Introduction
Multiple primary malignancies (MPM) in a single
patient are relatively rare but have increased in frequency in
recent decades (1). This may be a
result of medical advancements in diagnostic and therapeutic
strategies, a possible effect of new carcinogens in the industrial
environment and longer life span allowing another primary cancer to
develop (2,3). Among those patients with MPM, double
cancer is commonly observed, while triple cancers occur in 0.5% of
patients and quadruple or quintuple cancers occur in less than 0.1%
of patients (4). The present study
describes a case of a patient with double malignancy of an
undifferentiated pleomorphic sarcoma/pleomorphic malignant fibrous
histiocytoma and hereditary non-polyposis colorectal cancer
(HNPCC). The literature concerning HNPCC is also discussed.
Informed consent was obtained from the patient. The study was
approved by the Ethics Committee of The 6th People’s Hospital of
Shanghai, School of Medicine, Shanghai Jiaotong University,
Shanghai, China.
Case report
Patient
A 43-year-old male (proband IV12) with a past
medical history of penicillin allergy and syphilis was first
admitted to the hospital for a malignant histiocytoma (pG2T2bN0Mx,
stage III) of the right scapula in May 2009 (Fig. 1). The patient underwent 3 tumor
resections in situ due to the recurrence of the tumor in
July 2009, December 2009 and January 2010. The patient was
re-hospitalized in April 2010 due to a 1-month history of
