Introduction
Acute myeloid leukemia (AML) is the most common type
of hematological malignancy characterized by key properties,
including blocked differentiation, enhanced self-renewal and
increased proliferation. Although there have been great
developments in the understanding and treatment of AML, the
mortality of AML is still high. Due to the difficulties in
tolerance of treatment, the molecular mechanisms of uncontrolled
proliferation of AML have been a major research project all over
the world.
Gap junctions consist of arrays of intercellular
channels composed of two hemichannels or connexons, one of which is
formed by six protein subunits, termed connexins (Cxs). Cxs are a
conserved family of transmembrane proteins which regulate the
passage of biological molecules and allow the exchange of signaling
molecules smaller than 1 kDa between the cytoplasm of two
neighboring cells, e.g. Ca2+, cyclic adenosine
monophosphate (cAMP) and inositol triphosphate (IP3) (1–3). To
date, at least 21 different human Cxs have been identified and they
have been divided into two groups based on the primary amino acid
sequence homology (3). It suggests
that the dysregulation of Cx expression is related to uncontrolled
proliferation, embryogenesis, tissue homeostasis and carcinogenesis
(4). Recently, abnormal or
defective gap junction communication in various solid tumors
including liver, bladder, breast and prostate cancers was found
(5), and accumulating research has
also shown that restoring Cx gene expression and gap junction by
gene therapy in Cx-deficient tumor cells could decrease tumor cell
growth. However, despite much existing evidence, the exact
contribution of the Cx channel family still remains controversial,
as gap junction and cxs may furnish cell survival signals. The gap
junction and cxs could exert their effect on promoting cancer cell
proliferation (6,7). Each Cx shows tissue cell-specific
expression, and Cx43 is the major component of hematopoietic tissue
(8–11). Cx32 was also found to be important
in bone marrow stromal cells (12,13).
Until now, there have been few studies focusing on the expression
of the gap junction genes in the AML cell lines, and little is
known regarding the correlation between cell proliferation and Cxs.
In this study, the AML cell lines OCI-AML3 and OCIM2 were employed
to investigate the expression of Cx32 and Cx43 in AML and their
role in proliferation.
Materials and methods
Cell culture
The OCI-AML3 and OCIM2 were kindly provided by M.D.
Minden (Ontario Cancer Institute, Toronto, ON, Canada). OCI/AML3
(FAB M4) was established from a patient with AML, and OCIM2 (FAB
M6) from a patient with erythroleukemia. The cells were grown in
RPMI-1640 supplemented 10% fetal bovine serum (FCS). Cultures were
maintained in a humidified atmosphere with 5% CO2 at
37˚C.
Chemicals and antibodies
Thiazolyl blue tetrazolium bromide (MTT),
dimethysulfoxide (DMSO), prodidum iodide (PI) and Hoechst 33258
were bought from Sigma (Sigma-Aldrich, St. Louis, MO, USA).
Polyclonal rabbit antibody against Cx43 and Cx32 were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA) and PTG
(Chicago, IL, USA). γ-tubulin was bought from Jackson
ImmunoResearch (Jackson, WY, USA).
Cell growth curve
The OCI-AML3 and OCIM2 cells were seeded in 24-well
plates (2x104 per well), cultured in a humidified
atmosphere with 5% CO2 at 37˚C. The cells were harvested
and counted on days 1–4, three wells each time and three times each
well, to obtain an average to draw the growth curve.
Cell proliferation assay
The proliferative rate of OCI-AML3 and OCIM2 was
determined by MTT assay following the method of Mosmann (14). The OCI-AML3 and OCIM2 cells
(1x104 per well) were cultured for 24, 48 and 72 h in
96-well plates. Thereafter, 20 μl MTT solution was added to
each well. After continued incubation for 4 h, the supernatant was
discarded and 150 μl DMSO was added. Once the blue crystals
were dissolved, the optical density (OD) was measured at 490 nm
with background substraction of 630 nm using a plate microreader
(Tecan Spectra, Wetzlar, Germany). The experiments were performed
in triplicate. The proliferation rate was determined using the
following formula: Cell proliferation (%) = OD of the experimental
samples / OD of the control x 100% (n=3, mean ± SD).
