Introduction
Hepatocellular carcinoma (HCC) is the seventh most
prevalent cancer worldwide, accounting for 85–90% of all primary
liver cancers, and ranking as the fourth leading cause of
cancer-related mortality (1,2).
Despite great advances in the treatment of HCC, the 5-year survival
rate remains poor. In order to reduce the high mortality rate, it
is critical to clarify the precise mechanism underlying HCC
development and progression.
miRNAs are small non-coding RNAs of approximately
19–25 nucleotides that post-transcriptionally regulate gene
expression. miRNAs usually bind to the 3′ untranslated region (UTR)
of target mRNAs through their seed sequences, leading to the
suppression of translation, and occasionally mRNA degradation
(3,4). miRNAs are estimated to modulate 30% of
human protein-coding genes (5).
Increasing evidence has shown that miRNAs have significant roles in
diverse biological and pathological processes (6). Meanwhile, the deregulation of miRNAs
has been observed in diverse diseases, including cancer (7,8).
Numerous studies have shown that miRNAs are frequently deregulated
in HCC. For example, miR-21 is upregulated in HCC and negatively
regulates the expression of the tumor suppressor gene, phosphatase
and tensin homolog (PTEN), which promotes cancer progression and
metastasis (9). Wong et al
suggested that miR-222 functions as a metastatic activator in HCC
via the activation of the AKT signaling pathway by targeting
PPP2R2A (10). By contrast, certain
metastatic suppressive miRNAs including miR-34a, miR-23b, miR-122
and miR-124 are frequently downregulated in HCC and facilitate
tumor metastasis by regulating vital genes (11–14).
The abnormal expression of miR-224 has been reported
in several human cancers (15–19).
Wang et al found that miR-224 is upregulated in HCC patients
and HCC cell lines (20), and
promotes HCC cell apoptosis by targeting the transcript expression
levels of apoptosis inhibitor 5 (API-5). Simultaneously, miR-224
also promotes HCC cell proliferation, although its potential target
gene is currently unknown. More recently, miR-224 has been
confirmed to be involved in the malignant phenotype of HepG2 cells,
and is reported to be a significant factor in the regulation of the
migration and invasion of HepG2 cells (21); however, the authors did not propose
a potential target gene that may serve as an intermediary between
miR-224 and metastasis in HCC cells.
In the present study, TargetScan, PicTar and miRBase
Targets were employed to predict the putative targets of miR-224,
which has a pivotal role in cell proliferation and metastasis. We
selected the serine/threonine-protein phosphatase 2A 65 kDa
regulatory subunit A β isoform (PPP2R1B) as a target for our study,
since PP2A is a well-conserved and essential protein
serine/threonine phosphatase, as well as a critical regulator in
the control of certain key proteins of oncogenic signaling cascades
(22). It has also been suggested
that PP2A has an effect on the regulation of motility and invasion
of both normal and transformed cells (23). Hamano et al demonstrated that
miR-200c induces chemoresistance in esophageal cancer through the
activation of the AKT signaling pathway, and the authors proposed
that miR-200c stimulates the AKT signaling pathway by specifically
targeting PPP2R1B (24). Therefore,
we postulated that miR-224 might impact the proliferation and
metastasis of HCC cells through the activation of the AKT signaling
pathway by targeting the PPP2R1B tumor suppressor gene.
Materials and methods
Cell culture
The human HCC cell line HepG2 was purchased from the
Shanghai Institute of Cell Biology (Academia Sinica, Shanghai,
China). Cells were cultured in Dulbecco's modified Eagle's medium
(DMEM) high glucose supplemented with heat-inactivated 10% FBS, 100
U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified
incubator containing 5% CO2.
