among
179 mutant p53s. Cancer Res. 65:2108–2114. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldman SC, Chen CY, Lansing TJ, Gilmer TM
and Kastan MB: The p53 signal transduction pathway is intact in
human neuroblastoma despite cytoplasmic localization. Am J Pathol.
148:1381–1385. 1996.
|
|
19
|
Chen X, Ko LJ, Jayaraman L and Prives C:
p53 levels, functional domains, and DNA damage determine the extent
of the apoptotic response of tumor cells. Genes Dev. 10:2438–2451.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haupt Y, Rowan S, Shaulian E, Vousden KH
and Oren M: Induction of apoptosis in HeLa cells by
trans-activation-deficient p53. Genes Dev. 9:2170–2183. 1995.
View Article : Google Scholar : PubMed/NCBI
|