Cell cycle analysis
The OCI-AML3 and OCIM2 (1x106 cells) were
harvested. After being washed, the cells were fixed with 75%
ice-cold ethanol and maintained overnight at 4˚C. The cells were
collected and resuspended in PBS containing 40 μg/ml PI, 0.1
mg/ml RNase, and 5% Triton X-100, and then incubated at 37˚C for 30
min. The cells were evaluated by flow cytometry (FCM) using FACS
(BD, San Diego, CA, USA). At least 10,000 counts were made for each
sample. The percentage distribution in the cell cycle phases was
analyzed using CellQuest. The proliferation index (PI) was
determined using the following formula: PI = (S + G2/M) / (G0/G1 +
S + G2/M).
Semi-quantitative reverse
transcription-PCR (RT-PCR)
Total RNA was extracted from OCI-AML3 and OCIM2
cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA).
Following quantification by spectrophotometry, the first-strand
cDNA was synthesized from 2 μg of total RNA with the
RevertAid First-Strand cDNA Sythesis kit. Cx43: forward
5′-TCGCCTATGTCTCCTCCTGG-3′, reverse 5′-GCTGGCTCTGCTTGAAGGTC-3′,
with a PCR product of 270 bp; Cx32: forward 5′-AAATGCTACGGCTTGA
GGGC-3′, reverse 5′-CGGAACACCACGCTGATGAC-3′, which can amplify a
115 bp fragment; β-actin: forward 5′-G CG G GA A ATCGTG CGTGACAT
TA-3′, reverse 5′-GACTCGTCATACTCCTGCTTGCTGAT-3′, with an expected
PCR product of 480 bp. The products were electrophoresesed on 2%
agarose gel and the ratio between the target gene and β-actin gene
band density was used for quantitative evaluation.
Western blot analysis
For preparation of total cell lysates, cells were
collected and lysed in lysis buffer (50 mM HEPES, 150 mM NaCl, 1%
Triton X-100, 5 mM EGTA, 50 mM glycerophosphate, 20 mM NaF, 1 mM
Na3VO4, 2 mM phenylmethyl sulfonyl fluoride,
10 μg/ml leupeptin and 10 μg/ml aprotinin) by
incubating on ice for 30 min. Lysates were then centrifuged at
12,000 x g for 15 min at 4˚C. The supernatant was collected and the
total protein concentrations were determined using the BCA assay by
spectrophotometer (Biotech Instruments, NY, USA). Samples were
separated on 10% SDS-PAGE and transferred onto nitrocellulose
membranes. After blocking with 5% non-fat dry milk in blocking
buffer (25 mM Tris, pH 7.5, 150 mM NaCl, and 0.1% Tween-20), the
membranes were incubated with primary antibodies at 4˚C overnight.
The membranes were then incubated with appropriate
peroxidaseconjugated secondary antibodies, and the protein
expression was detected by ECL substrate solution (Thermo
Scientific, Rockford, IL, USA). Densitometric analysis was
performed using Quantity One software.
Fluorescent immunostaining
OCI-AML3 and OCIM2 cells were collected and washed
with PBS and then fixed with 40 mg/l paraformaldehyde for 10 min
before deposition on polylysine-coated coverslips. After washing,
samples were blocked with 10% goat serum albumin for 30 min. The
cells were reacted overnight at 4˚C with a drop of 1:100 diluted
Cx43 antibody, then washed and incubated with a drop of 1:500
diluted Cy3-labeled goat anti-rabbit IgG (Sigma, USA) for 1 h.
After that, cells were stained with Hoechst 33258 for 30 min at
37˚C. Finally, the slides were mounted with 50% glycerol and
observed by Olympus BH-2 fluorescence microscope (Tokyo,
Japan).
