Introduction
Gastric cancer (GC) is one of the most prevalent
malignancies worldwide and remains a leading cause of
cancer-related mortality (1,2). Since
the presence of lymph node metastases (LNM) is associated with a
significantly poorer prognosis of GC patients (3–5),
radical resection with free-margin gastrectomy and extended
lymphadenectomy are performed for patients with advanced GC to
eradicate LNM (6). Such an
aggressive resection of the lymph nodes is associated with higher
patient morbidity and/or mortality rates (7–9).
Alternatively, the absence of LNM allows for minimally invasive
surgery, which provides an improved quality of life following
treatment. Therefore, the accurate detection of LNM is useful for
the implementation of necessary and sufficient treatment.
To detect the presence of LNM, much effort has been
made in the fields of imaging and molecular markers. Imaging
modalities, including computed tomography (CT), endoscopic
ultrasonography (EUS) and 18F-fluorodeoxyglucose
positron emission tomography (FDG-PET) are used in clinical
practice. However, the sensitivities of these modalities are 77.2,
82.8 and 71%, respectively, and the specificities are 78.3, 74.2
and 74%, respectively (10–13). Moreover, these imaging modalities
are almost powerless to detect micrometastases (14,15).
With regard to molecular markers, analyses that targeted specific
RNA and protein expression have been made. Although a number of
these markers were associated with the presence of LNM of GCs
(16–19), their utility has not been confirmed
by independent studies. Therefore, genome-wide or comprehensive
analysis of molecular markers for LNM of GCs is required and
validation of the utility of the markers is essential for clinical
application.
As a molecular marker, DNA methylation is
advantageous, as its status is stable even if a cell is placed in
different environments (biologically stable) and DNA is chemically
stable, even in clinical materials. In addition, DNA methylation
profiles are not disturbed by the presence of a small population of
contaminating cells. As a strategy, we used metastatic lymph nodes
and primary GCs without LNM for genome-wide analysis as cells with
the abililty of LNM may constitute only a small population of the
cells in primary GCs with LNM. Differences in methylation levels
may be extremely small and may not be detected by the analysis
between primary GCs with and without LNM. Alternatively, in
metastatic lymph nodes, cancer cells are expected to possess the
aberrant DNA methylation following clonal selection. Moreover, the
methylation levels of appropriate marker CpG sites in the
metastatic lymph nodes are expected to be relatively high compared
with those in primary GCs with LNM.
In the present study, we aimed to identify CpG sites
with a methylation status associated with the presence of LNM of
GCs via a genome-wide methylation analysis using metastatic lymph
nodes and primary GCs without LNM and to validate the isolated
candidate markers.
Materials and methods
Patients, tissue samples and DNA
extraction
A total of 187 GC surgical samples were obtained
from patients who underwent gastrectomy with extended lymph node
dissection (D2) at the National Cancer Center Hospital (Tokyo,
Japan) and Aichi Cancer Center Hospital (Aichi, Japan) between 1994
and 2011 with informed consent. A total of three metastatic lymph
nodes were obtained from 3 of the 187 patients. No patients had
undergone prior chemotherapy or radiotherapy. Prognostic
information of 55 GC patients with LNM was available and the mean
follow-up period after surgery was 3,024 days. Disease grades were
classified according to the 6th edition of the TNM classification
by the UICC. Samples were stored at −80°C and a high molecular
weight DNA was extracted using the phenol/chloroform method. The
187 samples were divided into screening (28 GCs with LNM and 10
without) and validation (129 GCs with LNM and 20 without) sets in
advance, between which no significant differences in
clinicopathlogical data were observed (Table I). This study was conducted with the
approval of the Aichi Cancer Center and National Cancer Center.
 | Table IClinicopathological data of sample
sets. |
Table I
Clinicopathological data of sample
sets.
| N | Age (years) | P-value | Gender | N | P-value | T stage | N | P-value |
|---|
| Genome-wide analysis
seta |
| Meta (−) | 3 | 72±4 | 0.17 | Male | 2 | 1.0 | T1 | 0 | 0.51 |
| | | | | | | T2 | 1 | |
| | | | Female | 1 | | T3 | 1 | |
| | | | | | | T4 | 1 | |
| Meta (+) | 3 | 59±13 | | Male | | 2 | T1 | 0 | |
| | | | | | | T2 | 0 | |
| | | | Female | 1 | | T3 | 1 | |
| | | | | | | T4 | 2 | |
| Screening set |
| Meta (−) | 10 | 69±6 | 0.13 | Male | 7 | 0.53 | T1 | 0 | 0.17 |
| | | | | | | T2 | 1 | |
| | | | Female | 3 | | T3 | 6 | |
| | | | | | | T4 | 3 | |
| Meta (+) | 28 | 63±11 | | Male | 18 | | T1 | 0 | |
| | | | | | | T2 | 0 | |
| | | | Female | 10 | | T3 | 14 | |
| | | | | | | T4 | 14 | |
| Validation set |
| Meta (−) | 20 | 63±11 | 0.71 | Male | 13 | 0.6 | T1 | 0 | 0.14 |
| | | | | | | T2 | 3 | |
| | | | Female | 7 | | T3 | 8 | |
| | | | | | | T4 | 9 | |
| Meta (+) | 129 | 62±10 | | Male | 91 | | T1 | 0 | |
| | | | | | | T2 | 4 | |
| | | | Female | 38 | | T3 | 55 | |
| | | | | | | T4 | 70 | |
Genome-wide methylation analysis
Genome-wide screening of differentially methylated
CpG sites was performed using an Infinium HumanMethylation450
BeadChip array, which covers 485,577 CpG sites (Illumina, San
Diego, CA, USA) (20). Genomic DNA
(1 μg) was treated with sodium bisulfite using a Zymo EZ DNA
Methylation kit (Zymo Research, Irvine, CA, USA) and the
bisulfite-modified DNA was amplified prior to hybridization to the
array. The array was scanned with an iScan System (Illumina) and
the data were analyzed using GenomeStudio Methylation Module
Software (Illumina). A CpG site was considered to be informative if
the sum of the signals for methylated and unmethylated sequences at
the CpG site was significantly higher (at P<0.05) than signals
of the negative control probes on the same array. Methylation
levels were represented by β values, with a β value of 0
corresponding to no methylation and 1 corresponding to full
methylation.
