Introduction
Liver cancer is the fifth most commonly diagnosed
cancer and the second most frequent cause of cancer-related
mortality worldwide. Its rate is particularly high in Asia and
Africa. Of the liver cancers, hepatocellular carcinoma (HCC) is the
most common malignancy, and accounts for 70–85% of all liver
cancers (1). Several environmental
factors, such as hepatitis type B or C virus infections, aflatoxin
B1 and alcohol, are considered to be the main causes of HCC, but no
absolute cure for HCC has been devised (1). Many studies have suggested that the
losses of several tumor suppressor genes and aberrant regulations
of cell growth signaling pathways, including the ERK/MAPK pathway
and the Wnt/β-catenin pathway, are involved in hepatocarcinogenesis
(2–4), but the molecular mechanism underlying
the pathogenesis of HCC remains elusive.
Deleted in breast cancer-1 (DBC1, KIAA1967) was
originally suggested to be a putative tumor suppressor gene on
chromosome 8p21, which is frequently deleted in breast cancer
(5). Several researchers have shown
that DBC1 regulates cell survival by modulating its diverse binding
partners. DBC1 induces p53-mediated apoptosis by negatively
regulating silent mating type information regulation 2 homolog 1
(SIRT1) activity (6,7), and by directly interacting with
estrogen receptor-α (ERα) promotes breast cancer cell survival
(8). In addition, DBC1 regulates
cancer cell growth by mediating the transcriptional activation of
retinoic acid receptor α (RAR α) in breast cancer or androgen
receptor (AR) in prostate cancer cells (9,10).
DBC1 also regulates epigenetic mechanisms by inhibiting SUV39H1
methyltransferase and histone deacetylase 3 (HDAC3) (11,12).
These results indicate that DBC1 is a multifunctional protein that
may be involved in a variety of cellular pathways.
Sirtuins (SIRTs) are highly conserved mammalian
homologues of yeast SIR2 (silent mating type information regulation
2 homolog 1), which catalyze NAD+-dependent histone
deacetylation and ADP ribosylation. Many studies have shown that
SIRT1 levels are significantly elevated in prostate cancer, ovarian
cancer, gastric cancer, colorectal cancer and hepatocellular
carcinoma (13–17). Moreover, SIRT1 inhibition has been
reported to suppress cell growth and induce cell cycle arrest or
apoptosis in cancer cells (13–17).
Although SIRT1 has emerged as a key regulator in various cellular
pathways, the regulatory mechanisms responsible for SIRT1 activity
have not been determined.
Although DBC1 modulation of SIRT1 has been suggested
to be implicated in cancer cell death, its regulatory functions in
liver cancer have not been elucidated. Moreover, the correlation
between DBC1 and SIRT1 in liver cancer has not been studied.
Therefore, in this study, we investigated the aberrant regulations
of DBC1 and SIRT1 in HCC, and examined the association between DBC1
with SIRT1 in liver cancer cell lines.
Materials and methods
Tissue samples
Ten hepatocellular carcinoma tissues and
corresponding normal tissues were obtained from Yonsei University,
School of Medicine, Seoul. Informed consent was provided in
compliance with the Declaration of Helsinki, and the study was
approved by the Institutional Review of Board of Songeui Campus,
College of Medicine, The Catholic University of Korea (IRB approval
no. CUMC11U010).
Cell culture, transfections and
treatments
The human HCC cell lines, HepG2 (wt p53) and SNU-182
(mt p53), and the non-small lung carcinoma cell line A549 (wt p53)
were obtained from ATCC (American Type Culture Collection,
Manassas, VA, USA). Cell lines were maintained in RPMI-1640 medium
(Lonza, Walkersville, MD, USA) supplemented with 10% fetal bovine
serum (Sigma, St. Louis, MO, USA) and 100 units/ml of
penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA).