Statistical analysis
The statistical significance of difference between
control and treatment groups was determined by the Student’s
t-test. Values are shown as the mean ± SD, and P<0.05 was
considered to indicate a statistically significant difference.
Results
Growth curve and proliferation rate in
AML cell lines OCI-AML3 and OCIM2
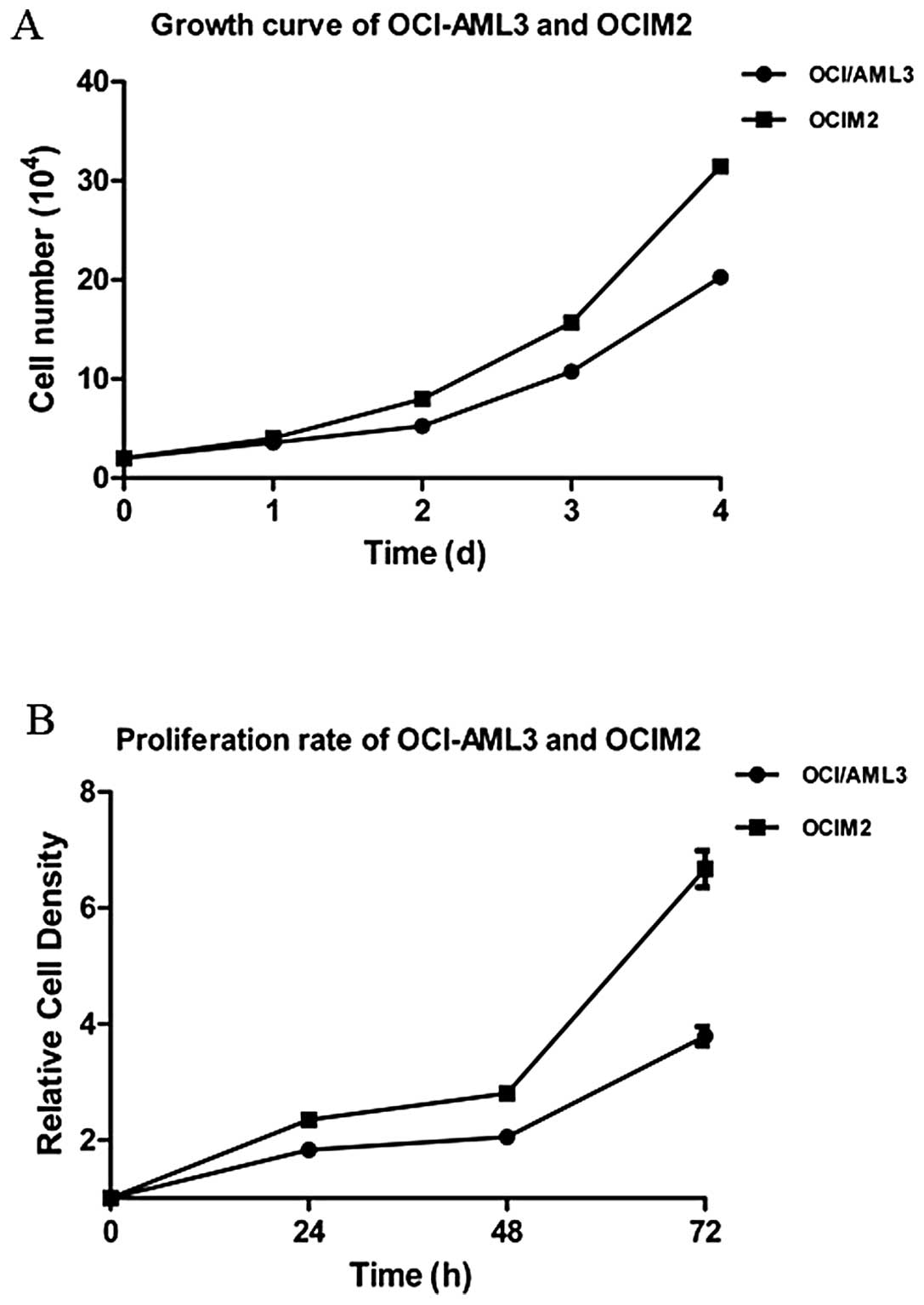
The OCI-AML3 and OCIM2 cells were seeded and
cultured for 1–4 days, and cells were harvested and counted each
day. According to the growth curve, as shown in Fig. 1A, the doubling time of OCI-AML3 and
OCIM2 was 48 and 36 h respectively, and OCIM2 grew faster than
OCI-AML3. To further confirm the proliferation rate of OCI-AML3 and
OCIM2, the cells were seeded and cultured for 24, 48 and 72 h and
detected by MTT. As shown in Fig.