Quantitative methylation-specific PCR
(qMSP)
Sample DNA was treated with sodium bisulfite and
purified as described previously (21). qMSP was performed using real-time
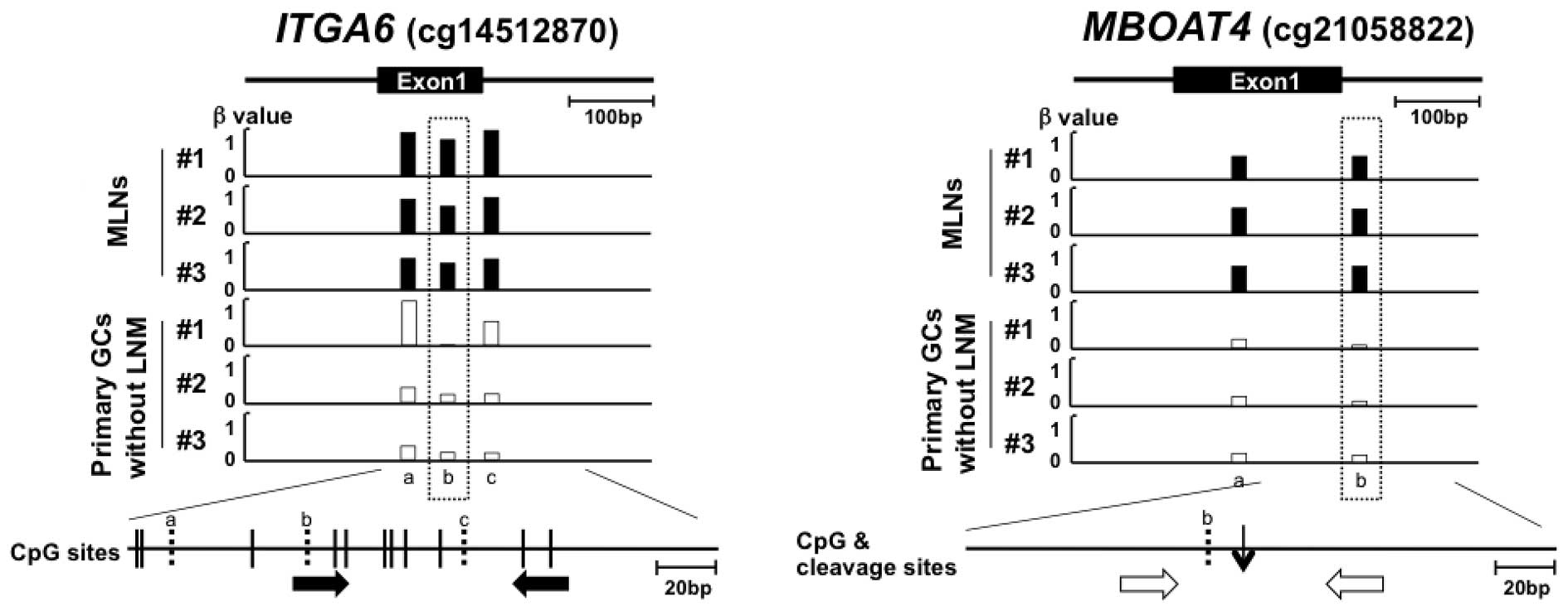
PCR with bisulfite-modified DNA and specific primers (Table II, Fig.
