Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common type of cancer worldwide, with 626,000 new cases reported in
2002 (1). Hepatitis B and C virus
infection accounts for the main cause in the majority of cases.
Chronic excessive alcohol consumption, environmental toxins,
aflatoxin B and nonalcoholic steatohepatitis (NASH) are other
common causes. The etiological factors also vary by geographical
location. The survival rate of HCC patients has been increased due
to surgical resection or transplantation, chemoembolization and a
combination of chemotherapy (2,3).
Further insight into the mechanism of hepatocarcinogenesis may
offer novel modalities to improve the outcome of HCC patients.
Arachidonoy lethanolamide (AEA), also called
anandamide, was the first identified endogenous agonist for
cannabinoid receptors (CB1 and CB2), and shares many of the
pharmacological effects of the psychotropic marijuana component
Δ9-tetrahydrocannabinol (Δ9-THC). Related acylethanolamines (AEs),
including dihomo-γ-linolenoyl ethanolamide,
7,10,13,16-docosatetraenoyl ethanolamide and mead ethanolamide have
also been identified as endogenous agonists for cannabinoid
receptors (4–6). The endogenous cannabinoid system,
comprising the cannabinoid receptors CB1 and CB2 as well as enzymes
regulating endocannabinoid biosynthesis and degradation, controls
several physiological and pathological conditions. Recent evidence
indicates that endocannabinoids influence the intracellular events
controlling the proliferation and apoptosis of a number of cancer
cell types, including breast cancer, prostate cancer, C6 glioma
cells, colorectal cancer, gastric cancer and cholangiocarcinoma,
thereby leading to antitumor effects both in vitro and in
vivo (7–13). Therefore, the endocannabinoid system
has been recommended as a target for the development of new drugs
for cancer therapy (14).
We have previously reported that the overexpression
of CB1 and CB2 receptors is correlated with dedifferentiation and
thrombus of the portal vein (15).
In the present study we investigated the inhibitory effects of AEA
on the proliferation of HCC cells. Our data demonstrates that AEA
induces cell cycle arrest and apoptosis, and may provide a novel
therapeutic strategy in the treatment of human HCC.
Materials and methods
Drugs and antibodies
AEA was purchased from Sigma Chemical Co. (St.
Louis, MO, USA). The final IC50 (the concentration that reduces
cell viability to 50 percent) of AEA was 0.11 μM. The polyclonal
antibodies to Bak, CDK4 and p21 were purchased from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA).
Cell culture
The human hepatocellular carcinoma cell line, Huh7,
was maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco
BRL Life Technologies, MD, USA) and supplemented with 1.0 mM
pyruvic acid. The media were also supplemented with 2 mM
l-glutamine, 10% inactivated fetal bovine serum and antibiotic
antimycotic solution (100 U/ml penicillin, 100 μg/ml streptomycin
and 250 ng/ml amphotericin B; Sigma Aldrich). The cells were
maintained in a humidified environment containing 5% CO2
and held at a constant temperature of 37°C. For the experiments,
the cells were seeded at 60–70% confluence, unless otherwise
indicated, and allowed to adhere overnight. The cells were then
kept in serum-free medium for at least 6 h prior to treatment.
Control cells were cultured in the presence of vehicle alone.
Cell viability by MTT assay
Cell viability of Huh7 was determined by the
3-[4,5-dimethylthiazolyl-2] 2,5-diphenyl-tetrazolium bromide (MTT;
Sigma Chemical Co.) assay as previously reported (16). Following treatment in 96-well plates
(triplicate wells for each sample), MTT stock solution was added to
each well (final concentration of 1 mg/ml) for 4 h. The medium was
then removed and 150 μl lysis buffer (DMSO) was added to dissolve
the formazan crystals. Finally, the absorbance at 570 nm (test
wavelength) and 630 nm (reference wavelength) was measured using an
ELISA microplate reader (Dynex Technologies). The cells were
treated with different doses of AEA at fixed molar ratios for 24 h.
Relative survival was assessed, and the IC50 was established.