progressive abdominal discomfort in the right lower quadrant,
increased bowel movement frequency and weight loss of 5 kg. A
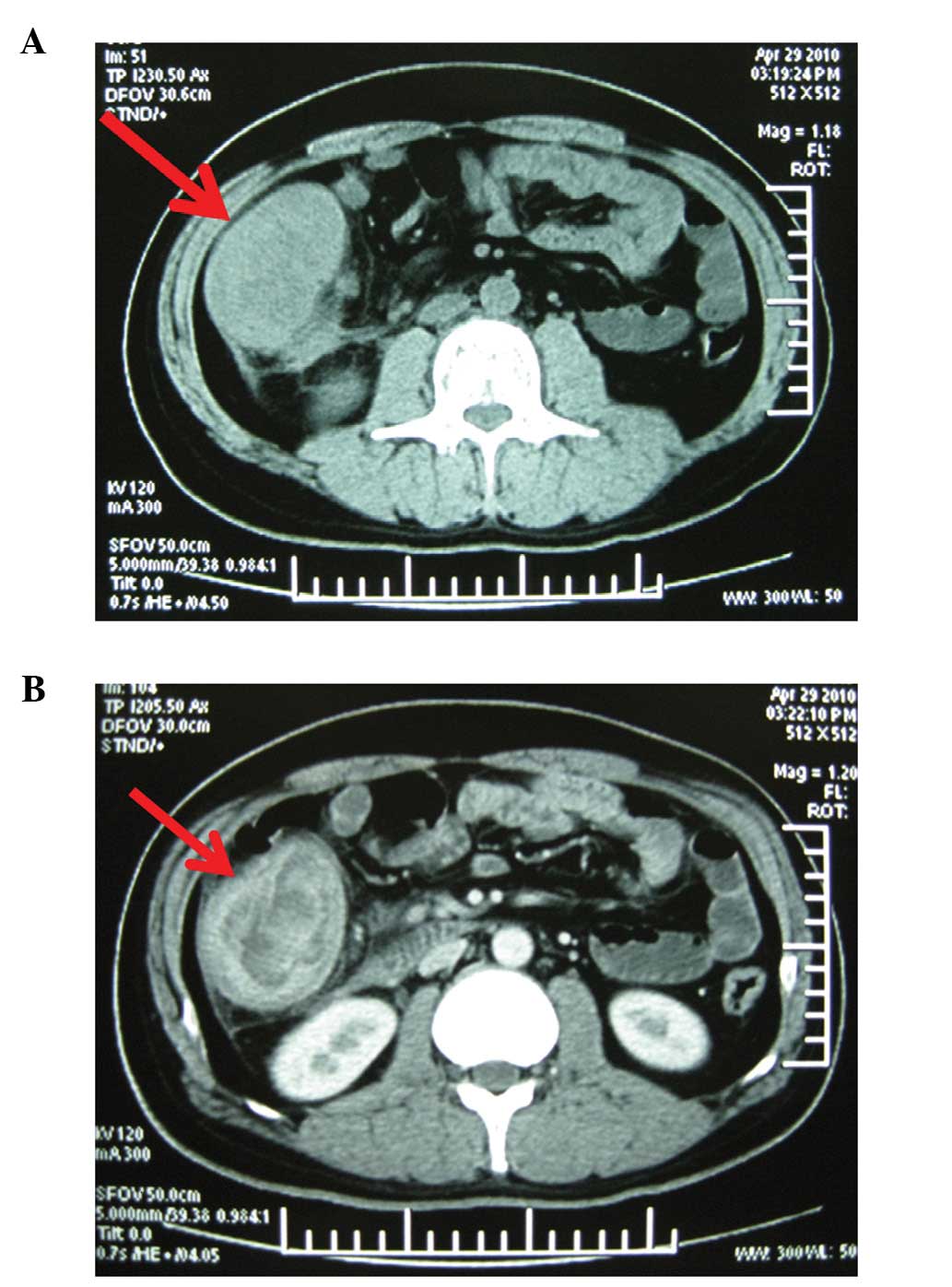
computed tomography scan of the abdomen revealed an irregularly
thickened wall of the ascending colon with localized stenosis and
regional mesenteric lymphadenopathy (Fig. 2). Colonoscopy revealed a 10x10-cm
obstructing tumor arising from the ileocecal valve to the hepatic
flexure. A biopsy specimen of the tumor suggested tubulovillous
adenoma with high grade dysplasia. Laboratory investigations
revealed an elevated serum level of carcinoembryonic antigen (CEA,
20.0 μg/l; normal range, 0.0–10.0 μg/l) and anemia
with low hemoglobin (Hb) concentration (83.0 μg/l; normal
range, 113.0–172.0 μg/l). Carbohydrate antigen (CA) 125 and
α fetoprotein (AFP) serum levels and other serum indices
were within normal limits. The patient underwent a right
hemicolectomy and an end-to-end ileocolostomy. After surgery, the
patient was discharged without any complications, and has been
well, without any evidence of recurrence or metastasis, for 15
months with outpatient follow-up every 1–2 months. The
postoperative course was uneventful, and the patient was referred
for chemotherapy and radiotherapy.
Pathological findings
Gross examination revealed a right hemicolectomy
specimen measuring 20 cm in length in the terminal ileum and 30 cm
in length in the colon. The colonic resection specimen included the
cecum, ascending colon and a proximal part of the transverse colon.