1A). A methylation level was expressed as a percentage of the
value of methylated DNA reference (PMR) calculated as the [(number
of fragments methylated at a target locus in sample/number of the
Alu sequences in sample)/(number of fragments methylated at
a target locus in SssI-treated DNA/number of the Alu
sequences in SssI-treated DNA)]×100 (22).
| Table IICpG sites identified by bead-chip
array analysis. |
Table II
CpG sites identified by bead-chip
array analysis.
| No. | Probe name
(IlmnID)a | Gene symbol | Location (Chr:
base) | Relation to CpG
island | Position to
gene | P-valueb | Cut-off (YI) | Primer sequences
(5′-3′) | Annealing
temp. | PCR type |
Mg2+c (μM) |
|---|
|
|
|---|
| Screening | Validation | Forward | Reverse |
|---|
| 1 | cg23218354 | - | Chr1:2885244 | Island | - | 0.05 | 0.17 | 10.1 (0.48) |
TGGTTTTTATACGGGGGATTTAC |
ACTAAACCAAAACGACGATTACG | 60 | qMSP | 1.5 |
| 2 | cg13239126 |
KIAA1026 | Chr1:15256136 | - | Body | 0.24 | - | - |
CTCCAGAGAGACAGGCATGGTT |
CAAGCCTGACCTTCCCTCTCC | 60 | qPTMR | 1.5 |
| 3 | cg16112880 | TMEM9 | Chr1:201123745 | Island | TSS200 | 0.41 | - | - |
CCCGCCCTCTCCTAGCTTCTAT |
GGCTGACGTTCCCTTTTCTGGT | 63 | qPTMR | 1.5 |
| 4 | cg14512870 | ITGA6 | Chr2:173330342 | - | Body | 0.01 | 0.07 | 32.5 (0.44) |
TATAGTTGCGATATTATCGTTC |
AAACTACCGAAATAAACGCT | 51 | qMSP | 2.5 |
| 5 | cg09866366 | ABCF3 | Chr3:183903315 | Shore | TSS1500 | 0.34 | - | - |
TCGTTAGATTACGGGTGTTTC |
CAAAACGCATATATAACGATAACG | 58 | qMSP | 2.5 |
| 6 | cg08812108 | - | Chr6:2515318 | - | - | 0.03 | 0.24 | 56.3 (0.44) |
AGCGTTGGCGTTAGGTAGGGTAGTTC |
CCAAATAACCACCTACGTCTTTACG | 63 | qMSP | 1.5 |
| 7 | cg06728252 | ABT1 | Chr6:26598149 | Island | Body | 0.24 | - | - |
CGCGTAGATCGGTTCGTGAGAC |
GCCACGCGCTTAACTATACG | 63 | qMSP | 1.5 |
| 8 | cg08972588 | TNXB | Chr6:32014674 | - | Body | 0.64 | - | - |
CCTGAGCAAGAATGAGGCCAGA |
GGGGACAAGGGGGAGATCACA | 65 | qPTMR | 2.5 |
| 9 | cg22126965 | COX19 | Chr7:1015501 | Shore | TSS1500 | 0.50 | - | - |
GGTTTAGAAAGGTTTAGCGAATTGTTC |
AACAACCGCAAACAACG | 62 | qMSP | 2.5 |
| 10 | cg18450582 | DYNC1I1 | Chr7:95546539 | - | Body | 0.32 | - | - |
ACCTTGGCCTCTGGATTGTGGA |
GCACTGCCTGCCTGAAAGGAGA | 64 | qPTMR | 1.5 |
| 11 | cg02005782 | - | Chr7:105857664 | - | - | 0.59 | - | - |
GAAGTCAGCCAGGCATTGGAAG |
CCCAGCTGCCTTTCTGATCTCT | 65 | qPTMR | 1.5 |
| 12 | cg06436185 | PRKAG2 | Chr7:151442351 | - | Body | 0.04 | 0.03 | 28.8 (0.24) |
ATTTAGTTTTTTGTACGGTTGC |
CCCAATAAAACGACGTAACG | 55 | qMSP | 2.5 |
| 13 | cg21058822 | MBOAT4 | Chr8:30002223 | - | TSS200 | 0.38 | - | - |
GGCTGTCTCTGGTCTTTTTATC |
AGAAAGCCAGTTTTTATTCTGC | 61 | qPTMR | 1.5 |
| 14 | cg12089032 | - | Chr8:72881203 | - | - | 0.03 | 0.09 | 40.6 (0.41) |
GCAAGTTAAGGCATCGTAGGAAAGC |
GGCAGAGAGGAACAGCTCCTAAG | 66 | qPTMR | 1.5 |
| 15 | cg23170346 | - | Chr8:134863880 | - | - | 0.95 | - | - |
CTAGCCACATCCATAGCAGACAGG |
CACTCAGCAATGCAAACAGTCTTG | 66 | qPTMR | 1.5 |
| 16 | cg19878482 | C8orf73 | Chr8:144655026 | Shore | TSS200 | 0.10 | - | - |
GGAGTTTTTCGGGTTCGGTTTC |
CAAAAACCCATTATAAACACGTCCGT | 65 | qMSP | 2.5 |
| 17 | cg01263942 | DIP2C | Chr10:695859 | - | Body | 0.01 | 0.12 | 23.2 (0.38) |
GTTCGTTATTTGCGTTTTCGTGC |
CAACGAAAAAACTCCATAAACCG | 59 | qMSP | 2.5 |
| 18 | cg03015672 |
ARHGAP12 | Chr10:32216066 | Shore | 5′UTR | 0.88 | - | - |
AGAACAGTGGAGCCGCATGCAA |
CCAAAGCAGGCAGTGAAAGCGT | 66 | qPTMR | 1.5 |