Cell cycle analysis
Cell cycle analysis was performed on Huh7 cells
plated in 6-well plates, initially seeded at a density of
50×103, 100×103 or 150×103
cells/well, and incubated for 24, 48 or 72 h, respectively, with
vehicle or AEA at 0.11 μM. When specified, the cells were
synchronized with 30 ng/ml nocodazole for 14 h prior to the
addition of the tested compounds. Following treatment, the cells
were harvested by trypsinization, washed with phosphate-buffered
saline (PBS), pelleted and fixed by rapid submersion in ice-cold
80% ethanol with vigorous vortexing. After overnight fixation at
-20°C, the cells were washed with PBS, pelleted, resuspended and
incubated for 20 min in a saponin-based permeabilization solution
containing 1% BSA, 0.2 mg/ml ribonuclease A and 20 μg/ml propidium
iodide. Data were collected on a FACSCalibur flow cytometer (BD
Biosciences) and then analysis was performed using ModFit LT v3.0
from Verity Software House, Inc. (Topsham, ME, USA). A total of
10,000 events were collected and corrected for debris and aggregate
populations.
Apoptosis assay
Terminal deoxynucleotidyl transferase-mediated dUTP
nick-end labeling (TUNEL) was employed to detect DNA fragmentation
of cells using an in situ cell death detection kit POD
(Roche Applied Science, Mannheim, Germany) in accordance with the
manufacturer’s instructions (17).
Briefly, the cells were seeded in a 6-well plate for 24 h and then
treated with vehicle or AEA at 0.11 μM. After treatment, the cells
were fixed with 4% paraformaldehyde for 15 min at room temperature,
and permeabilized with 0.1% Triton X-100 for 2 min (on ice).
Endogenous peroxidase activity was quenched with 3%
H2O2 for 10 min. After rinsing with PBS, the
cells were exposed for 60 min at 37°C in a humid atmosphere to the
TUNEL reaction mixture containing 0.135 U/ml calf thymus terminal
deoxynucleotidyl transferase (TdT), 0.0044 nmol/ml
digoxigenin-11-2′-deoxy-uridine-5′-triphosphate (DIG-dUTP), and 1
mM cobalt chloride in 1X reaction buffer in distilled water. After
washing with PBS, the cells were treated for 30 min at room
temperature with streptavidin-horseradish peroxidase, followed by
washing and 0.05% 3-3′-diaminobenzidine tetrahydrochloride (DAB)
color reaction. Counterstaining was performed with Harris’s
hematoxylin and the morphologic features were visualized by light
microscopy. Routine H&E staining was also conducted. A negative
control was obtained by omitting TdT. As a positive control, the
cells were incubated with 1,000 U/ml DNase I recombinant
(Fermentas) prior to the labeling procedures (18).
Real-time polymerase chain reaction (PCR)
analysis
Total RNA was isolated from the cells using RNeasy
Mini kit (Qiagen, Valencia, CA, USA) with a DNase digestion step
using the RQ1 RNase-free DNase (Promega). cDNA samples were
amplified using IQ SYBR-Green Supermix (Bio-Rad Laboratories)
according to the manufacturer’s instructions. The following primers
were used: Bak: 5′-CAGGGCTTA GGACTTGGTTT-3′ (forward),
5′-TTTTTTCAGGGTGAG GGGAT-3′ (reverse), 400 bp; p21: 5′-GCAGCGGAACAA
GGAGT-3′ (forward), 5′-GGAGAAACGGGAACCAG-3′ (reverse), 251 bp;
CDK4: 5′-GTGGTGGAACAGTCAAG-3′ (forward), 5′-AGCCCAATCAGGTCA-3′
(reverse), 247 bp. All reactions were performed in triplicate. For
each PCR, we checked the linear range of a standard curve of serial
dilutions. The relative quantification of p21, Bak and CDK4 gene
expression was evaluated following normalization with the GAPDH
gene as the endogenous control. Data processing and statistical
analysis were performed using IQ5 Cycler software (Bio-Rad
Laboratories).
Western blot analysis
After treatment with the compounds, protein extracts
were prepared by washing the cells in PBS and incubating for 20 min
in ice-cold lysis buffer supplemented with protease inhibitor
cocktail as reported previously (19). After performing sonication three
times for 10 sec and evaluating the protein concentration, equal
amounts of protein were electrophoresed and then electrotransferred
to a nitrocellulose membrane, which was developed using the
alkaline phosphatase colorimetric system. The correct protein
loading was verified by means of red Ponceau staining and
immunoblotting for GAPDH. All of the antibodies used were purchased
from Santa Cruz Biotechnology, Inc.
Statistical analysis
Cell viability data were expressed as the mean ± SE
and evaluated using the Student’s t-test. P<0.05 was considered
to indicate a statistically significant result.
Results
Cell growth and the cell cycle
AEA significantly inhibited the growth of Huh7
cells. Statistical significance between cultures treated with AEA
versus vehicle is shown in Fig. 1.