Small interfering RNA duplexes (siRNAs) specific to
SIRT1 (sense, 5′-GGAUAGAGCCUCACAUGCA-3′; anti-sense,
5′-UGCTUGUGAGGCUCUAUCC-3′) and DBC1 (sense,
5′-CAGCUUGCAUGACUACUUU-3′; antisense, 5′-AAAGUAGUCAUGCAAGCUG-3′)
and negative control siRNA were purchased from Ambion (Austin, TX,
USA). SIRT1 expression plasmid, pcDNA 3.1-SIRT1-myc-His was kindly
donated by Dr. Kouzarides (University of Cambridge, UK). For gene
silencing, siRNAs against SIRT1 and DBC1 were transfected at final
concentrations of 50 and 200 nM, respectively. Two micrograms of
mock (empty vector) and SIRT1 expression plasmids were used to
induce SIRT1 overexpression. All transfections were performed using
Lipofectamine™ 2000 reagents (Invitrogen) according to the
manufacturer’s instructions. To induce DNA damage, cells were
treated with 20 μM etoposide (Sigma) for 12 h.
Western blotting and
co-immunoprecipitation
Tissue proteins and cell lysates were prepared using
Radio Immunoprecipitation (RIPA) buffer (50 mM Tris-HCl, pH 7.4,
150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate)
supplemented with 1 mM phenylmethane-sulfonylfluoride (PMSF, Sigma)
and 1X complete protease inhibitor cocktail tablets (Roche,
Mannheim, Germany). Proteins (10 μg) were separated by
SDS-PAGE and transferred onto polyvinylidene difluoride membranes
(PVDF, Bio-Rad Laboratories, Hercules, CA, USA). Proteins were
analyzed using anti-SIRT1, -p53 and -GAPDH (Santa Cruz
Biotechnology, Santa Cruz, CA, USA), anti-DBC1 (Bethyl
Laboratories, Montgomery, TX, USA) and anti-acetylated p53 (Cell
Signaling, Danvers, MA, USA). An ECL Plus western blotting
detection system (Millipore, Billerica, MA, USA) was used to detect
signals. Membranes were evaluated using a LAS 3000 image analyzer
(Fuji Photo Film, Japan). For immunoprecipitations, cells were
lysed with sodium deoxycholate-free RIPA buffer. Cell lysates (1∼2
mg) were incubated with 1∼2 μg anti-His (Applied Biological
Materials, BC, Canada) or anti-SIRT1 at 4°C overnight, and then
rotated with 50 μl protein-A-agarose (Millipore) at 4°C for
4 h. Samples were analyzed by western blotting with appropriate
primary antibodies.
3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) assay
SNU-182 cells were seeded into 6-well plates and
transfected with each siRNA. Twenty-four hours later, cells were
treated with 20 μM etoposide for 12 h. After etoposide
treatment, cells were incubated with 0.5 mg/ml MTT for 1 h at 37°C
for the indicated times. The formazan crystals produced were
dissolved with 500 μl DMSO and absorbances were read at 570
nm using a VICTOR3™ Multilabel Plate Reader (PerkinElmer, Foster
City, CA, USA). All measurements were performed in triplicate and
each experiment was repeated at least three times.
Results
DBC1 and SIRT1 are aberrantly
overexpressed in a subset of human HCCs
It was recently suggested that SIRT1 is expressed at
very low levels in normal liver tissue, but that it is
overexpressed in HCC cell lines and in a subset of HCCs, and that
it is essential for tumorigenesis in HCC (17). Conversely, an analysis of SIRT1 mRNA
levels in HCC patients by real-time qPCR revealed that average
SIRT1 mRNA levels in tumor and non-tumoral liver tissues do not
differ significantly, which suggests that the tumor-specific
overexpression of SIRT1 is regulated in a transcription-independent
manner (17). In addition, it has
been demonstrated that SIRT1 activity is negatively regulated by
DBC1 by direct binding (6,7). However, how SIRT1 is regulated by DBC1
in liver cancer remains unclear. Therefore, to explore this
association in liver cancer, we examined SIRT1 and DBC1 gene
expressions available from the National Center for Biotechnology
Information (NCBI) Gene Expression Omnibus (GEO) database
(accession numbers GSE14520 and GSE25097) in a large cohort of HCC
patients. As shown previously, SIRT1 and DBC1 gene expression was
not significantly different in the two large HCC cohorts (Fig. 1A and B). We then examined SIRT1
protein overexpression by using western blotting in 10 randomly
selected human HCC tissues and paired non-cancerous surrounding
liver tissues. As expected, SIRT1 was markedly upregulated in HCCs
compared to non-cancerous tissues (Fig.