| 19 | cg10326726 | MSMB | Chr10:51549505 | - | TSS200 | 0.16 | - | - |
CAACCCTCTGTAAACACTCAAT |
TATAGACAGGTACATCCAGGCA | 57 | qPTMR | 2.5 |
| 20 | cg19864370 | - | Chr10:80354592 | - | - | 0.00 | 0.29 | 70.6 (0.69) |
GAATAGCTTAGGCCCCTGTCAT |
GATAGTGCTAGCCCTTGGGAAT | 60 | qPTMR | 1.5 |
| 21 | cg03850986 | ABLIM1 |
Chr10:116408382 | - | Body | 0.38 | - | - |
TGATAAAAATGCTCTGGAATTAG |
TGGAGATGTAATGTAGTACACCATA | 51 | qPTMR | 1.5 |
| 22 | cg25885280 | SHANK2 | Chr11:70760166 | - | Body | 0.34 | - | - |
GCGGTGGGGGATTTCTGTAAGGA |
GAGCAGGGTGTGCCTTCTCAGGG | 68 | qPTMR | 1.5 |
| 23 | cg26894278 | CRYL1 | Chr13:21016241 | - | Body | 0.22 | - | - |
GTTAAGTTTAAATGGAGCCTTG |
TGACAGGATTACAATAAGGCTA | 56 | qPTMR | 1.5 |
| 24 | cg04339360 | KLF5 | Chr13:73635568 | Shore | Body | 0.04 | 0.31 | 25.4 (0.43) |
TAGTCAAGAAAAGAAACCTGTGCAA |
TGCCAAACTACCTCAATTCTGTTTA | 61 | qPTMR | 1.5 |
| 25 | cg16206504 | - |
Chr13:114917223 | Shelf | - | 0.02 | 0.35 | 35.2 (0.41) |
CGAGATTGTAGGCGGTTGTTC |
CCTAACTATTACAACAATACCGAACG | 63 | qMSP | 1.5 |
| 26 | cg14851578 | - |
Chr14:106187192 | Shore | - | 0.08 | - | - |
GGAGTGTGGGTTACGTGTGATTAC |
CAATCTCGCCCACTCACG | 66 | qMSP | 1.5 |
| 27 | cg02990302 |
C16orf80 | Chr16:58155189 | - | Body | 0.04 | 0.45 | 56.2 (0.65) |
TCCTTTCCTTAGCTCCTTCCAG |
AAAAACAGTCGGCTCTTTGTGA | 63 | qPTMR | 1.5 |
| 28 | cg08292959 | MGAT5B | Chr17:74878420 | Island | Body | 0.97 | - | - |
GGCACCTGCCACTCCATCCG |
TGCACTCTGGGCTGTACCACAGTG | 63 | qPTMR | 1.5 |
| 29 | cg15645685 | PBX4 | Chr19:19730175 | Shore | TSS1500 | 0.26 | - | - |
CTAATGCTCCCTGCATCCTCAG |
TAAACAAGCGAGGTCACTCTTCAGC | 64 | qPTMR | 1.5 |
| 30 | cg14571622 | NLRP8 | Chr19:56499348 | - | 3′UTR | 0.01 | - | - |
TGGGGCTTGATTGATCAGTTCC |
CCAGGGTTCAAAGCTGAGGTTC | 62 | qPTMR | 1.5 |
| 31 | cg27050343 | OTC | ChrX:38211596 | - | TSS200 | 0.15 | - | - |
AATTTTTGGGTTTAAGTGATTCGTTC |
AAAAAAATAATTACTAACCGAACACG | 62 | qMSP | 1.5 |
Quantitative PCR following treatment with
a methylation-dependent restriction enzyme (qPTMR)
A fully unmethylated control was prepared by
amplifying human blood genomic DNA with phi29 DNA polymerase
(Illustra GenomiPhi HY kit, GE Healthcare, Buckinghamshire, UK)
(23). DNA (1 μg)was treated with
MspJI (New England Biolabs, Beverly, MA, USA), which cleaves
DNA 9 bp downstream from the mCNNR sequence (24,25),
in a 30 μl reaction [4 U of MspJI, 1× NEB buffer 4 (New
England Biolabs) and 0.1 mg/ml BSA] at 37°C for 20 h. Following
purification, the DNA was treated with MspJI again and
dissolved in TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA) at a
concentration of 5 ng/μl without purification. Using 1 μl of the
solution, quantitative PCR (qPCR) was performed by real-time PCR
with primers that encompassed a target MspJI site (Fig. 1B). To normalize the quantity of
input DNA, the number of copies of a standard sequence, which may
be amplified with a primer pair (5′-TTGCTTGAAGTTTTGTTGCTGTAGT-3′
and 5′-AATAAACTCAGTTGTGACATGGACA-3′) and contains no MspJI
site, was measured by qPCR. A percentage of the value of
unmethylated reference (PUR) was calculated as the [(number of
fragments at target locus in sample/number of the standard sequence
in sample)/(number of fragments at target locus in
GenomiPhi-amplified DNA/number of the standard sequences in
GenomiPhi-amplified DNA)]×100. For convenience, the methylation
level was expressed as 100-PUR.