2, the proliferation rate of OCIM2 was nearly twice that of
OCI-AML3.
Cell cycle distribution and PI in AML
cell lines OCI-AML3 and OCIM2
As shown in Fig. 2A,
the cell cycle distribution of AML cell lines OCI-AML3 and OCIM2
was determined by FCM. As shown in Fig.
2B, the percentage of OCI-AML3 cells in the S phase fraction
was 24.95±5.8%; however, the percentage of OCIM2 cells was much
higher, at OCI-AML3, at 59.47±9.6%. At the same time, the PI of
OCI-AML3 and OCIM2 was calculated to further confirm the role of
the cell cycle in the proliferation rate of these two cell lines.
As shown in Fig. 2B, that of OCIM2
was 78.12±8.9%, however, the PI of OCI-AML3 was only 35.21±6.7%.
There were significant differences in S phase distribution and PI
between OCI-AML3 and OCIM2, as shown in Fig. 2C (P<0.01).
Expression of Cx32, Cx43 mRNA and
proteins in AML cell lines OCI-AML3 and OCIM2
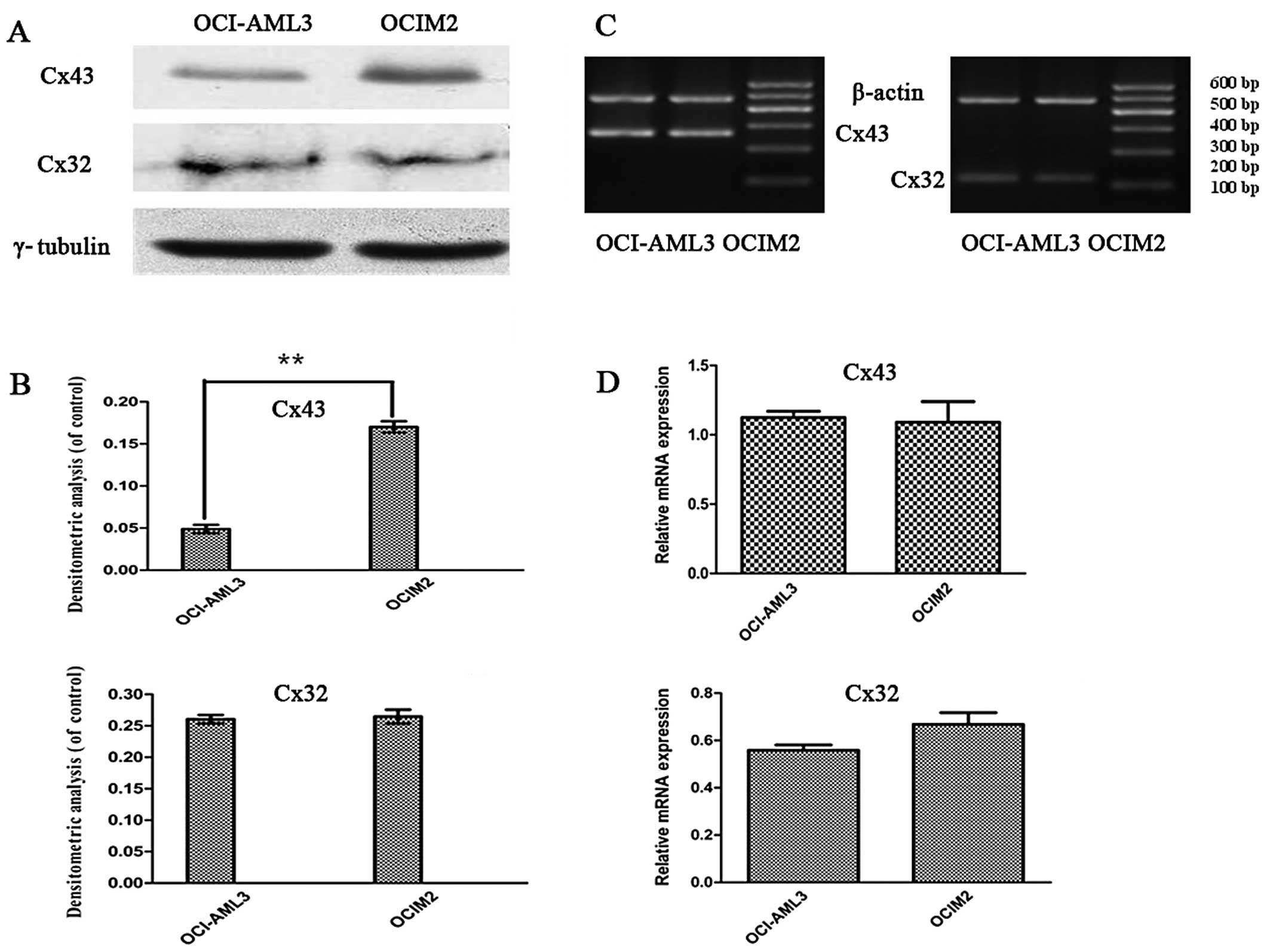
To investigate whether there was a difference in the
expression of Cxs in AML cell lines OCI-AML3 and OCIM2, the
expression of Cx32, and Cx43 mRNA in OCI-AML3 and OCIM2 was
detected by RT-PCR. The protein levels of Cx32 and Cx43 were
determined by western blot assay and immunofluorescence. As shown
in Fig. 3C and D, the RT-PCR assay
showed that the expression of Cx43 and Cx32 mRNA in these two cell
lines was not significantly different (P>0.05). In contrast, a
clear difference was noted between OCI-AML3 and in OCIM2 in the
