Introduction
Ewing’s sarcoma/primitive neuroectodermal tumour
(ES/PNET) is a member of the Ewing’s sarcoma family of tumours
(ESFT). Extraskeletal ES is a manifestation of ES within the soft
tissues, and an ES tumour presenting as a breast mass is unusual.
ES typically occurs in adolescents and young adults aged between 10
and 20 years (1). The diagnosis of
ES/PNET requires panels of immunohistochemical study and the
presence of a t(11;22) translocation detected through fluorescent
in situ hybridization (FISH). This case report examines a
rare case of ES/PNET observed in a 46-year-old woman, who developed
a painless and progressive tumour presenting as a breast mass. The
patient’s family provided their consent to the study.
Case report
In 2010, a 46-year-old woman presented a painless
and progressive mass in her right breast for 1 month. Upon
diagnostic mammography and ultrasonography, a suspicious mass of 4
cm in size was identified (Fig. 1A and
B). Following this, a biopsy was performed and the initial
diagnosis was a neuroendocrine carcinoma, as the tumour was
positive for synaptophysin, but negative for ER, PR, HER2/neu and
CK8/18.
Following a core needle biopsy, the tumour increased
rapidly in size and became 12×12 cm within 1 month. The patient
received chemotherapy containing cyclophosphamide, adriamycin and
vincristine for 6 cycles, to which the tumour responded well and
shrank to 1.5×2.5 cm. Whole breast radiation was continued at a
dose of 50 Gy; however, following completion, the tumour progressed
quickly to 6×7 cm within 1 month. A CT scan of the chest revealed a
0.6 cm sized nodule in the right lung. Therefore, platinum and
etoposide were commenced for 3 cycles, followed by paclitaxel and
carboplatin. Despite this treatment, the tumour did not respond to
the chemotherapy administered and the disease progressed; the
patient was then referred to our institute.
While the patient was in our institute, the tumour
invaded through the skin and grew to 20×15 cm. A review of a
previous biopsy demonstrated ES/PNET as a malignant small round
cell neoplasm (Fig. 2A).
Immunohistochemical staining was performed for vimentin (clone V9,
dilution 1:500), CD20 (clone L26, dilution 1:2,000), CD99 (clone
12E7, ready-to-use), desmin (clone D33, dilution 1:1,000), S-100
(polyclonal, dilution 1:10,000) from DakoCytomation (Glostrup,
Denmark); FLI-1 (clone MRQ-1, ready-to-use), CD56 (clone 123C3.D5,
ready-to-use), AE1/AE3 (ready-to-use), chromogranin A (LK2H10,
ready-to-use), CD30 (clone Ber-H2, ready-to-use) from Cell Marque
(Rocklin, CA, USA); CK8/18 (clone 5D3, dilution 1:300) from
NEOMARKERS (Fremont, CA, USA); synaptophysin (clone SP11, dilution
1:500) from Diagnostic Biosystems (Pleasanton, CA, USA); CD45
(clone PD7/26/16&2B11, dilution 1:2,000) from BioGenex
(Fremont, CA, USA); CD3 (clone LN10, ready-to-use), TdT (clone
NPT26, ready-to-use), TTF-1 (clone SPT24, ready-to-use) from
Novocastra (Newcastle, UK) and Pax-5 (clone SP34, ready-to-use)
from Ventana (Tucson, AZ, USA). An automated immunostainer,
BenchMark XT (Ventana), with a polymer-based DakoEnVision detection
system (DakoCytomation), were used. The tumour cells marked with
vimentin, FLI-1 (diffuse nuclear staining), CD99 (membrane
staining; intense) and CD56 were negative for desmin, AE1/AE3,
CK8/18, chromogranin A, synaptophysin, S100, CD45, CD3, CD20, PAX5,
CD30, TdT and TTF-1 (Fig. 2B and
C). These results were consistent with ES/PNET. Following this,
two cycles of paclitaxel and carboplatin were initiated.