Statistical analysis
Statistical analyses were conducted using PASW
statistics version 18.0.0 (SPSS Japan Inc., Tokyo, Japan). The
difference between the mean values of the two groups of samples was
evaluated using Welch’s t-test. The Fisher’s exact test was used to
evaluate the significant difference in relative frequency of the
phenomena between two independent groups. Survival curves were
computed according to the Kaplan-Meier method and the log-rank test
was employed to evaluate the level of significant difference.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Genome-wide screening using metastatic
lymph nodes and GCs without LNM
To isolate the CpG sites that are hypermethylated
specifically in GCs with LNM, genome-wide methylation analysis was
performed using metastatic lymph nodes (n=3) and GCs without LNM
(n=3) using an Infinium HumanMethylation450 BeadChip array. The
samples used for this analysis were prepared from 6 patients in the
screening set (Table I). The mean
number of informative CpG sites was 485,170 (SD 209) in the
metastatic lymph nodes and 485,001 (SD 514) in the GCs without LNM
(P=0.63). We searched for CpG sites that were highly methylated in
the three metastatic lymph nodes [β value > a) 0.6, b) 0.5 and
c) 0.4] and hardly methylated in the three primary GCs without LNM
(β value <0.2) and the number of hypermethylated CpG sites was
a) 1, b) 31 and c) 209, respectively. To obtain a practicable
number of candidate CpG sites, we adopted a cut-off β value of 0.5
and the 31 CpG sites were selected for further analysis (Table II).
Selection of informative candidate
genomic regions among primary GCs
Using primary GCs with and without LNM (screening
set, Table I), the methylation
levels of genomic regions around the 31 CpG sites were measured by
qMSP or qPTMR, which are accurate and sensitive enough to detect
aberrant DNA methylation in a small population of cells. Of the 31
regions, 10 regions exhibited higher methylation levels in GCs with
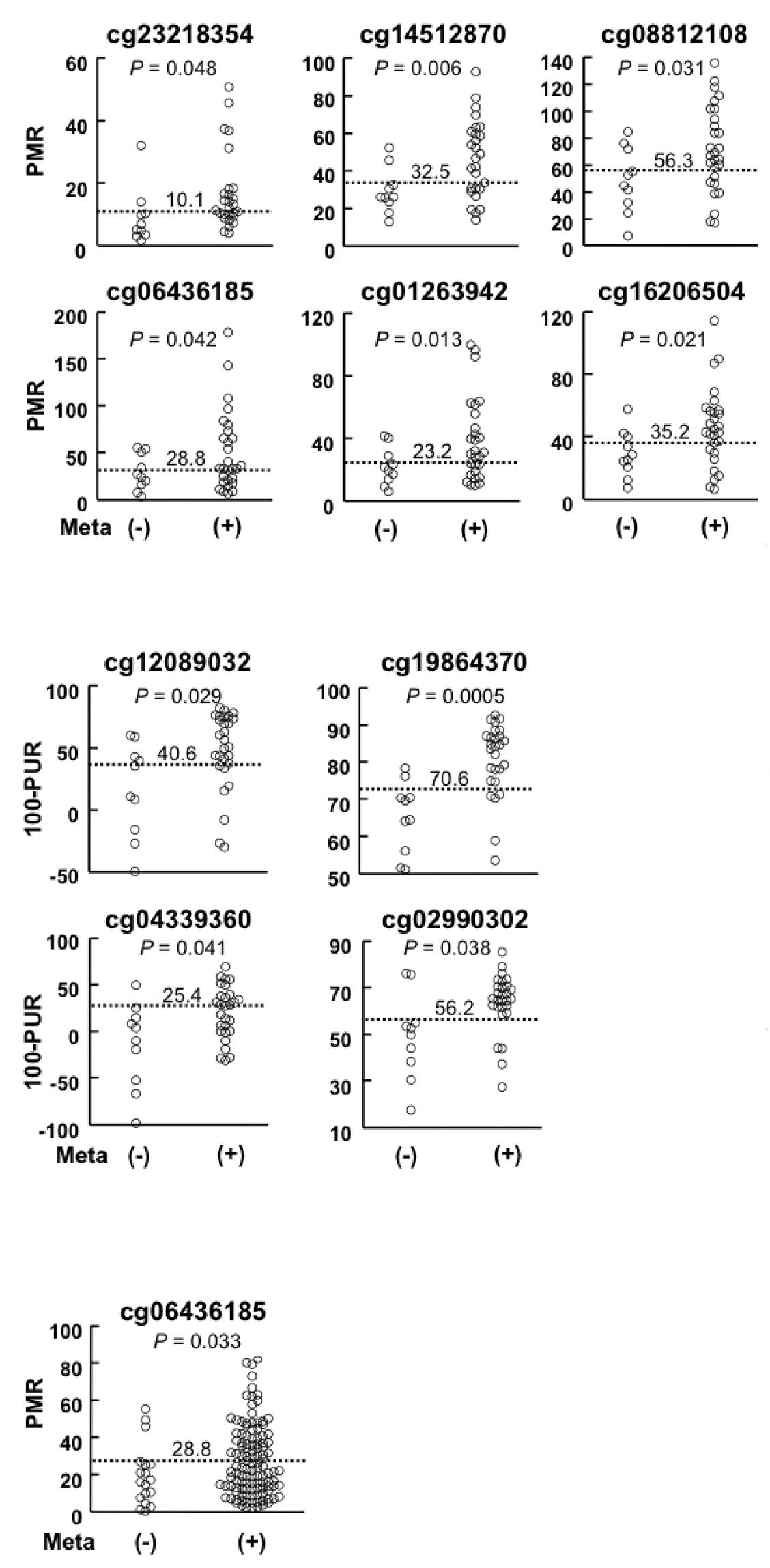
LNM (1.4- to 1.9-fold) than in those without LNM (Table II and Fig. 2A and B). For each of the 10 genomic
regions, a cut-off methylation level was established in order that
the Youden index (sensitivity + specificity − 1) would be maximized
(Table II and Fig. 2).