Analysis of miR-224 expression by
qRT-PCR
Reverse transcription and quantitative real-time PCR
(qPCR) were employed to measure the expression levels of miR-224 in
HepG2 cells. Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA). Whole-cell cDNA was synthesized,
then miR-224 expression was analyzed using a real-time PCR
instrument (ABI7000) and an All-in-One™ miRNA qRT-PCR detection kit
(GeneCopoeia, Rockville, MD, USA). The reverse transcription
reaction parameters were 37°C for 60 min followed by 85°C for 5
min. The PCR parameters were as follows: 95°C for 10 min, followed
by 40 cycles of 95°C for 10 sec, 60°C for 20 sec, and 72°C for 3
sec. The expression levels of miR-224 were normalized to RNU6B
levels. All reactions were performed in triplicate and included
negative control reactions without cDNA.
miRNAs and transfection
The miRNAs were designed and synthesized by
GenePharma (Shanghai, China). The following Homo sapiens
(has)-miR-224 mimics were synthesized: sense, 5′-CAA GUC ACU AGU
GGU UCC GUU-3′; antisense, 5′-CGG AAC CAC UAG UGA CUU GUU-3′;
negative control: sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′;
antisense, 5′-ACG UGA CAC GUU CGG AGA ATT-3′; has-miR-224
inhibitor: 5′-AAC GGA ACC ACU AGU GAC UUG-3′; has-miR-224 inhibitor
negative control: 5′-CAG UAC UUU UGU GUA GUA CAA-3′. miRNA
transfection was performed using FuGENE HD transfection reagent
(Indianapolis, IN, USA). In brief, 4×105 cells were
plated in 2 ml medium in the wells of a six-well plate. Cells were
incubated overnight to achieve >80% confluence at the time of
transfection. In each well, 5 μl miRNA was added to 100 μl
serum-free medium. Separately, 5 μl FuGENE HD transfection reagent
was added to 100 μl serum-free medium and mixed gently. The
transfection complex was incubated for 15 min at room temperature,
added to cells, and incubated for 6 h after which the
serum-containing medium was replaced. Following transfection, the
cells were incubated for 48 and 72 h prior to qPCR and western blot
analysis, respectively.
Western blot analysis
Total protein from HepG2 cells was extracted with 1%
RIPA lysis buffer (Beyotime, Shanghai, China) containing 1 mM
phenylmethanesulfonylfluoride (PMSF). The supernatants were
collected, and protein concentration was determined using the BCA
assay kit (Beyotime). Proteins were resolved on 12% SDS-PAGE gels
using a mini-gel apparatus (Bio-Rad Laboratories, Hercules, CA,
USA) and transferred to nitrocellulose membranes. The membranes
were blocked for 1 h at room temperature and incubated overnight at
4°C with primary antibody, followed by incubation with
HRP-conjugated secondary antibody after three 10-min washes in
TBST. After incubation with secondary antibody, the membrane was
washed three times for 10 min in TBST, and the protein was detected
by chemiluminescence with the ECL detection reagent (Pierce
Biotechnology, Inc., Rockford, IL, USA). The data were normalized
to β-actin.
Luciferase reporter assay
Human PPP2R1B 3′UTR containing the miR-224 target
sequence (position 141–147) and mutant sequences were chemically
synthesized (GenScript, Nanjing, China) and cloned into the PGL3
control vector (Promega, Madison, WI, USA) downstream of the
luciferase gene using the XbaI site. The resulting plasmids
were designated pPPP2R1B 3′UTR-luci-WT and pPPP2R1B 3′UTR-luci-MUT,
respectively. HepG2 cells were transfected with either pPPP2R1B
3′UTR-luci-WT or pPPP2R1B 3′UTR-luci-MUT in 24-well plates using
FuGENE HD transfection reagent according to the manufacturer's
instructions, and co-transfected with either 30 nM of has-miR-224
mimics (GenePharma) or the same concentration of negative control
(GenePharma). Each well was also co-transfected wth 50 ng of the
pRL-TK plasmid (Promega) to determine transfection efficiency.
Then, firefly and Renilla luciferase activity levels were measured
in cells that were harvested 24 h post-transfection, using the
Dual-Luciferase Reporter assay kit (Promega). Each transfection was
performed in triplicate.
Cell proliferation assay
The viability of cells was determined using the Cell
Counting Kit-8 (Dojindo Laboratories, Japan). Cells were typsinized
and seeded into 96-well plates at a density of 5×103
cells/well 6 h after transfection. After incubation in a humidified
incubator (37°C, 5% CO2) for different time periods, 10
μl CCK-8 was added to each well, followed by incubation at 37°C for
another 1 h. The absorbance was measured at 450 nm using a
microplate reader to evaluate the number of viable cells.