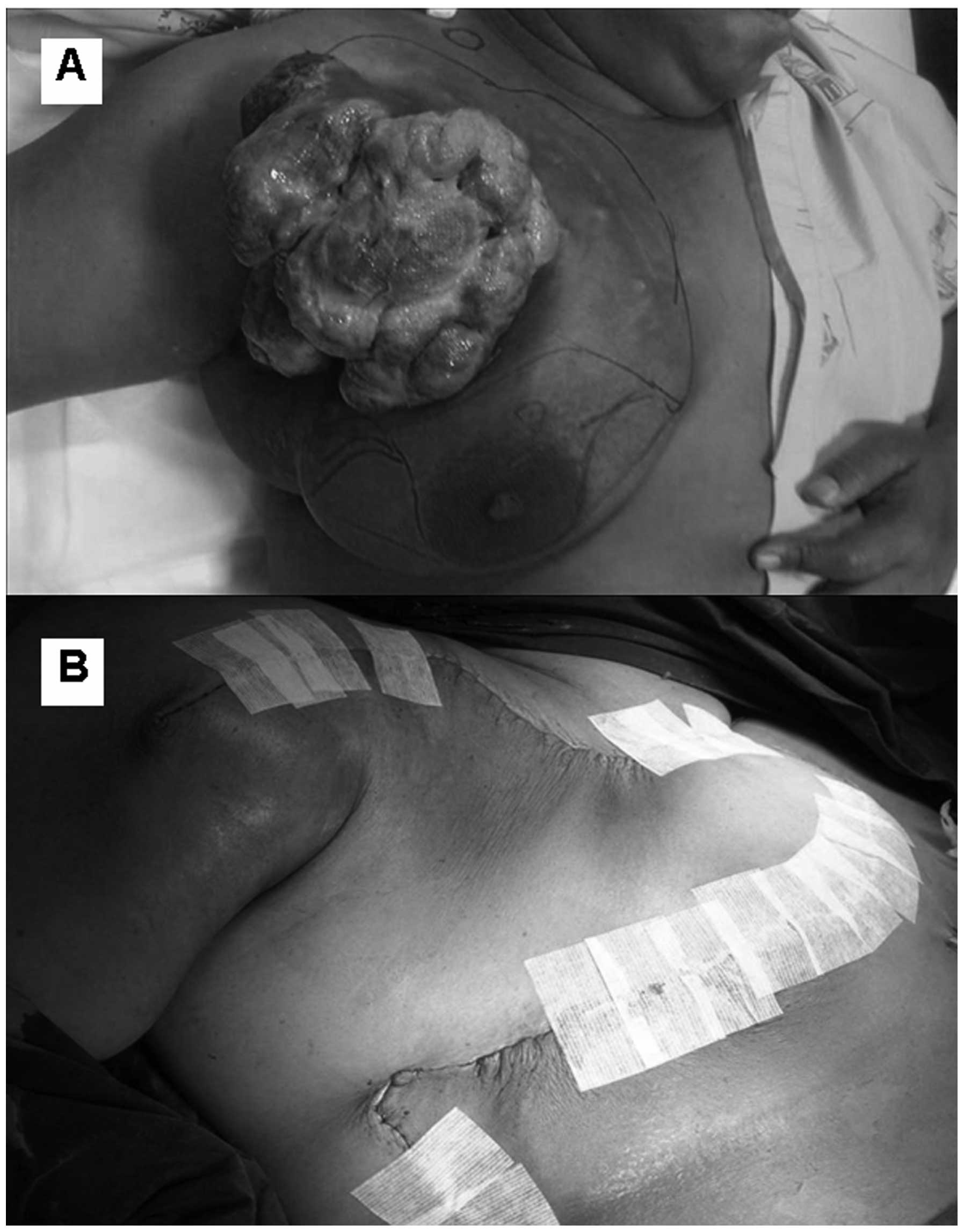
Progression of the disease was observed and a modified radical
mastectomy with local skin flap coverage was performed (Fig. 3A and B). The tumour was localized in
the upper outer part of the right breast, with involvement of the
underlying skeletal muscle, measuring 20.5×15.5×12 cm. The surgical
margins were free from the tumour, and lymphovascular invasion and
metastatasis to the axillary lymph nodes (3 of 24 nodes), were
reported. An additional FISH study confirmed the presence of
EWSR1 gene translocation (Fig.
2D). Thus, ES/PNET was the final diagnosis.