expression of Cx43 protein. As shown in Fig. 3A and B, the protein level of Cx43 in
OCIM2 was notably higher than that of OCI-AML3 (P<0.01).
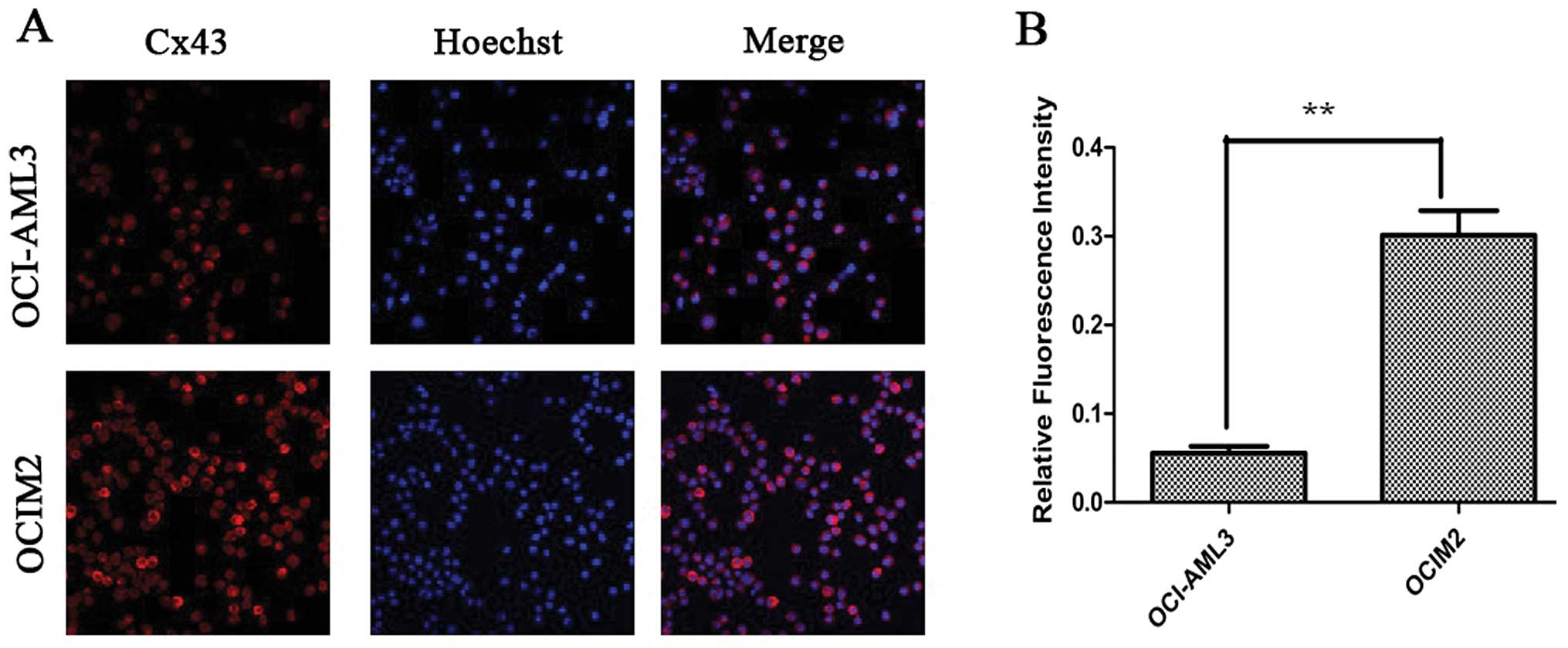
Cx43 proteins appeared as a collection of red spots
intracytoplasmically and in certain parts of the plasma membrane,
as shown in Fig. 4A. The
immunofluorescence assay showed that the fluorescence intensity of
Cx43 protein in OCIM2 was higher than that in OCI-AML3. OCIM2 cells
showed both stronger western blot and fluorescence intensity. These
results suggested that the expression of Cx43 in OCIM2 was much
higher than OCI-AML3 at the protein level, but there was no
difference at the mRNA level.
Discussion
Deviation of physiological cell cycling, as a
consequence of homeostatic imbalance, may lead to uncontrolled cell
proliferation activity (15–17).
The S phase (synthesis phase) is the part of the cell cycle in
which DNA is replicated, occurring between the G1 and G2 phase,
which is a reliable marker to indicate how fast a tumor is growing.
In clinical practice, the S phase fraction could be used as an
independent prognostic factor in node-negative invasive breast
carcinoma (18). The proliferative
index (PI) is a measure of the number of proliferated cells in a
tumor, which is also an indicator of the cell proliferation rate.
Our results revealed that OCIM2 grew much faster than OCI-AML3 and
that the proliferation ability of OCIM2 was much stronger than that