Validation of the candidate genomic
regions in a different set of samples
To validate the hypermethylation of the 10 candidate
genomic regions in GCs with LNM, the methylation levels were
analyzed in an independent sample set (validation set, Table I). A region around the cg06436185
CpG site revealed significantly higher methylation levels in GCs
with LNM (1.5-fold) than those without (P=0.033, Fig. 2C), whereas the other nine regions
were not validated (Table II). The
region was located in the gene body of the PRKAG2 gene and
did not belong to a CpG island (Table
II). Therefore, it was unlikely that the methylation status of
the region around cg06436185 affected the transcription of a gene.
Using a cut-off level established in the analysis of the screening
set (28.8%), the presence of LNM was detected at a sensitivity of
43% and a specificity of 85%. This result indicated that a
methylation level of this region is a candidate marker for the
detection of the presence of LNM.
Association between the methylation level
of the genomic region around the cg06436185 CpG site and
clinicopathological characteristics
Associations between the methylation level of the
genomic region around cg06436185 and clinicopathological
characteristics (age, gender and T category) were analyzed in 157
GC patients with LNM and 30 without LNM. No difference in
methylation levels according to age, gender or T category was found
(Table III). Using 55 of the 157
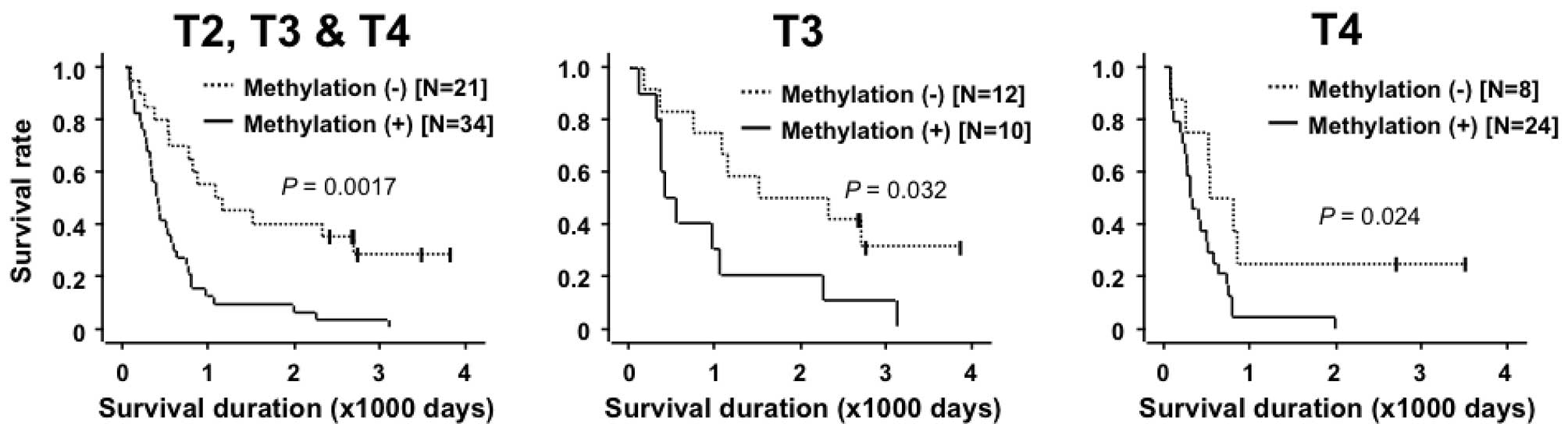
GC patients with LNM, whose prognostic information was available
(T2, one patient; T3, 22 patients; T4, 32 patients), a correlation
between the methylation level and survival rate was analyzed.
Patients with high methylation levels (>28.8%; the value used to
detect the presence or absence of LNM) had a significantly poorer
overall survival rate compared to those with low methylation levels
(P=0.0017; Fig. 3A). Since the T
category is known to be the major prognostic factor in GC patients
(26), patients in the T3 and T4
categories were analyzed separately. In the T3 and T4 subgroups,
the patients with high methylation levels demonstrated a
significantly poorer overall survival rate than those with low
methylation (P=0.032 and 0.024, respectively; Fig. 3B and C). These results revealed that
the high methylation level of the genomic region around cg06436185
was associated with an unfavorable prognosis, regardless of the
depth of tumor invasion.
| Table IIIAssociation between methylation
levels of the genomic region around cg0643618 and clinical
characteristics. |
Table III
Association between methylation
levels of the genomic region around cg0643618 and clinical
characteristics.