We then performed flow cytometry analysis at 12, 24, 48 and 72 h
after treatment. In the Huh7 cells tested, treatment with AEA for
24 h increased the G0-G1 phase fraction and decreased the S phase
fraction when compared to the vehicle-treated cultures. Repeat
experiments of cell growth and cell cycle analysis produced similar
results. Representative results are shown in Fig. 2, with the following fraction values
for cultures treated with AEA versus vehicle, respectively: G0-G1
phase, 64.98 and 57.81%; S phase, 26.13 and 21.04%; G2-M phase,
8.89 and 21.15% (Fig. 2).
Induction of apoptosis by AEA
We examined the contribution of apoptosis to the
observed growth inhibition in Huh7 cells induced by AEA. The rate
of Huh7 cells with apoptosis following AEA treatment was
substantially increased compared with that following vehicle
treatment: at 12 h, 0.20 and 6.77%; at 24 h, 0.55 and 10.69%; at 48
h, 0.72 and 16.31%; at 72 h, 0.85 and 43.03%, respectively
(p<0.001; Fig. 3).
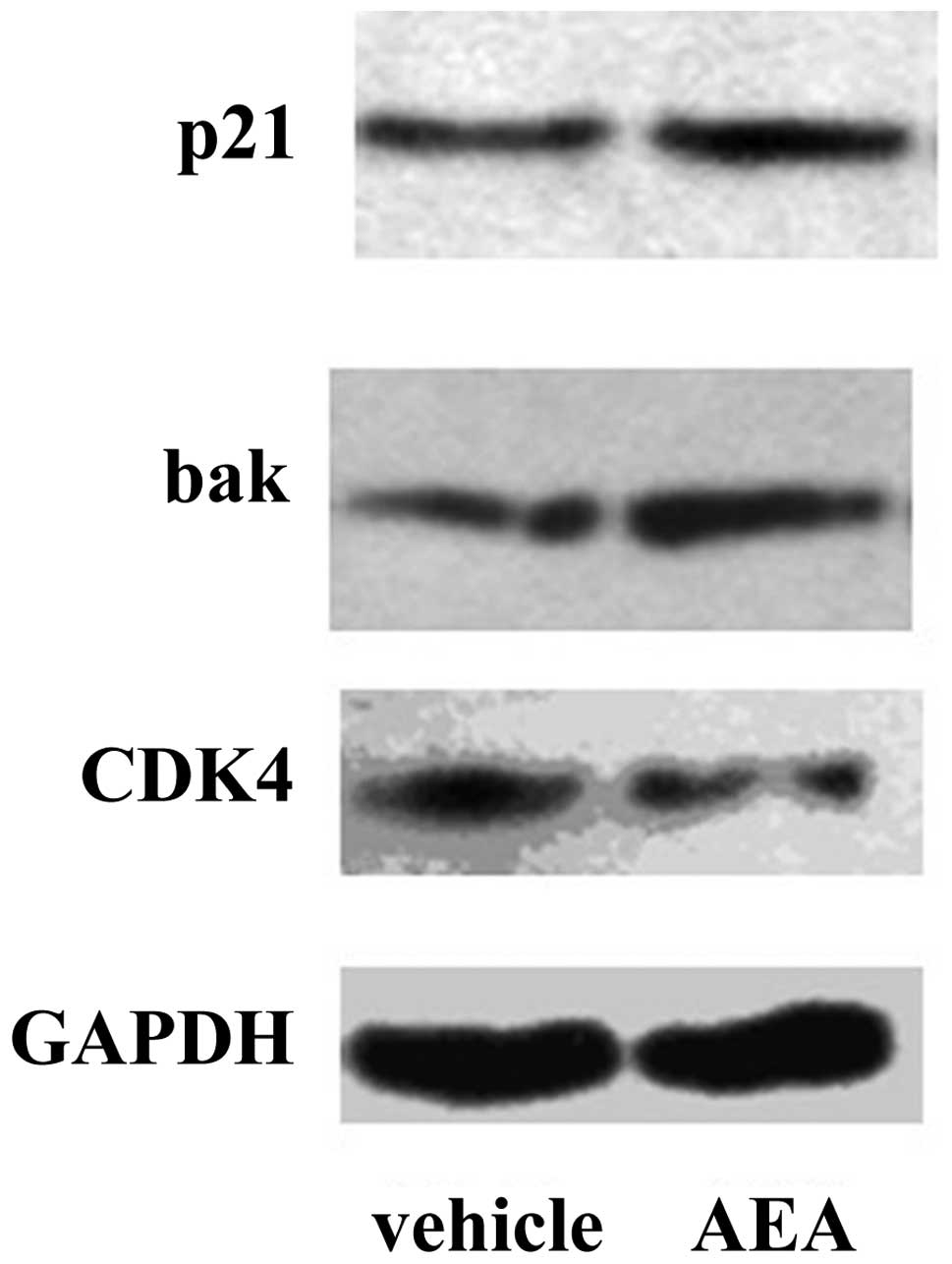
Downregulation of CDK4 and upregulation
of Bak and p21 by AEA
We then investigated the effects of AEA treatment on
the expression of molecules involved in the G1 phase of the cell
cycle and apoptosis at the mRNA and protein level. Real-time PCR
revealed that Huh7 cells treated with AEA had decreased expression
of CDK4 (57.1%), and increased expression of p21 (3.75-fold) and
Bak (1.38-fold) at the mRNA level (Fig.