Transwell migration and invasion
assays
The Transwell chamber with 8-μm pore filters (6.5 mm
in diameter, 8 μm pore size, Corning Inc., Lowell, MA, USA) was
used for the migration and invasion assays. For the migration
assay, HepG2 cells were transfected with the desired miRNAs, and
then trypsinized 48 h after transfection. The cells were then
resuspended in serum-free medium, and the concentration was
adjusted to 1×105, followed by the addition of 200 μl
cell suspension to the upper chamber. Next, 700 μl medium
containing 20% FBS was added to the lower chamber. Cells were
incubated for 24 h at 37°C in a humidified incubator containing 5%
CO2. Then, cells that migrated to the bottom of the
membrane were fixed in 4% paraformaldehyde and stained with crystal
violet for 10 min at room temperature, followed by visualization
under a microscope after washing with distilled water. For the
invasion assay, the protocol was the same as the migration assay,
except that the upper chamber was precoated with 1 mg/ml Matrigel
(BD Biosciences, Bedford, MA, USA), followed by prolonged
incubation for 36 h.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD) from at least three separate experiments.
Differences among experimental groups were analyzed by the
Student's t-test using the SPSS Graduate Pack statistical software,
version 11.0. P<0.05 was considered to indicate a statistically
significant result.
Results
miR-224 positively regulates HCC cell
proliferation in vitro
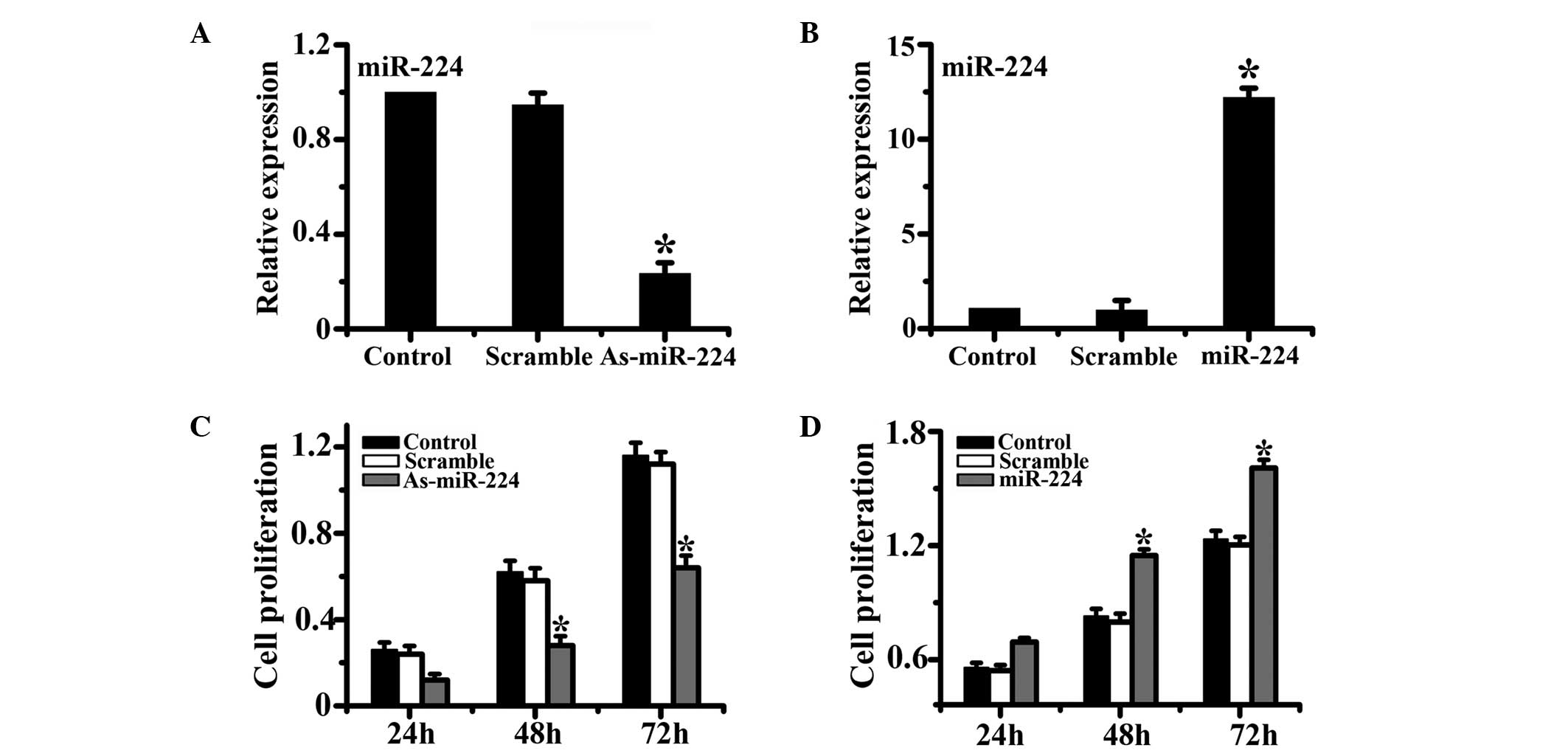
To assess the role of miR-224 in the proliferation
of HCC cells, both loss-of-function and gain-of-function approaches
were employed in our study. HepG2 cells were treated with miR-224
mimics or miR-224 inhibitors and control scramble oligonucleotides.
Results from qPCR demonstrated that the relative expression of
miR-224 was successfully modulated in both the miR-224 mimic and
miR-224 inhibitor groups compared with the control group. Cell
proliferation was evaluated by the CCK-8 assay at different time
points. The results showed that the downregulation of miR-224
significantly inhibited cell proliferation compared with the
control group, while relative to the control group, the
upregulation of miR-224 significantly enhanced the cell
proliferation capability of HCC cells (Fig. 1).