1C). Notably, we also observed that DBC1 protein was highly
overexpressed in the same HCCs. Since DBC1 has been reported to
function as a negative regulator of SIRT1 in other cancers, we had
expected an inverse correlation between DBC1 and SIRT1. Thus, we
investigated the association between DBC1 and SIRT1 expression in a
liver cancer cell line by immunoprecipitation and western blot
analysis.
As shown in Fig. 2A,
to elucidate whether DBC1 is associated with SIRT1, we introduced
His-tagged SIRT1 expression plasmid into SNU-182 cells. In the
following immunoprecipitation with anti-His antibody, endogenous
DBC1 was detected by western blotting. SIRT1 was simultaneously
detected in the same anti-His immunoprecipitates. In addition,
endogenous DBC1 and SIRT1 were also co-immunoprecipitated in
SNU-182 cells (Fig. 2B). These
results demonstrated that DBC1 interacts with SIRT1 in liver cancer
cells in vitro.
Targeted inhibition of DBC1 and
SIRT1-mediated growth in liver cancer cell lines
Next, to explain the biological consequences of the
aberrant expressions of SIRT1 and DBC1 in hepatocarcinogenesis,
SIRT1 and DBC1 expression was abrogated using the RNA
interference-mediated protein knockdown method in HepG2 and SNU-182
cells (two different HCC cell lines). As previously observed
(17), SIRT1 depletion resulted in
a significant reduction in tumor cell growth in both cell lines
(Fig. 3A and B). Notably, when DBC1
protein was inactivated by its siRNA, both cell lines exhibited
significantly less cell viability. However, double knockdown of
SIRT1 and DBC1 did not exhibit a synergistic effect on cell
viability (Fig. 3A and B). These
results suggest that DBC1 does not negatively regulate SIRT1, but
that it does contribute to the neoplastic property of liver cancer
cells.
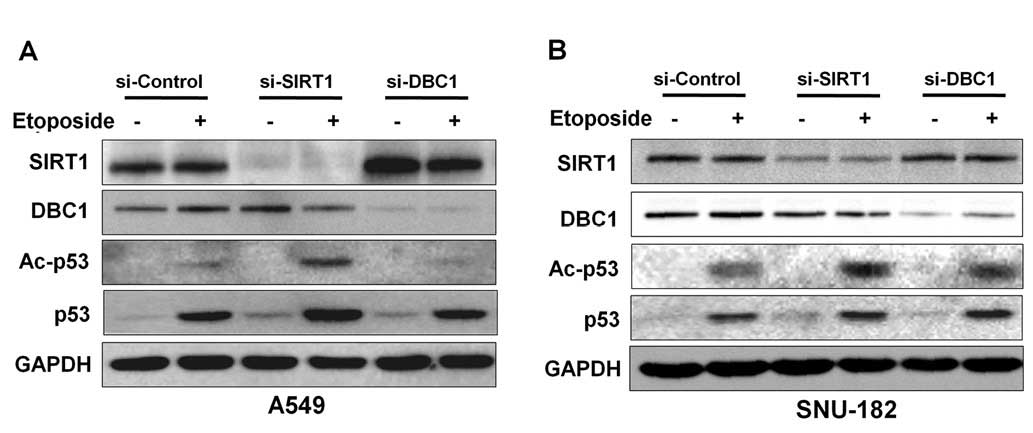
In previous studies, inactivation of DBC1 by siRNA
attenuated etoposide-induced p53 acetylation by inhibiting SIRT1
deacetylase activity in A549 and U2OS cell lines (6,7). To
determine whether DBC1 inhibits p53, we analyzed acetylated p53 in
the absence or presence of DBC1 protein in liver cancer cells.