There was an ulcerating, circumferential mass in the cecum, ∼9x6 cm
in size, which extended through the entire bowel wall, infiltrating
the surrounding fatty tissue. The immunohistochemical results of
the tumor tissue were: CK(+), EMA(+), P53(+), CerbB-2, EGFR(+),
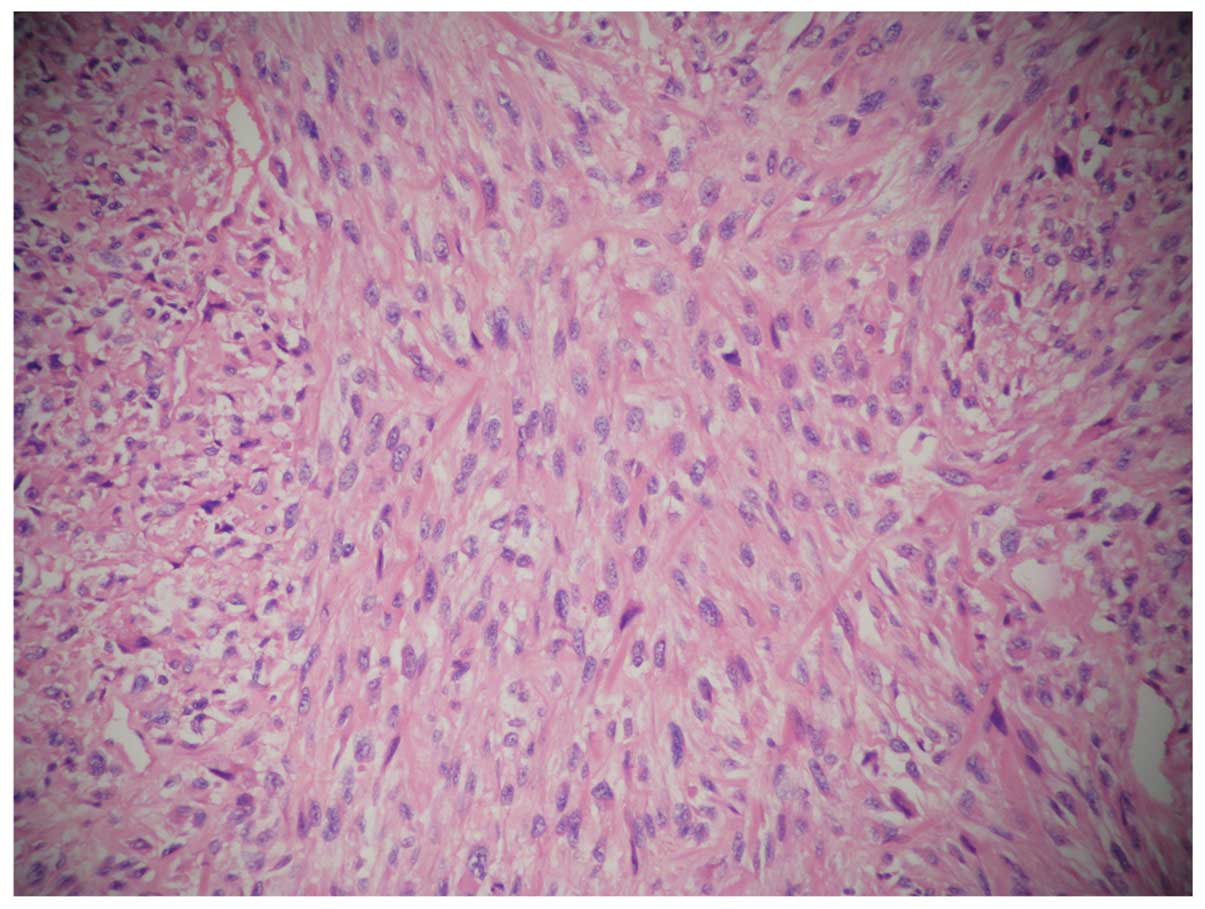
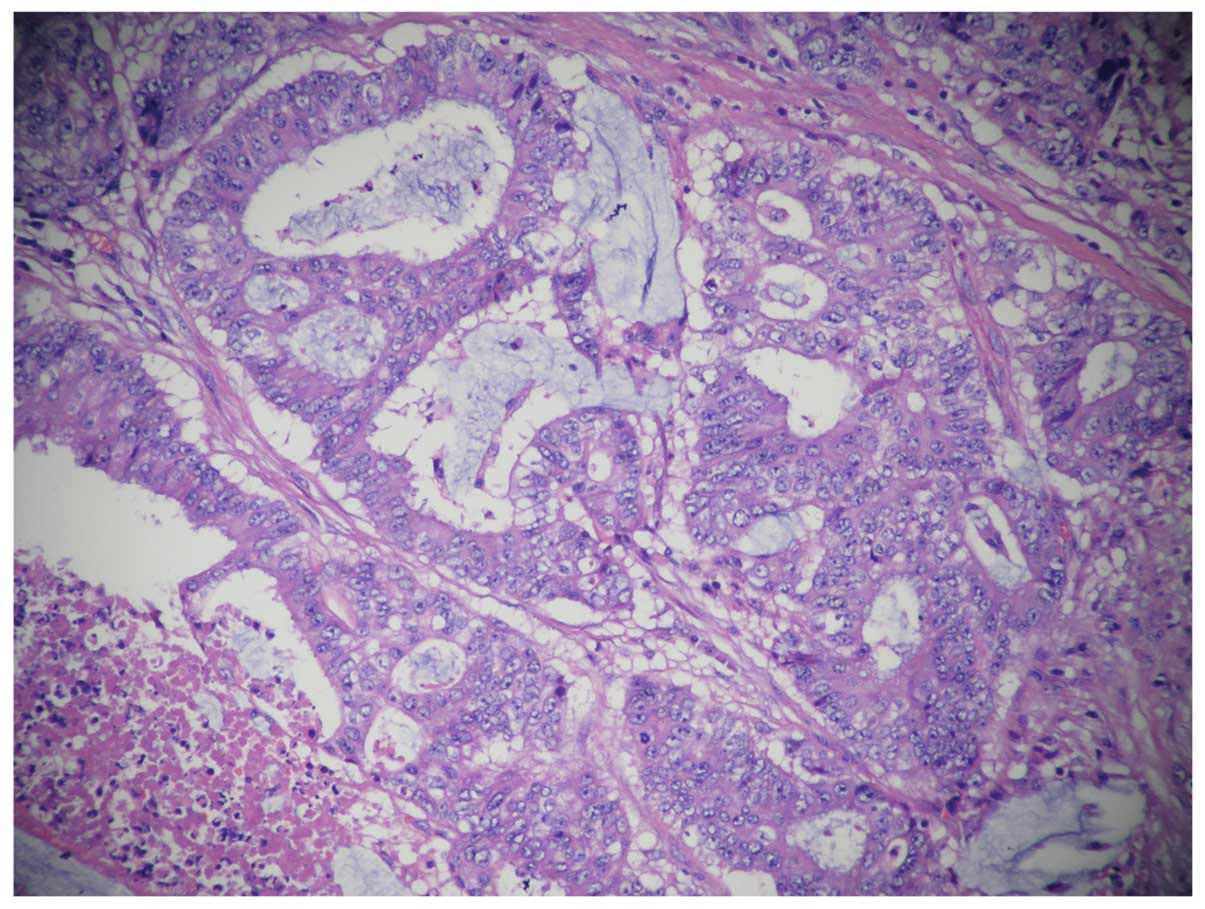
Ki67(60%+), CD34 (−), S-100(−). Histologically, specimens from the
tumor showed mucinous adenocarcinoma and invasion of the right
renal adipose capsule, lymphatic vessel and perineural region
(Fig. 3). Of the 14 peritumoral
lymph nodes dissected, 1 contained metastases of the tumor. Nine
mesenteric lymph nodes and 2 lymph nodes at the mesenteric root
were also dissected and no metastasis was found. The immunological
response of the dissected lymph nodes was SH(+), PH(+), GH(+).
Specimens which were free of tumor cells exhibited chronic
inflammation of the mucosa.
Family history investigation (Fig. 4)
I1 succumbed to gastrointestinal cancer, the details
of which could not be verified. II1 was diagnosed with colonic
cancer at the age of 60 years and succumbed to the disease. III1
succumbed to colonic cancer at the age of 50 years. III2 was
diagnosed with colonic cancer at the age of 70 years and succumbed
to the disease 7 years after colorectomy. III3 was diagnosed with
colonic cancer at the age of 56 years and succumbed to the disease
3 years after colorectomy. III4 was diagnosed with colonic cancer