Cell migration and invasion promoted by
miR-224
In order to determine the effect of miR-224 on cell
migration and invasion, HepG2 cells were transfected with synthetic
oligonucleotides and divided into four groups: miR-224 mimics,
miR-224 inhibitors, scrambled oligonucleotides and control groups.
The results from the Transwell migration assay revealed a
significant difference in the experimental groups compared to the
controls. Specifically, the upregulation of miR-224 significantly
increased the number of cells that migrated through the membrane;
in contrast, downregulation of miR-224 significantly inhibited the
migratory ability of the HCC cells. In parallel, we analyzed the
effect of miR-224 on HCC cell invasion using the Matrigel invasion
assay. The results were similar to the migration assay mentioned
above (Fig. 2).
PPP2R1B is a candidate target of
miR-224
To explore the mechanism by which miR-224 regulates
cell proliferation and metastasis, we performed a miRNA target
search using PicTar and TargetScan. The 3′UTR of PPP2R1B was found
to contain a highly conserved putative miR-224 binding site. In the
present study, the level of endogenous miR-224 was up- or
downregulated by transfection with a miR-224 mimic or miR-224
inhibitor, respectively; scrambled oligonucleotides were used as
controls. Western blot analysis showed that the expression of
PPP2R1B was elevated in HepG2 cells upon transfection of miR-224
inhibitor, relative to the control groups. Compared to the control
groups, the expression of PPP2R1B was reduced in HepG2 cells upon
transfection of miR-224 mimics. In addition, we generated the
pPPP2R1B 3′UTR-luci-WT and pPPP2R1B 3′UTR-luci-MUT plasmids to
investigate the interaction between miR-224 and its putative target
sequences. Data from the luciferase reporter assay showed that the
downregulation of miR-224 resulted in a marked increase in the
luciferase activity of pPPP2R1B 3′UTR-luci-WT, without obvious
changes in the luciferase activity of pPPP2R1B 3′UTR-luci-MUT. By
contrast, the luciferase activity of pPPP2R1B 3′UTR-luci-WT
decreased upon transfection of miR-224 mimics compared with the
control groups. These results indicate that miR-224 directly
modulates PPP2R1B expression by binding to the 3′UTR of
PPP2R1B.