Initially, to confirm the suppressive effect of DBC1 on p53
acetylation by SIRT1, we performed etoposide-induced
hyperacetylation of p53 in A549 cells. As shown in Fig. 4A, DBC1 silencing reduced the
acetylation status of p53 in response to etoposide treatment in
A549 cells (Fig. 3B). However,
inactivation of DBC1 by its siRNA did not affect the
etoposide-mediated p53 hyperacetylation status in SNU-182 cells
(Fig. 4B). These results suggest
that DBC1 does not functionally influence SIRT1 activity in liver
cancer cells.
Discussion
Accumulating evidence indicates that the interaction
between DBC1 and SIRT1 is implicated in metabolic disease and
cancers. For example, DBC1 knockout mice were found to be protected
against high-fat diet-induced liver steatosis with increased SIRT1
activity (18). It has also been
reported that interactions between DBC1 and SIRT1 are dysregulated
in breast cancer cell lines (19).
In terms of the regulation of estrogen receptor α (ERα) activity,
DBC1 and SIRT1 reciprocally interacted with the cell cycle and
apoptosis regulator 1 (CCAR1), and modulate its co-activator
function to ERα (20). Furthermore,
the inactivation of SIRT1 or the introduction of DBC1 promoted
c-MYC-induced apoptosis in different cell lines (21). In the present study, we found DBC1
is associated with SIRT1, but that it does not function as a
negative regulator of SIRT1 in HCC.
The functions of DBC1 and SIRT1 in tumorigenesis
remain controversial. In an early publication, the pro-apoptotic
and tumor-suppressive functions of DBC1 were proposed to be due to
the involvement of the caspase-mediated activation of DBC1 in
apoptotic induction by TNF-α (22).
On the other hand, DBC1 was found to be highly upregulated and
associated with poor survival in gastric carcinoma and breast
cancer (15,23). Notably, and in agreement with
previous results in studies of gastric carcinoma and breast cancer,
our results show that both DBC1 and SIRT1 proteins are highly
overexpressed in human HCC tissues (Fig. 1C). Furthermore, the mRNA expression
of the SIRT1 gene in a large cohort of HCC patients in the GEO
(Gene Expression Omnibus) database showed no significant
differences between tumor and matched non-tumor tissues (Fig. 1A). However, it was of note to find
that DBC1 overexpression was also observed in HCC tissues that
showed SIRT1 overexpression. Thus, it appears that both proteins
are aberrantly regulated at a post-transcriptional level in liver
cancer, and positively associated within cells. Indeed, our results
show that the expressions of DBC1 and SIRT1 are positively
correlated in HCC cells (Fig.
2).
Many proteins positively or negatively regulate
SIRT1 activity by direct binding in vitro. The active
regulator of SIRT1, AROS, activates SIRT1 and enhances the
p53-suppressive function of SIRT1 (24). In addition, casein kinase 2 (CK2)
mediated SIRT1 phosphorylation and increased deacetylation of p53
by SIRT1 and protected cells from apoptosis following DNA damage
(25). On the other hand,
sentrin-specific proteinase 1 (SENP1) inhibited SIRT1 activity by
interacting and desumoylating SIRT1, thereby promoting
stress-induced apoptosis (26). Of
these, CK2 and SENP1 have been shown to regulate p53 activity
either directly or indirectly (27,28).