of OCI-AML3. In addition, the results showed that the S phase
percentage and PI of OCIM2 was higher than OCI-AML3. Since acute
erythroleukemia is an uncommon subtype of AML associated with a
very poor prognosis compared to other subtypes, such as OCI-AML3,
these results support the use of the S phase fraction as a
parameter to predict the proliferation rate in AML cells.
It is well known that gap junctions, a group of
specialized cell-to-cell junctions composed of Cx proteins
(19–21), are critical gatekeepers of cell
proliferation, controlling the intercellular exchange of essential
growth regulators (22). The role
of Cxs in leukemogenesis and leukemic cell functions are not well
defined. Previous studies have clearly shown that soluble mediators
alter AML cell proliferation and apoptosis both when the leukemic
cells are cultured alone and when co-cultured with fibroblasts,
endothelial cells and osteoblasts (23). Furthermore, direct cell-cell
interactions between AML cells also alter AML cell function. Even
though the function of Cx building blocks affects the regulation of
several cellular functions, including growth and apoptosis of
normal and malignant cells, and also appears to be important for
chemosensitivity, little is known about their role in leukemic cell
functions. Certain reports demonstrate that overexpression or
genetic mutation of Cx could induce tumor cell growth and inhibit
G1/S cell cycle arrest (12,24,25).