Only 1 month following the surgery, the patient
developed recurrent disease on the chest wall, as well as multiple
lung nodules. The disease progressed rapidly and two months
following surgery, the patient succumbed to respiratory failure due
to pulmonary metastasis.
Discussion
We report a rare case of extraskeletal ES/PNET,
presenting with a rapidly growing palpable breast lump. Age at
presentation, radiological findings and immunohistochemical
findings were of interest and documented in this report.
ES and PNET are typically undifferentiated (1,2).
Translocation t(11;22)(q24;q12) resulting in EWS/FLI1
fusion, which can be identified in more than 90% of ES/PNET cases,
is the genetic hallmark of ES/PNET (3). In cases with classic morphology where
other small round cell neoplasms have been excluded
immunohistochemically, the expression of CD99 cell surface antigen
(product of the MIC-2 gene) is required to support ES/PNET
diagnosis (4,5).
The majority of patients with ES/PNET are 10–20
years old (1), and other small
studies of adult ES/PNET from the Royal Marsden, the Memorial
Sloan-Kettering and the Dana-Faber Cancer Centers have reported a
median age of 24–27 years(1,6,7).
However, our patient was 46 years of age at the time of diagnosis,
which is unusual.
ES/PNET development within breast tissue was
unlikely to be diagnosed upon first presentation. Findings from
mammography and ultrasonography breast images may vary as they
could be from a hypoechoic mass with posterior enhancement or
heterogeneous mass with a necrotic area (8,9). In
this patient, a well-circumscribed mass was documented (Fig. 1A and B), which possibly arose from
the soft tissue beneath the breast. Precise identification of the
tumour location (chest wall soft tissue) and recognition of
variation in radiographic findings described in this tumour may be
a clue for radiologists to avoid misinterpreting the tumour as a
breast mass that might preclude the diagnosis of soft tissue
tumours.
Diagnosis of ES/PNET in this patient was based on
the immunohistochemical staining results, which documented a
positive expression of CD56, CD99, FLI-1, synaptophysin and
vimentin, but a negative expression of AE1/AE3, CK8/18, EMA,
chromogranin A, CD45 (LCA), HER-2, ER and PR. An additional FISH
study confirmed the presence of EWSR1 gene translocation.
The positive expression of CD99 (MIC2), a cell surface glycoprotein
involved in cell adhesion, plays a crucial role in the diagnosis of
ES/PNET (10). However, CD99 may
also be expressed in other tumours, including metaplastic carcinoma
of the breast, neuroendocrine carcinoma, lymphoma and
rhabdomyosarcoma (11). In our
patient, small cell carcinoma, neuroendocrine carcinoma and
malignant lymphoma were excluded by negative staining for
cytokeratins, chromogranin A and LCA.
ES/PNET is an aggressive tumour with a high
incidence of local recurrence and distant metastasis. A combination
of multiple modalities, including surgery, chemotherapy and
radiation therapy, was the most appropriate treatment for our
patient (1). All members of the
ESFT tend to share the propensity for metastatic spread. Consistent
use of systemic chemotherapy to treat localised ESFT effectively
improved the 5-year survival rate from 5 to 10% up to 65%, which is
primarily due to the elimination of micrometastases (12–14).
Although the optimum combination chemotherapy has not yet been
established, a regimen containing vincristine, adriamycin,
cyclophosphamide and actinomycin D, was the standard first-line
treatment for patients with localized disease (12). In patients with unresectable or
metastatic disease, palliative chemotherapy may be useful. In this
patient, vincristine, adriamycin and cyclophosphamide were utilized
at the first instance. The tumour appeared to respond well
initially, but became resistant to various chemotherapeutic agents,
which resulted in progression of the disease.
The role of radiation therapy in the treatment of
ES/PNET is unclear. However, the use of radiation therapy combined
with surgery, in order to control local disease, is proving to be
helpful (1). Our patient did not
respond to radiation therapy treatment and the tumour grew rapidly
following cessation of radiation. This is similar to a previous
study where a 78% disease progression rate was observed in 14
patients treated with radiotherapy plus chemotherapy (15).
In conclusion, we report a rare case of ES/PNET
presenting as a breast mass. Clinical presentation mimicked
invasive breast cancer. The tumour did not respond well to
multimodality treatment and local and distant metastasis occurred
less than 2 years after first diagnosis.