Our results show that the inactivation of DBC1 or SIRT1 reduced HCC
cell viability and that DBC1 does not negatively regulate SIRT1
activity in HCC cells (Figs. 3 and
4). Many studies have shown that
SIRT1 is overexpressed in various human cancers, and that the
targeted disruption of SIRT1 inhibits cancer cell growth. Recently,
DBC1 was also suggested to be a new candidate gene with a potential
role in colon cancer (29). These
reports suggest that DBC1 acts during tumor progression or
development, and that the interaction between DBC1 and SIRT1 plays
a crucial role in tumorigenesis. Taken together, our results
suggest that the overexpression of DBC1 is associated with HCC and
that DBC1 does not function as a negative regulator of SIRT1
activity in HCC.
Acknowledgements
This study was supported by the
National Research Foundation of Korea (NRF) which is funded by the
Korean government (MEST) (Grant No. 2011-0010705), by the Korean
Ministry of the Environment ‘The Converging-Technology Project’
(Grant No. 212 101 003), and by the Korean Science and Engineering
Foundation via the ‘Cancer Evolution Research Center’ at The
Catholic University of Korea.
References
|
1.
|
A JemalF BrayMM CenterJ FerlayE WardD
FormanGlobal cancer statisticsCA Cancer J
Clin616990201110.3322/caac.20107
|
|
2.
|
S WhittakerR MaraisAX ZhuThe role of
signaling pathways in the development and treatment of
hepatocellular
carcinomaOncogene2949895005201010.1038/onc.2010.23620639898
|
|
3.
|
Y ItoY SasakiM HorimotoActivation of
mitogen-activated protein kinases/extracellular signal-regulated
kinases in human
hepatocellularcarcinomaHepatology27951958199810.1002/hep.5102704099537433
|
|
4.
|
CM WongST FanIO NgBeta-catenin mutation
and overexpression in hepatocellular carcinoma: clinicopathologic
and prognostic
significanceCancer92136145200110.1002/1097-0142(20010701)92:1%3C136::AID-CNCR1301%3E3.0.CO;2-R11443619
|
|
5.
|
M HamaguchiJL MethC von KlitzingDBC2, a
candidate for a tumor suppressor gene involved in breast cancerProc
Natl Acad Sci USA991364713652200210.1073/pnas.212516099
|
|
6.
|
JE KimJ ChenZ LouDBC1 is a negative
regulator of
SIRT1Nature451583586200810.1038/nature0650018235501
|
|
7.
|
W ZhaoJP KruseY TangSY JungJ QinW
GuNegative regulation of the deacetylase SIRT1 by
DBC1Nature451587590200810.1038/nature0651518235502
|
|
8.
|
AM TrauernichtSJ KimNH KimTG
BoyerModulation of estrogen receptor α protein level and survival
function by DBC-1Mol Endocrinol21152615362007
|
|
9.
|
J FuJ JiangJ LiDeleted in breast cancer 1,
a novel androgen receptor (AR) coactivator that promotes AR
DNA-binding activityJ Biol
Chem28468326840200910.1074/jbc.M80898820019126541
|
|
10.
|
S GarapatyCF XuP TrojerMA MahajanTA
NeubertHH SamuelsIdentification and characterization of a novel
nuclear protein complex involved in nuclear hormone
receptor-mediated gene regulationJ Biol
Chem28475427552200910.1074/jbc.M80587220019131338
|
|
11.
|
Z LiL ChenN KabraC WangJ FangJ
ChenInhibition of SUV39H1 methyltransferase activity by DBC1J Biol
Chem2841036110366200910.1074/jbc.M90095620019218236
|
|
12.
|
CC ChiniC EscandeV NinEN ChiniHDAC3 is
negatively regulated by the nuclear protein DBC1J Biol
Chem2854083040837201010.1074/jbc.M110.15327021030595
|
|
13.