at the age of 60 years, underwent colorectomy 8 years ago and is
currently well. IV1 was diagnosed with colonic cancer at the age of
30 years and succumbed to the disease 8 years after colorectomy.
IV2 was diagnosed with endometrial carcinoma 8 years ago at the age
of 50 years and she is currently well after surgery. IV4 was
diagnosed with pancreatic cancer 2 years ago at the age of 52 years
and is now well after surgery. IV6 was diagnosed with colonic
cancer 4 years ago at the age of 42 years and is currently well
after surgery. IV7 was diagnosed with colonic and pancreatic
cancers at the age of 40 years and succumbed to the disease 6 years
after surgery. IV12 was diagnosed with colonic cancer at the age of
41 years and is currently well after colorectomy 15 months ago.
IV15 was diagnosed with colonic cancer at the age of 42 years,
underwent colorectomy 1 year ago and is currently well. V5 was
diagnosed with colonic cancer at the age of 32 years, underwent
colorectomy 1 year ago and is currently well. IV9, 10, 11 and 13
and V22 and 23 have not been diagnosed with any cancer but have
chronic gastritis, constipation and indigestion. The mean age of
the patients when cancer was diagnosed in two generations (III, IV)
was 56.5±12.5 and 41.7±7.6 years, respectively. Four generations
(II, III, IV and V) underwent colonoscopy and there was no evidence
of colonic polyps. There was no history of familial adenomatous
polyposis (FAP) and no family spouse diagnosed with cancer.