| | Methylation
level |
|---|
| |
|
|---|
| Parameters | N | Mean | SD | P-value |
|---|
| Age |
| ≤60 | 77 | 32.2 | 24.0 | 0.26 |
| >60 | 110 | 28.1 | 25.2 | |
| Gender |
| Female | 58 | 34.9 | 26.4 | 0.07 |
| Male | 129 | 27.6 | 23.6 | |
| T category |
| T3 | 83 | 26.8 | 27.1 | 0.07 |
| T4 | 96 | 33.6 | 22.5 | |
Discussion
Using a genome-wide methylation analysis using
metastatic lymph nodes and primary GCs without LNM, a genomic
region (around cg06436185) whose methylation level in primary GCs
was associated with the presence of LNM was successfully
identified. Notably, the association was also significant in an
independent validation set (P=0.033). Generally, markers isolated
by genome-wide analyses need to be validated in a different set of
samples due to the overfitting issues caused by multiple testing
(27). Even in the present study, 9
of the 10 candidate genomic regions that revealed significant
hypermethylation in GCs with LNM in the screening set
(P=0.0005–0.048) were not reproduced in the validation set. This
observation emphasizes the value of the methylation level of the
genomic region around cg06436185. Since it had a sensitivity of 43%
and specificity of 85%, the combined use of this novel methylation
marker with imaging tools is predicted to improve the diagnostic
accuracy of LNM of GCs.
The mean methylation levels of GCs with and without
LNM were 18.7 and 27.5%. This small difference is extremely
difficult to detect by a genome-wide screening method. Our strategy
in the present study was to benefit from the monoclonal growth of
cells in metastatic lymph nodes and compare metastatic lymph nodes
and GCs without LNM. The methylation levels of the genomic regions
around cg06436185 were 13.2 and 54.3%, respectively, in these
samples. This relatively significant difference was identified
using genome-wide screening, which has a relatively low accuracy in
the analysis of methylation levels. Using a more accurate and
sensitive method, qMSP, the small difference between GCs with and
without LNM (18.7 and 27.5%, respectively) was clearly
demonstrated.
A method to measure methylation levels in CpG-poor
genomic regions, qPTMR, was developed using a combination of
digestion with a methylation-dependent restriction enzyme and qPCR.
qPTMR had an error range of 5% in this study. It is difficult to
measure methylation levels in CpG-poor genomic regions by qMSP, a
well-established method with a high accuracy, due to the difficulty
in designing primers. Alternatively, MspJI, a recently
developed methylation-sensitive restriction enzyme, recognizes
mCNNR (N=A, T, G or C; R=G or C) sequences and cleaves
DNA when the C is methylated (24,25).
Since the recognition sequence is applicable to the majority of CpG
sites and cytosines in non-CpG sites are not methylated in somatic
cells, the positive cleavage by MspJI is used to determine
methylation status of most CpG sites. Using qPTMR, the methylation
levels of all the 19 candidate regions with few CpG sites were
quantified. This new method is predicted to have various
applications.
The methylation status of the genomic region around
cg06436185 was unlikely to affect transcription of a known nearby
gene (PRKAG2). However, its high methylation level in GCs,
namely large fractions of cancer cells with methylation in cancer
tissue, was associated with the presence of LNM and also with a
poorer prognosis of the GC patients. One possible reason is that
the region is located in a promoter region of unknown genes,
including microRNA genes, or in enhancer regions whose methylation
is critical for the regulation of gene expression levels. Another
possible reason is that the methylation of the region is caused by
an abnormality of unknown methylation regulation and that this
abnormality is critical for tumor metsastasis or malignancy. In
this case, other genomic regions are likely to be methylated in GCs
with LNM or a poorer prognosis.
In conclusion, we identified one genomic region with
a methylation status in primary GCs that was associated with the
presence of LNM and a poorer prognosis of GC patients.
Acknowledgements
We thank Dr Michihiro Ishida, Dr Yukie Yoda and Dr
Masahiro Maeda for their assistance during sample preparation. This
study was supported by the Third-term Comprehensive Cancer Control
Strategy from the Ministry of Health, Labour and Welfare, Japan, by
the JSPS A3 Foresight Program and by the National Cancer Center
Research and Development Fund. Y.S. is a recipient of Research
Resident Fellowships from the Foundation for Promotion of Cancer
Research.
References
|
1
|
Matsuda A and Matsuda T: Time trends in
stomach cancer mortality (1950–2008) in Japan, the USA and Europe
based on the WHO mortality database. Jpn J Clin Oncol. 41:932–933.
2011.
|
|
2
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
3
|
Yokota T, Ishiyama S, Saito T, et al:
Lymph node metastasis as a significant prognostic factor in gastric
cancer: a multiple logistic regression analysis. Scand J
Gastroenterol. 39:380–384. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen JH, Wu CW, Lo SS, et al: Lymph node
metastasis as a single predictor in patients with Borrmann type I
gastric cancer. Hepatogastroenterology. 54:981–984. 2007.PubMed/NCBI
|
|
5
|
Saito H, Fukumoto Y, Osaki T, et al:
Prognostic significance of level and number of lymph node
metastases in patients with gastric cancer. Ann Surg Oncol.
14:1688–1693. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sano T and Aiko T: New Japanese
classifications and treatment guidelines for gastric cancer:
revision concepts and major revised points. Gastric Cancer.