AKT signaling pathway is modulated by
miR-224
We postulated that AKT signaling is modulated by
miR-224, since PPP2R1B has been implicated in the negative control
of kinase and phosphatase function, and therefore plays an integral
role in numerous signal transduction pathways, including the AKT
signaling pathway. To ascertain the effect of miR-224 on the
regulation of the AKT signaling pathway, western blot analysis was
performed to investigate the expression of AKT in the presence of
miR-224. The results revealed that AKT phosphorylation was
upregulated in HepG2 cells treated with miR-224 mimics, and
downregulated in HepG2 cells upon transfection of miR-224
inhibitors compared to cells treated with negative control RNA
sequences or transfection reagent alone. The expression of total
AKT protein was also assessed, but there was no significant change
upon transfection with miR-224 inhibitors or mimics. These data
suggest that miR-224 impacts the AKT signaling pathway through
modulation of AKT phosphorylation levels by regulation of PPP2R1B
expression (Fig. 3).
Taken together, the data above suggest that miR-224
serves as a potent oncogenic microRNA in HCC progression by
impacting cell proliferation, migration and invasion, possibly
through the activation of AKT signaling by targeting PPP2R1B.
Discussion
Current data reveal that many miRNAs are frequently
deregulated in human cancers (10),
and play essential roles in cancer development and progression, as
well as the invasion-metastasis cascade by targeting certain
critical genes involved in HCC.
miR-224 is reported to be overexpressed in various
types of cancer (15–20), and it plays critical roles in
malignant phenotypes. Consistent with these previous observations,
miR-224 was found to be upregulated in HepG2 cells in our study.
Furthermore, in order to elucidate the precise function of miR-224,
proliferation, migration and invasion assays were performed in HCC
cells in the present study. Our results demonstrated that miR-224
positively regulates HCC cell proliferation, migration and
invasion, which suggests that miR-224 might function in liver
carcinogenesis and progression as an oncogenic regulator, although
a previous study stated that miR-224 promotes HCC cell apoptosis by
targeting the API-5 transcript (20). Despite great advances in determining
that several deregulated miRNAs are involved in HCC, their precise
roles in the malignant phenotypes of HCC cells are largely unknown,
partly due to each miRNA having several putative targets with
disparate functions.
To better understand the roles that miRNAs play in
cells, certain pioneer researchers hypothesized that signal
transduction pathways may be prime candidates for miRNA modulation,
rather than individual putative genes (25).
In concordance with this theory, increasing data has
shown that a number of aberrant miRNAs indeed modulate certain
signaling pathways that have been extensively documented in HCC
cells. In light of this recent evidence, we postulated that miR-224
might also impact certain key signaling pathways that are
significant in HCC.
In the present study, data from the western blot
analysis revealed that the phosphorylation state of AKT correlated
with miR-224 levels; more specifically, miR-224 appeared to
activate AKT phosphorylation. In addition, since the levels of
total AKT remained unchanged in the presence of miR-224 mimics and
inhibitors, we reasoned that miR-224 modulates AKT activity in the
process of phosphorylation.
AKT, also known as protein kinase B (PKB), is a
serine/threonine protein kinase that plays a key role in multiple
cellular processes, and is associated with tumor cell survival,
proliferation and invasion. The activation of AKT is also one of
the most frequent alterations observed in human cancers (29). In fact, recent evidence suggests
that a correlation exists between the AKT signaling pathway and
deregulated miRNAs in human cancers, as mentioned above. In the
present study, we demonstrated for the first time that miR-224
plays an essential role in regulating HCC cell functions, partly
through modulation of the AKT signaling pathway, which most likely
occurs via the inhibition of PPP2R1B, a putative target of
miR-224.
PPP2R1B was selected as the focus of the study, as a
result of analyzing the list of putative target genes identified by
online software. PPP2R1B is a regulatory subunit of PP2A, which is
a conserved and ubiquitous serine/threonine phosphatase in
eukaryotes. A previous study (22)
suggested that PP2A functions as a suppressive regulator in various
human cancers through the modulation of several oncogenic signaling
cascades, including the AKT signaling pathway. Wong et al
(10) suggested that the
overexpression of miR-222 confers HCC cells with stronger
metastasis abilities, through enhancing AKT signaling by targeting
PPP2R2A, another subunit of PP2A.