Discussion
Ever since Billroth’s report, in 1889, of a patient
with multiple tumors, a gastric carcinoma that developed after the
removal of a spinocellular epithelioma of the right ear, MPM have
been objects of medical curiosity (5). Until 1932, only a few such cases had
been recognized, when Warren and Gates classified 1,259 such
patients from literature reports and post mortem examinations and
modified the diagnostic criteria for MPM, which included the
following (6,7): i) each tumor must present a definite
picture of malignancy; ii) each tumor must be histologically
distinct; and iii) the possibility that one is a metastasis of
another must be excluded. A number of theories have been proposed
to explain MPM but, to date, none have been proven, although the
key risk factors appear to be smoking and family history (8).
In review of the literature regarding MPM, several
common points may be concluded (2).
First, the Japanese population appears to have a higher likelihood
of developing MPM. Yamamoto et al reported that 15 to 20% of
Japanese patients with colorectal carcinoma developed MPM (9). This may be caused by genetic
susceptibility, longer average life span or medical advances in
chemotherapy and radiotherapy. Second, most patients with MPM are
geriatric. Third, smoking-related cancers, prostate cancers and
renal cell carcinoma are most commonly associated with MPM
(10). Fourth, head and neck cancer
survivors are at an increased risk of developing another cancer of
the respiratory or digestive tract (11). A ‘field cancerization effect’ was
assumed to explain this phenomenon, with carcinogens to which the
organ has been exposed initiating the proliferation of numerous
clones of cells (12). Carcinogenic
insults, such as tobacco and alcohol, may increase the likelihood
of multiple independent malignant foci developing in the mucosa
epithelium. The patient (IV12, proband) described in the present
study never smoked tobacco or drank alcohol, but he received
chemotherapy and radiotherapy for the first primary cancer.
The frequency of MPM depends on the length of the
observation period, applied diagnostic and prognostic criteria,
exposure to environmental factors, genetically defined individual
susceptibility, diagnostic accuracy, follow-up and administered
treatment. There was no family history of FAP in this case, and two
family members (IV7 and IV12) have multiple malignancies. Our
patient (IV12, proband) was diagnosed with malignant histiocytoma
and colonic cancer, which have different histopathological
criteria. Family members in this case had a high incidence of colon
cancer, suggesting that certain genetic features may induce
tumorgenesis.
In 15–20% of patients with colon cancer there is
evidence of a family history (13).
HNPCC is the most common form of hereditary colorectal cancers,
with conservative estimates of 5–10% of the total colorectal cancer
burden (14,15). Predisposed individuals have an
increased lifetime risk of developing colorectal, endometrial and
other types of cancer. HNPCC may present with differing phenotypes
and a detailed family history is required going back at least 3
generations (13). The first report
of a cancer family that represented what is now known as HNPCC was
published by Aldred Scott Warthin, a renowned pathologist, in 1913
(16). Over time, as general
knowledge about HNPCC improved, more and more affected families
were reported. The characteristic presentation of HNPCC is
frequently right-sided localization, the presence of synchronous
and metachronous colorectal carcinomas and its association with
other HNPCC-related extra-colonic tumors, including gastric,
endometrial and urinary and biliary tract cancers in afflicted
families (13). Pancreatic tumors
have been reported in relatives with HNPCC (17). Colorectal adenocarcinoma occurring
in patients with HNPCC frequently exhibits medullary features. We
report a case of MPM involving tubulovillous adenoma in the
ascending colon and malignant histiocytoma in the scapular with
family history of colorectal carcinoma. In this family, 11 members
were diagnosed with colonic cancer. The family history is notable
for a high incidence of right-sided colonic cancer with no evidence
of FAP. HNPCC has an earlier age of onset, Crohn’s disease-like
lymphocytic infiltration of tumor tissues, increased mucin
production and a lower degree of histological differentiation
compared with FAP (13). In this
case report, 11 patients were male and 3 were female, and the
fourth generation was markedly younger than the third generation
when their cancers were first diagnosed. This family fulfills
Amsterdam criteria I, with high incidence in males and early age at
onset of cancer.
We are aware of some limitations in this study.