In contrast, several studies have demonstrated that the presence of
Cxs is indispensable for the enrollment of cell proliferation
(26–29). Our research demonstrated a lower
expression of Cx43 in OCI-AML3 than OCIM2, but RT-PCR results
revealed no decrease of Cx43 mRNA in these two cell lines. It is of
note, however, that the same level of Cx32 mRNA and proteins was
detected in OCIM2 and OCI-AML3 cell lines. It appears that Cx43
gene transcription was not responsible for the aberrant expression
of Cx43 proteins. Although there was no difference in the
expression of Cx32 both at the transcriptional and protein level,
we found that there was a significant difference in the expression
of Cx43 at the protein level. Therefore, the results of the present
study indicate that Cx43 could be considered as a potential tumor
promoter which exerts its effect by promoting the exchange of
growth factors or facilitating proliferation by itself as a
pro-survival signal, and may also have a correlation with malignant
AML cell proliferation. Further investigation is required to
understand the precise mechanism in which intercellular
communication participates in neoplastic growth in the
hematopoietic process.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (no.
81070429).
Reference
|
1.
|
M OyamadaY OyamadaT TakamatsuRegulation of
connexin expressionBiochim Biophys
Acta1719623200510.1016/j.bbamem.2005.11.00216359940
|
|
2.
|
M KonoplevaS KonoplevW HuAY ZaritskeyBV
AfanasievM AndreeffStromal cells prevent apoptosis of AML cells by
up-regulation of anti-apoptotic
proteinsLeukemia1617131724200210.1038/sj.leu.240260812200686
|
|
3.
|
DW LairdLife cycle of connexins in health
and diseaseBiochem J394527543200610.1042/BJ2005192216492141
|
|
4.
|
L CronierS CrespinPO StraleN DefamieM
MesnilGap junctions and cancer: new functions for an old
storyAntioxid Redox
Signal11323338200910.1089/ars.2008.215318834328
|
|
5.
|
M MesnilS CrespinJL AvanzoML
Zaidan-DagliDefective gap junctional intercellular communication in
the carcinogenic processBiochim Biophys
Acta1719125145200510.1016/j.bbamem.2005.11.00416359943
|
|
6.
|
SF GiardinaM MikamiF GoubaevaJ
YangConnexin 43 confers resistance to hydrogen peroxide-mediated
apoptosisBiochem Biophys Res
Commun362747752200710.1016/j.bbrc.2007.08.06617761141
|
|
7.
|
M FreidinS AscheTA BargielloMV BennettCK
AbramsConnexin 32 increases the proliferative response of Schwann
cells to neuregulin-1 (Nrg1)Proc Natl Acad Sci
USA10635673572200910.1073/pnas.081341310619218461
|
|
8.
|
X LiYB XuQ WangLeukemogenic AML1-ETO
fusion protein upregulates expression of connexin 43: the role in
AML 1-ETO-induced growth arrest in leukemic cellsJ Cell
Physiol208594601200610.1002/jcp.2069516741927
|
|
9.
|
Y LiuX ZhangZJ LiXH ChenUp-regulation of
Cx43 expression and GJIC function in acute leukemia bone marrow
stromal cells post-chemotherapyLeuk
Res34631640201010.1016/j.leukres.2009.10.01319910046
|
|
10.
|
Y LiuX ZhangYJ SiL GaoXH Chen[Connexin 43
expression and interacellular communicating function in acute
leukemia bone marrow stroma cells]Zhongguo Shi Yan Xue Ye Xue Za
Zhi156796822007
|
|
11.
|
T KrenacsM RosendaalConnexin43 gap
junctions in normal, regenerating, and cultured mouse bone marrow
and in human leukemias: their possible involvement in blood
formationAm J Pathol152993100419989546360
|
|
12.