More recently, Hamano et al (24) demonstrated that miR-200c is the
responsible miRNA for chemoresistance in esophageal cancer.
Furthermore, these authors showed that miR-200c-induced resistance
is mediated through the activation of the AKT pathway, largely
through the direct binding and suppression of PPP2R1B mRNA
translation.
These two experiments suggested that PP2A might act
as the important mediator between AKT signaling and aberrant
miRNAs, which are able to inhibit the expression of the subunits
that are indispensable for PP2A functions. In the present study,
using a dual luciferase assay and western blotting, we demonstrated
that miR-224 directly binds and inhibits PPP2R1B expression. Given
that the reported role of PPP2R1B is the negative regulation of AKT
phosphorylation, we proposed that miR-224 indirectly regulates AKT
signaling by targeting and suppressing PPP2R1B expression.
In summary, we demonstrated that miR-224 is
upregulated in HepG2 cells, and the effects of miR-224 in HCC cells
were also shown. Together, our data demonstrated that miR-224 plays
a role in the regulation of various cell functions, in addition to
proliferation, cell migration and invasion. Moreover, we not only
demonstrated that miR-224 functions as an oncogenetic regulator of
HCC cells through the activation of AKT signaling, but also that
the likely intermediary is PPP2R1B, which was identified as an
miR-224 target in the present study. These results may lead to a
better understanding of miR-224 function in HCC, and also suggest a
new direction for future studies aimed towards investigating
aberrant miRNA functions in human cancers.
Acknowledgements
This study was supported by the China Postdoctoral
Science Foundation (20080440910) and the Heilongjiang Provincial
Health Office (2009-149).
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: GLOBOCAN 2008 v1.2, Cancer Incidence and
Mortality Worldwide: IARC CancerBase No. 10. Lyon, France:
International Agency for Research on Cancer; 2010
|
|
2
|
Di Bisceglie AM: Issues in screening and
surveillance for hepatocellular carcinoma. Gastroenterology.
127:S104–S107. 2004.PubMed/NCBI
|
|
3
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bushati N and Cohen SM: MicroRNAs
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007. View Article : Google Scholar
|
|
5
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
He L and Hannon GJ: MicroRNAs: small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sayed D and Abdellatif M: MicroRNAs in
development and disease. Physiol Rev. 91:827–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Calin GA and Croce CM: MicroRNAs
signatures in human cancers. Nat Rev Cancer. 6:857–866. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meng F, Henson R, Wehbe-Janek H, Ghoshal
K, Jacob ST and Patel T: MicroRNA-21 regulates expression of PTEN
tumor suppressor gene in human hepatocellular cancer.
Gastroenterology. 133:647–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong QW, Ching AK, Chan AW, Choy KW, To
KF, Lai PB and Wong N: MiR-222 overexprssion confers cell migratory
advantages in hepatocellular carcinoma through enhancing AKT
signaling. Clin Cancer Res. 16:867–875. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li N, Fu H, Tie Y, Hu Z, Kong W, Wu Y and
Zheng X: MiR-34a inhibits migration and invasion by down-regulation
of c-Met expression in human hepatocellaular carcinoma cells.
Cancer Lett. 275:44–53. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Salvi A, Sabelli C, Moncini S, Venturin M,
Arici B, Riva P, Portolani N, Giulini SM, De Petro G and Barlati S:
MicroRNA-23b mediates urokinase and c-met downmodulation and a
decreased migration of human hepatocellular carcinoma cells. FEBS
J. 276:2966–2982. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW,
Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, et al: MicroRNA-122, a
tumor suppressor microRNA that regulates intrahepatic metastasis of
hepatocellular carcinoma. Hepatology. 49:1571–1582. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Furuta M, Kozaki KI, Tanaka S, Arii S,
Imoto I and Inazawa J: MiR-124 and miR-203 are epigenetically
silenced tumor-suppressive microRNAs in hepatocellular carcinoma.