4). Western blot analysis also revealed that Huh7 cells treated
with AEA had increased expression of p21 and Bak and decreased
expression of CDK4 at the protein level (Fig. 5).
Discussion
The pharmacological effects of endocannabinoids in
cancer cells have been increasingly reported. In our earlier study
of cannabinoid receptor expression in human HCC tissue samples, we
found that high expression of CB1 and CB2 was associated with
portal vein invasion and dedifferentiated histology, which
highlighted the role of cannabinoids in human hepatocarcinogenesis
(15). The aim of this study was to
investigate the effect of AEA on suppression of growth in HCC
cells. The data presented in this study demonstrate the
anti-proliferative effect induced by AEA in HCC cells.
The first endogenous ligand of the cannabinoid
receptor to be identified was anandamide (AEA), which was isolated
in 1992. AEA is an arachidonic acid derivative which appears to act
on the central nervous system as a neuromodulator or retrograde
messenger inhibiting the release of classical neurotransmitters
(5,20). The antitumor effect of cannabinoids
was first reported in 1975 when Δ9-THC, Δ8-THC and cannabinol were
found to inhibit the growth of Lewis lung adenocarcinoma cells
in vitro and in vivo, following oral administration
in mice (21). Similar antitumor
effects have recently been reported in a variety of cancers
including breast cancer, prostate cancer, C6 glioma cells,
colorectal cancer, gastric cancer and cholangiocarcinoma (7–13). In
HCC, Δ(9)-tetrahydrocannabinol
(Δ(9)-THC), the main active
component of Cannabis sativa and JWH-015 (a cannabinoid
receptor 2 (CB2)-selective agonist) were suggested to demonstrate
inhibitory effects on growth both in vitro and in
vivo (22). Concordant with
these findings, the data in the present study demonstrate that the
endogenous cannabinoid AEA reduced the viability of HCC cells.
Cell cycle checkpoints control the proper timing of
cell cycle events by enforcing the dependency of late events on the
completion of early events. Consequently, checkpoint blocking may
result in cell cycle arrest and significantly alter cell
proliferation activity (23). We
then investigated the action mechanism of AEA as it was effective
in reducing cell viability. Treatment with AEA slowed the Huh7 cell
cycle progression and reduced transition through the G1-S
checkpoint, causing an accumulation of cells in the G1 phase. The
concordant effects were observed in hepatoma HepG2 cells, epidermal
growth factor-stimulated PC3 prostate cancer cells, the breast
cancer cell line EFM-19, and gastric cancer cells (7,11,24,25).
From the data of the present study, it was suggested that AEA
inhibited the proliferation of Huh7 cells through cell cycle arrest
in the G1-S phase. The molecules involved in cyclin-CDK complexes,
including the cyclin-dependent kinase (CDK) and cyclin kinase
inhibitor (CKI) families, are modulated in the G0-G1 phase of the
cell cycle in human hepatocarcinogenesis (26,27).
Hence, we further screened the dysregulation natures of these
relevant molecules. Huh7 cells treated with AEA demonstrated
upregulation of p21 and downregulation of CDK4 at the mRNA and
protein levels. In human prostate carcinoma cells, the
endocannabinoid 2-arachidonylglyceryl ether (noladin ether, NE)
induced cell cycle arrest in the G0-G1 phase in PC3 cells, and
downregulated the expression of cyclin D1 and cyclin E in PC3 cells
(28). In human breast cancer cell
lines, AEA led to cell cycle arrest and inhibited the expression of
CDK2 (29). Our findings further
supported the hypothesis that AEA arrests the cell cycle in the
G1-S phase via the upregulation of p21 and downregulation of CDK4
in HCC cells.