|
H SatoH HagiwaraY OhdeH SenbaN VirgonaT
YanoRegulation of renal cell carcinoma cell proliferation, invasion
and metastasis by connexin 32 geneJ Membr
Biol2161721200710.1007/s00232-007-9020-517565422
|
|
13.
|
Y HirabayashiBI YoonI TsuboiProtective
role of connexin 32 in steady-state hematopoiesis, regeneration
state, and leukemogenesisExp Biol Med
(Maywood)232700712200717463168
|
|
14.
|
DM SpinnerMTT growth assays in ovarian
cancerMethods Mol Med39175177200121340769
|
|
15.
|
M MalumbresM BarbacidCell cycle, CDKs and
cancer: a changing paradigmNat Rev
Cancer9153166200910.1038/nrc260219238148
|
|
16.
|
R SuryadinataM SadowskiB SarcevicControl
of cell cycle progression by phosphorylation of cyclin-dependent
kinase (CDK) substratesBiosci
Rep30243255201010.1042/BSR2009017120337599
|
|
17.
|
S van den HeuvelCell-cycle
regulationWormBook11620053318688
|
|
18.
|
L Moureau-ZabottoC BouchetD CesariCombined
flow cytometry determination of S-phase fraction and DNA ploidy is
an independent prognostic factor in node-negative invasive breast
carcinoma: analysis of a series of 271 patients with stage I and II
breast cancerBreast Cancer Res
Treat916171200510.1007/s10549-004-7047-1
|
|
19.
|
M VinkenT VanhaeckeP PapeleuS SnykersT
HenkensV RogiersConnexins and their channels in cell growth and
cell deathCell
Signal18592600200610.1016/j.cellsig.2005.08.01216183253
|
|
20.
|
M VinkenT HenkensE De RopJ FraczekT
VanhaeckeV RogiersBiology and pathobiology of gap junctional
channels in
hepatocytesHepatology4710771088200810.1002/hep.2204918058951
|
|
21.
|
M VinkenE De RopE DecrockEpigenetic
regulation of gap junctional intercellular communication: more than
a way to keep cells quiet?Biochim Biophys
Acta17955361200918801412
|
|
22.
|
E DecrockM VinkenE De
VuystConnexin-related signaling in cell death: to live or let
die?Cell Death Differ16524536200910.1038/cdd.2008.19619197295
|
|
23.
|
K HatfieldA RyningenM CorbascioO
BruserudMicrovascular endothelial cells increase proliferation and
inhibit apoptosis of native human acute myelogenous leukemia
blastsInt J Cancer11923132321200610.1002/ijc.22180
|
|
24.
|
C RogerB MograbiD ChevallierDisrupted
traffic of connexin 43 in human testicular seminoma cells:
overexpression of Cx43 induces membrane location and cell
proliferation decreaseJ Pathol202241246200410.1002/path.1509
|
|
25.
|
H SatoK FukumotoS HadaEnhancing effect of
connexin 32 gene on vinorelbine-induced cytotoxicity in A549 lung
adenocarcinoma cellsCancer Chemother
Pharmacol60449457200710.1007/s00280-006-0406-317569045
|
|
26.
|
CE ChadjichristosCM MatterI RothReduced
connexin43 expression limits neointima formation after balloon
distension injury in hypercholesterolemic
miceCirculation11328352843200610.1161/CIRCULATIONAHA.106.62770316769907
|
|
27.
|
H OzawaH MutaiT MatsunagaPromoted cell
proliferation by connexin 30 gene transfection to head-and-neck
cancer cell lineAnticancer Res2919811985200919528455
|
|
28.
|
HH WangCI KungYY TsengActivation of
endothelial cells to pathological status by down-regulation of
connexin43Cardiovasc Res79509518200810.1093/cvr/cvn11218445604
|
|
29.
|
X LiuT FuruyaD LiConnexin 26 expression
correlates with less aggressive phenotype of intestinal
type-gastric carcinomasInt J Mol Med25709716201020372813
|