Carcinogenesis. 31:766–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Prueitt RL, Yi M, Hudson RS, Wallace TA,
Howe TM, Yfantis HG, Lee DH, Stephens RM, Liu CG, Calin GA, et al:
Expression of microRNAs and protein-coding genes associated with
perineural invasion in prostate cancer. Prostate. 68:1152–1164.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mees ST, Mardin WA, Sielker S, Willscher
E, Senninger N, Schleicher C, Colombo-Benkmann M and Haier J:
Involvement of CD40 targeting miR-224 and miR-486 on the
progression of pancreatic ductal adenocarcinomas. Ann Surg Oncol.
16:2339–2350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arndt GM, Dossey L, Cullen LM, Lai A,
Druker R, Eisbacher M, Zhang C, Tran N, Fan H, Retzlaff K, et al:
Characterization of global microRNA expression reveals oncogenic
potential of miR-145 in metastatic colorectal cancer. BMC Cancer.
9:3742009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
White NM, Chow TF, Mejia-Guerrero S,
Diamandis M, Rofael Y, Faragalla H, Mankaruous M, Gabril M, Girgis
A and Yousef GM: Three dysregulated miRNAs control kallikrein 10
expression and cell proliferation in ovarian cancer. Br J Cancer.
102:1244–1253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boguslawska J, Wojcicka A,
Piekielko-Witkowska A, Master A and Nauman A: MiR-224 targets the
3′UTR of type 1 5′-iodothyronine deiodinase possibly contributing
to tissue hypothyroidism in renal cancer. PLoS One.
6:245412011.
|
|
20
|
Wang Y, Lee AT, Ma JZ, Wang J, Ren J, Yang
Y, Tantoso E, Li KB, Ooi LL, Tan P and Lee CG: Profiling microRNA
expression in hepatocellular carcinoma reveals microRNA-224
up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific
target. J Biol Chem. 283:13205–13215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Q, Wang G, Shan JL, Yang ZX, Wang HZ,
Feng J, Zhen JJ, Chen C, Zhang ZM, Xu W, et al: MicroRNA-224 is
upregulated in HepG2 cells and involved in cellular migration and
invasion. J Gastroenterol Hepatol. 25:164–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ory S, Zhou M, Conrads TP, Veenstra TD and
Morrison DK: Protein phosphatase 2A positively regulates Ras
signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3
binding sites. Curr Biol. 13:1356–1364. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Basu S: PP2A in the regulation of cell
motility and invasion. Curr Protein Pept Sci. 12:3–11. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hamano R, Miyata H, Yamasaki M, Kurokawa
Y, Hara J, Moon J, Nakajima K, Takiguchi S, Fujiwara Y, Mori M and
Doki Y: Overexpression of miR-200c induces chemoresistance in
esophageal cancers mediated through activation of the AKT signaling
pathway. Clin Cancer Res. 17:26032011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Inui M, Martello G and Piccolo S: MicroRNA
control of signal transduction. Nat Rev Mol Cell Biol.
11:2252–2262. 2010. View
Article : Google Scholar
|
|
26
|
Datta J, Kutay H, Nasser MW, Nuovo GJ,
Wang B, Majumder S, Liu CG, Volinia S, Croce CM, Schmittgen TD, et
al: Methylation mediated silencing of microRNA-1 gene and its role
in hepatocellular carcinogenesis. Cancer Res. 68:5049–5058. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garofalo M, Di Leva G, Romano G, Nuovo G,
Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P,
et al: miR-221 & 222 regulate TRAIL resistance and enhance
tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell.
16:498–509. 2009.
|
|
28
|
Wang B, Hsu SH, Majumder S, Kutay H, Huang
W, Jacob ST and Ghoshal K: TGFbeta-mediated upregulation of hepatic
miR-181b promotes hepatocarcinogenesis by targeting TIMP3.
Oncogene. 29:1787–1797. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wendel HG, De Stanchina E and Fridman JS:
Survival signalling by Akt and eIF4E in oncogenesis and cancer
therapy. Nature. 428:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|