In addition, this study observed the effects of AEA
on apoptosis in Huh7 cells. AEA was found to induce apoptosis in
Huh7 cells compared with vehicle. In HepG2 cells, AEA or the
synthetic cannabinoid WIN induced hepatocyte apoptosis through the
activation of proapoptotic signaling pathways (i.e., p38 MAPK and
JNK) and inhibition of antiapoptotic signaling pathways (i.e.,
PKB/Akt), or via transcription factor PPARgamma (25,30).
Furthermore, our data demonstrated that AEA upregulated Bak, which
is a well-known cell death initiator in the apoptotic signaling
cascade (31,32). The role of Bak in
hepatocarcinogenesis or for HCC treatment has also been documented.
The level of Bak is reduced or even non-detectable in HCC cells
(33–35). The pro-apoptotic role of Bak in HCC
cells is further supported by studies in which different agents
induced apoptosis in HCC cells by stimulating Bak expression
(36,37). In accordance with these findings,
our results demonstrated that AEA induced apoptosis via the
upregulation of Bak in Huh7 cells.
In conclusion, the results of the present study
revealed that AEA inhibited the proliferation of Huh7 cells,
arrested the cell cycle in the G1-S transition via the upregulation
of p21 and downregulation of CDK4, and induced apoptosis via the
upregulation of Bak. These findings suggest that AEA has antitumor
potential in HCC cells.
Acknowledgements
This study was funded by the National Natural
Science Foundation of China (grant no. 30750005).
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
AEA
|
anandamide
|
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar
|
|
3
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schuel H, Burkman LJ, Lippes J, Crickard
K, Forester E, Piomelli D and Giuffrida A: N-Acylethanolamines in
human reproductive fluids. Chem Phys Lipids. 121:211–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Devane WA, Hanus L, Breuer A, et al:
Isolation and structure of a brain constituent that binds to the
cannabinoid receptor. Science. 258:1946–1949. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mechoulam R and Hanus L: A historical
overview of chemical research on cannabinoids. Chem Phys Lipids.
108:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
De Petrocellis L, Melck D, Palmisano A,
Bisogno T, Laezza C, Bifulco M and Di Marzo V: The endogenous
cannabinoid anandamide inhibits human breast cancer cell
proliferation. Proc Natl Acad Sci USA. 95:8375–8380.
1998.PubMed/NCBI
|
|
8
|
Melck D, De Petrocellis L, Orlando P,
Bisogno T, Laezza C, Bifulco M and Di Marzo V: Suppression of nerve
growth factor Trk receptors and prolactin receptors by
endocannabinoids leads to inhibition of human breast and prostate
cancer cell proliferation. Endocrinology. 141:118–126.
2000.PubMed/NCBI
|
|
9
|
Fowler CJ, Jonsson KO, Andersson A, et al:
Inhibition of C6 glioma cell proliferation by anandamide,
1-arachidonoylglycerol, and by a water soluble phosphate ester of
anandamide: variability in response and involvement of arachidonic
acid. Biochem Pharmacol. 66:757–767. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ligresti A, Bisogno T, Matias I, et al:
Possible endocannabinoid control of colorectal cancer growth.
Gastroenterology. 125:677–687. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyato H, Kitayama J, Yamashita H, Souma
D, Asakage M, Yamada J and Nagawa H: Pharmacological synergism
between cannabinoids and paclitaxel in gastric cancer cell lines. J
Surg Res. 155:40–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeMorrow S, Glaser S, Francis H, Venter J,
Vaculin B, Vaculin S and Alpini G: Opposing actions of
endocannabinoids on cholangiocarcinoma growth: recruitment of Fas
and Fas ligand to lipid rafts. J Biol Chem. 282:13098–13113. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bifulco M, Laezza C, Pisanti S and
Gazzerro P: Cannabinoids and cancer: pros and cons of an antitumour
strategy. Br J Pharmacol. 148:123–135. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bifulco M and Di Marzo V: Targeting the
endocannabinoid system in cancer therapy: a call for further
research. Nat Med. 8:547–550. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu X, Liu Y, Huang S, et al:
Overexpression of cannabinoid receptors CB1 and CB2 correlates with
improved prognosis of patients with hepatocellular carcinoma.