|
DM HuffmanWE GrizzleMM BammanSIRT1 is
significantly elevated in mouse and human prostate cancerCancer
Res6766126618200710.1158/0008-5472.CAN-07-008517638871
|
|
14.
|
KY JangKS KimSH HwangExpression and
prognostic significance of SIRT1 in ovarian epithelial
tumoursPathology41366371200910.1080/0031302090288445119404850
|
|
15.
|
EJ ChaSJ NohKS KwonExpression of DBC1 and
SIRT1 is associated with poor prognosis of gastric carcinomaClin
Cancer Res1544534459200910.1158/1078-0432.CCR-08-332919509139
|
|
16.
|
W StunkelBK PehYC TanFunction of the SIRT1
protein deacetylase in cancerBiotechnol
J213601368200710.1002/biot.20070008717806102
|
|
17.
|
J ChenB ZhangN WongSirtuin 1 is
upregulated in a subset of hepatocellular carcinomas where it is
essential for telomere maintenance and tumor cell growthCancer
Res7141384149201110.1158/0008-5472.CAN-10-427421527554
|
|
18.
|
C EscandeCC ChiniV NinDeleted in breast
cancer-1 regulates SIRT1 activity and contributes to high-fat
diet-induced liver steatosis in miceJ Clin
Invest120545558201010.1172/JCI3931920071779
|
|
19.
|
JE KimZ LouJ ChenInteractions between DBC1
and SIRT 1 are deregulated in breast cancer cellsCell
Cycle837843785200910.4161/cc.8.22.1005519855164
|
|
20.
|
E Ji YuSH KimK HeoCY OuMR StallcupJH
KimReciprocal roles of DBC1 and SIRT1 in regulating estrogen
receptor α activity and co-activator synergyNucleic Acids
Res3969326943201121596782
|
|
21.
|
A MenssenP HydbringK KapelleThe c-MYC
oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the
SIRT1 deacetylase form a positive feedback loopProc Natl Acad Sci
USA201122190494
|
|
22.
|
R SundararajanG ChenC MukherjeeE
WhiteCaspase-dependent processing activates the proapoptotic
activity of deleted in breast cancer-1 during tumor necrosis
factor-α-mediated death signalingOncogene2449084920200515824730
|
|
23.
|
H LeeKR KimSJ NohExpression of DBC1 and
SIRT1 is associated with poor prognosis for breast carcinomaHum
Pathol42204213201010.1016/j.humpath.2010.05.02321056897
|
|
24.
|
EJ KimJH KhoMR KangSJ UmActive regulator
of SIRT1 cooperates with SIRT1 and facilitates suppression of p53
activityMol
Cell28277290200710.1016/j.molcel.2007.08.03017964266
|
|
25.
|
H KangJW JungMK KimJH ChungCK2 is the
regulator of SIRT1 substrate-binding affinity, deacetylase activity
and cellular response to DNA-damagePLoS
One4e6611200910.1371/journal.pone.000661119680552
|
|
26.
|
Y YangW FuJ ChenSIRT1 sumoylation
regulates its deacetylase activity and cellular response to
genotoxic stressNat Cell
Biol912531262200710.1038/ncb164517934453
|
|
27.
|
L McKendrickD MilneD MeekProtein kinase
CK2-dependent regulation of p53 function: evidence that the
phosphorylation status of the serine 386 (CK2) site of p53 is
constitutive and stableMol Cell
Biochem191187199199910.1023/A:1006854109926
|
|
28.
|
R BennettY PanJ ChristianT HuiWS May JrThe
RAX/PACT-PKR stress response pathway promotes p53 sumoylation and
activation, leading to G1 arrestCell
Cycle11407417201210.4161/cc.11.2.1899922214662
|
|
29.
|
JF ReidM GariboldiV SokolovaIntegrative
approach for prioritizing cancer genes in sporadic colon
cancerGenes Chromosomes
Cancer48953962200910.1002/gcc.2069719672874
|