14:97–100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonenkamp JJ, Songun I, Hermans J, et al:
Randomised comparison of morbidity after D1 and D2 dissection for
gastric cancer in 996 Dutch patients. Lancet. 345:745–748. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cuschieri A, Fayers P, Fielding J, et al:
Postoperative morbidity and mortality after D1 and D2 resections
for gastric cancer: preliminary results of the MRC randomised
controlled surgical trial. The Surgical Cooperative Group. Lancet.
347:995–999. 1996. View Article : Google Scholar
|
|
9
|
Degiuli M, Sasako M, Ponti A, Soldati T,
Danese F and Calvo F: Morbidity and mortality after D2 gastrectomy
for gastric cancer: results of the Italian Gastric Cancer Study
Group prospective multicenter surgical study. J Clin Oncol.
16:1490–1493. 1998.
|
|
10
|
Seevaratnam R, Cardoso R, McGregor C, et
al: How useful is preoperative imaging for tumor, node, metastasis
(TNM) staging of gastric cancer? A meta-analysis. Gastric Cancer.
Aug 12–2011.(E-pub ahead of print).
|
|
11
|
Ganpathi IS, So JB and Ho KY: Endoscopic
ultrasonography for gastric cancer: does it influence treatment?
Surg Endosc. 20:559–562. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshioka T, Yamaguchi K, Kubota K, et al:
Evaluation of 18F-FDG PET in patients with advanced,
metastatic, or recurrent gastric cancer. J Nucl Med. 44:690–699.
2003.PubMed/NCBI
|
|
13
|
Ha TK, Choi YY, Song SY and Kwon SJ:
F18-fluorodeoxyglucose-positron emission tomography and computed
tomography is not accurate in preoperative staging of gastric
cancer. J Korean Surg Soc. 81:104–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Natsugoe S, Mueller J, Stein HJ, Feith M,
Hofler H and Siewert JR: Micrometastasis and tumor cell
microinvolvement of lymph nodes from esophageal squamous cell
carcinoma: frequency, associated tumor characteristics, and impact
on prognosis. Cancer. 83:858–866. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kojima N, Yonemura Y, Bando E, et al:
Optimal extent of lymph node dissection for T1 gastric cancer, with
special reference to the distribution of micrometastasis, and
accuracy of preoperative diagnosis for wall invasion.
Hepatogastroenterology. 55:1112–1117. 2008.PubMed/NCBI
|
|
16
|
Motoyama K, Inoue H, Mimori K, et al:
Clinicopathological and prognostic significance of PDCD4 and
microRNA-21 in human gastric cancer. Int J Oncol. 36:1089–1095.
2010.PubMed/NCBI
|
|
17
|
Tanaka M, Kitajima Y, Edakuni G, Sato S
and Miyazaki K: Abnormal expression of E-cadherin and beta-catenin
may be a molecular marker of submucosal invasion and lymph node
metastasis in early gastric cancer. Br J Surg. 89:236–244.
2002.PubMed/NCBI
|
|
18
|
Arigami T, Natsugoe S, Uenosono Y, et al:
CCR7 and CXCR4 expression predicts lymph node status including
micrometastasis in gastric cancer. Int J Oncol. 35:19–24. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen Z, Ye Y, Dong L, et al: Kindlin-2: a
novel adhesion protein related to tumor invasion, lymph node
metastasis, and patient outcome in gastric cancer. Am J Surg.
203:222–229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bibikova M, Barnes B, Tsan C, et al: High
density DNA methylation array with single CpG site resolution.
Genomics. 98:288–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oka D, Yamashita S, Tomioka T, et al: The
presence of aberrant DNA methylation in noncancerous esophageal
mucosae in association with smoking history: a target for risk
diagnosis and prevention of esophageal cancers. Cancer.
115:3412–3426. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niwa T, Tsukamoto T, Toyoda T, et al:
Inflammatory processes triggered by Helicobacter pylori
infection cause aberrant DNA methylation in gastric epithelial
cells. Cancer Res. 70:1430–1440. 2010.
|
|
23
|
Niwa T, Yamashita S, Tsukamoto T, et al:
Whole-genome analyses of loss of heterozygosity and methylation
analysis of four tumor-suppressor genes in
N-methyl-N′-nitro-N-nitrosoguanidine-induced rat stomach
carcinomas. Cancer Sci. 96:409–413. 2005.PubMed/NCBI
|
|
24
|
Zheng Y, Cohen-Karni D, Xu D, et al: A
unique family of Mrr-like modification-dependent restriction
endonucleases. Nucleic Acids Res. 38:5527–5534. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cohen-Karni D, Xu D, Apone L, et al: The
MspJI family of modification-dependent restriction endonucleases
for epigenetic studies. Proc Natl Acad Sci USA. 108:11040–11045.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yokota T, Ishiyama S, Saito T, et al: Is
tumor size a prognostic indicator for gastric carcinoma? Anticancer
Res. 22:3673–3677. 2002.PubMed/NCBI
|
|
27
|
Simon R, Radmacher MD, Dobbin K and
McShane LM: Pitfalls in the use of DNA microarray data for
diagnostic and prognostic classification. J Natl Cancer Inst.
95:14–18. 2003. View Article : Google Scholar : PubMed/NCBI
|