Cancer Genet Cytogenet. 171:31–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emanuele S, D’Anneo A, Bellavia G, et al:
Sodium butyrate induces apoptosis in human hepatoma cells by a
mitochondria/caspase pathway, associated with degradation of
beta-catenin, pRb and Bcl-XL. Eur J Cancer. 40:1441–1452. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Youm YS, Lee SY and Lee SH: Apoptosis in
the osteonecrosis of the femoral head. Clin Orthop Surg. 2:250–255.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Labat-Moleur F, Guillermet C, Lorimier P,
Robert C, Lantuejoul S, Brambilla E and Negoescu A: TUNEL apoptotic
cell detection in tissue sections: critical evaluation and
improvement. J Histochem Cytochem. 46:327–334. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miao X, Liu G, Xu X, et al: High
expression of vanilloid receptor-1 is associated with better
prognosis of patients with hepatocellular carcinoma. Cancer Genet
Cytogenet. 186:25–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wilson RI and Nicoll RA: Endocannabinoid
signaling in the brain. Science. 296:678–682. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Munson AE, Harris LS, Friedman MA, Dewey
WL and Carchman RA: Antineoplastic activity of cannabinoids. J Natl
Cancer Inst. 55:597–602. 1975.PubMed/NCBI
|
|
22
|
Vara D, Salazar M, Olea-Herrero N, Guzmán
M, Velasco G and Díaz-Laviada I: Anti-tumoral action of
cannabinoids on hepatocellular carcinoma: role of AMPK-dependent
activation of autophagy. Cell Death Differ. 18:1099–1111. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Biswas KK, Sarker KP, Abeyama K, et al:
Membrane cholesterol but not putative receptors mediates
anandamide-induced hepatocyte apoptosis. Hepatology. 38:1167–1177.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mimeault M, Pommery N, Wattez N, Bailly C
and Hénichart JP: Anti-proliferative and apoptotic effects of
anandamide in human prostatic cancer cell lines: implication of
epidermal growth factor receptor down-regulation and ceramide
production. Prostate. 56:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Greenbaum LE: Cell cycle regulation and
hepatocarcinogenesis. Cancer Biol Ther. 3:1200–1207. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hui AM, Makuuchi M and Li X: Cell cycle
regulators and human hepatocarcinogenesis. Hepatogastroenterology.
45:1635–1642. 1998.PubMed/NCBI
|
|
28
|
Nithipatikom K, Isbell MA, Endsley MP,
Woodliff JE and Campbell WB: Anti-proliferative effect of a
putative endocannabinoid, 2-arachidonylglyceryl ether in prostate
carcinoma cells. Prostaglandins Other Lipid Mediat. 94:34–43. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laezza C, Pisanti S, Crescenzi E and
Bifulco M: Anandamide inhibits Cdk2 and activates Chk1 leading to
cell cycle arrest in human breast cancer cells. FEBS Lett.
580:6076–6082. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giuliano M, Pellerito O, Portanova P, et
al: Apoptosis induced in HepG2 cells by the synthetic cannabinoid
WIN: involvement of the transcription factor PPARgamma. Biochimie.
91:457–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cory S and Adams JM: The Bcl2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Griffiths GJ, Dubrez L, Morgan CP, et al:
Cell damage-induced conformational changes of the pro-apoptotic
protein Bak in vivo precede the onset of apoptosis. J Cell Biol.
144:903–914. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu LX, Jiang HC, Liu ZH, et al: Gene
expression profiles of hepatoma cell line BEL-7402.
Hepatogastroenterology. 50:1496–1501. 2003.PubMed/NCBI
|
|
34
|
Rousseau B, Ménard L, Haurie V, et al:
Overexpression and role of the ATPase and putative DNA helicase
RuvB-like 2 in human hepatocellular carcinoma. Hepatology.
46:1108–1118. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Madesh M and Hajnóczky G: VDAC-dependent
permeabilization of the outer mitochondrial membrane by superoxide
induces rapid and massive cytochrome c release. J Cell Biol.
155:1003–1015. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu R, Zhai Q, Liu W and Liu X: An insight
into the mechanism of cytotoxicity of ricin to hepatoma cell: roles
of Bcl-2 family proteins, caspases, Ca(2+)-dependent proteases and
protein kinase C. J Cell Biochem. 81:583–593. 2001.PubMed/NCBI
|
|
37
|
Wang QF, Chen JC, Hsieh SJ, Cheng CC and
Hsu SL: Regulation of Bcl-2 family molecules and activation of
caspase cascade involved in gypenosides-induced apoptosis in human
hepatoma cells. Cancer Lett. 183:169–178. 2002. View Article : Google Scholar : PubMed/NCBI
|