References
|
1
|
Baldini EH, Demetri GD, Fletcher CD, Foran
J, Marcus KC and Singer S: Adults with Ewing’s sarcoma/primitive
neuroecto-dermal tumor: adverse effect of older age and primary
extraosseous disease on outcome. Ann Surg. 230:79–86. 1999.
|
|
2
|
Dehner LP: Primitive neuroectodermal tumor
and Ewing’s sarcoma. Am J Surg Pathol. 17:1–13. 1993.
|
|
3
|
Jambhekar NA, Bagwan IN, Ghule P, Shet TM,
Chinoy RF, Agarwal S, et al: Comparative analysis of routine
histology, immunohistochemistry, reverse transcriptase polymerase
chain reaction, and fluorescence in situ hybridization in diagnosis
of Ewing family of tumors. Arch Pathol Lab Med. 130:1813–1818.
2006.
|
|
4
|
De Alava E and Gerald WL: Molecular
biology of the Ewing’s sarcoma/primitive neuroectodermal tumor
family. J Clin Oncol. 18:204–213. 2000.
|
|
5
|
Turc-Carel C, Philip I, Berger MP, Philip
T and Lenoir GM: Chromosome study of Ewing’s sarcoma (ES) cell
lines. Consistency of a reciprocal translocation t(11;22)(q24;q12).
Cancer Genet Cytogenet. 12:1–19. 1984.
|
|
6
|
Martin RC and Brennan MF: Adult soft
tissue Ewing sarcoma or primitive neuroectodermal tumors:
predictors of survival? Arch Surg. 138:281–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verrill MW, Judson IR, Harmer CL, Fisher
C, Thomas JM and Wiltshaw E: Ewing’s sarcoma and primitive
neuroectodermal tumor in adults: are they different from Ewing’s
sarcoma and primitive neuroectodermal tumor in children? J Clin
Oncol. 15:2611–2621. 1997.
|
|
8
|
Da Silva BB, Lopes-Costa PV, Pires CG,
Borges RS and da Silva RG Jr: Primitive neuroectodermal tumor of
the breast. Eur J Obstet Gynecol Reprod Biol. 137:248–249.
2008.PubMed/NCBI
|
|
9
|
Maxwell RW, Ghate SV, Bentley RC and Soo
MS: Primary primitive neuroectodermal tumor of the breast. J
Ultrasound Med. 25:1331–1333. 2006.PubMed/NCBI
|
|
10
|
Tamura G, Sasou S, Kudoh S, Kikuchi J,
Ishikawa A, Tsuchiya T, et al: Primitive neuroectodermal tumor of
the breast: immunohistochemistry and fluorescence in situ
hybridization. Pathol Int. 57:509–512. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Milanezi F, Pereira EM, Ferreira FV,
Leitao D and Schmitt FC: CD99/MIC-2 surface protein expression in
breast carcinomas. Histopathology. 39:578–583. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paulussen M, Ahrens S, Dunst J, Winkelmann
W, Exner GU, Kotz R, et al: Localized Ewing tumor of bone: final
results of the cooperative Ewing’s Sarcoma Study CESS 86. J Clin
Oncol. 19:1818–1829. 2001.PubMed/NCBI
|
|
13
|
Rosito P, Mancini AF, Rondelli R, Abate
ME, Pession A, Bedei L, et al: Italian Cooperative Study for the
treatment of children and young adults with localized Ewing sarcoma
of bone: a preliminary report of 6 years of experience. Cancer.
86:421–428. 1999.PubMed/NCBI
|
|
14
|
Verrill MW, Judson IR, Wiltshaw E, Thomas
JM, Harmer CL and Fisher C: The use of paediatric chemotherapy
protocols at full dose is both a rational and feasible treatment
strategy in adults with Ewing’s family tumours. Ann Oncol.
8:1099–1105. 1997.PubMed/NCBI
|
|
15
|
Kushner BH, Hajdu SI, Gulati SC, Erlandson
RA, Exelby PR and Lieberman PH: Extracranial primitive
neuroectodermal tumors. The Memorial Sloan-Kettering Cancer Center
experience. Cancer. 67:1825–1829. 1991. View Article : Google Scholar : PubMed/NCBI
|