First, we report only one family history investigation with the
charted family tree, which limits the ability to provide robust
evidence. More cases should be investigated. Second, we were unable
to demonstrate a detailed explanation for the mechanism of MPM and
HNPCC. Further basic research is thus needed. Despite these
limitations, we believe that the present study provides evidence to
improve the recognition of MPM and HNPCC, which are not well-known
among practicing physicians, leading to not recognizing families at
risk.
In summary, cases with MPM have been reported more
often in the recent literature, but clinicians should also keep in
mind that the prevalence of MPM is increasing. The assumption of a
new lesion in a patient with a previous history of cancer as
metastasis could possibly change the treatment strategies and delay
the management of a curable neoplasm. The early detection of
additional primary malignancies will enable prompt management and
is likely to increase the cure rate of the disease. Multiplicity of
primary malignancies itself does not necessarily indicate a poor
prognosis, as long as adequate diagnosis and management are
performed (2). This study suggests
that family history may be a key risk factor for MPM and HNPCC,
whose detailed molecular mechanism remains to be elucidated. This
study, with an investigation of family history, may improve the
clinical recognition of HNPCC and MPM, which is important for
successful surgical treatment.
Acknowledgements
We thank Yong Hua Xu, Department of
Radiology, the Central Hospital of Shanghai Xuhui District,
Shanghai, China, for expert technical assistance. This research
project was supported by grants from the Foundation of Shanghai
Health Bureau, Shanghai, China (no. 2010L059A), and the National
Natural Science Foundation of China (no. 81272401)
References
|
1.
|
DR YouldenPD BaadeThe relative risk of
second primary cancers in Queensland, Australia: a retrospective
cohort studyBMC Cancer1183201110.1186/1471-2407-11-8321342533
|
|
2.
|
NC HuSC HsiehTJ ChenJY ChangMultiple
primary malignancies including colon, stomach, lung, breast, and
liver cancer: a case report and literature reviewChin Med J
(Eng)12230913093200920137508
|
|
3.
|
O LandgrenA ThomasS MailankodyMyeloma and
second primary cancersN Engl J
Med36522412242201110.1056/NEJMc111101022150057
|
|
4.
|
Z NémethJ CzignerL IvánM UjpálJ BarabásG
SzabóQuadruple cancer, including triple cancers in the head and
neck regionNeoplasma494124142002
|
|
5.
|
A RendaN CarlomagnoNosographyMultiple
Primary MalignanciesAndrea
RendaSpringer-VerlagItaly200910.1007/978-88-470-1095-6_1
|
|
6.
|
S WarrenO GatesMultiple primary malignant
tumors: a survey of the literature and statistical studyAm J
Cancer16135814141932
|
|
7.
|
M YamasakiM HiguchiAn autopsy case of
synchronous quadruple
cancerStrahlentherapie14027527919705509810
|
|
8.
|
SL AnguranaR KapoorP KumarD KhoslaN
KumarSC SharmaFD PatelQuadruple malignancy in a single patient: a
case report and comprehensive review of literatureJ Cancer Res
Ther6230232201010.4103/0973-1482.6523720622376
|
|
9.
|
S YamamotoK YoshimuraS RiS FujitaT AkasuY
MoriyaThe risk of multiple primary malignancies with colorectal
carcinomaDis Colon Rectum4910
SupplS30S36200610.1007/s10350-006-0600-817106813
|
|
10.
|
A EngelandT BjørgeT HaldorsenS TretliUse
of multiple primary cancers to indicate associations between
smoking and cancer incidence: an analysis of 500,000 cancer cases
diagnosed in Norway during 1953–1993Int J
Cancer7040140719979033646
|
|
11.
|
S MussariM AmichettiL TomioQuadruple
cancer in a single patient: a report of four casesEur J Surg
Oncol26614616200010.1053/ejso.2000.095811034817
|
|
12.
|
DP SlaughterHW SouthwickW SmejkalField
cancerization in oral stratified squamous epithelium; clinical
implications of multicentric
originsCancer6963968195310.1002/1097-0142(195309)6:5%3C963::AID-CNCR2820060515%3E3.0.CO;2-Q13094644
|
|
13.
|
M TanyiJ OlaszE KámoryO CsukaJL TanyiZ
RessL DamjanovichDifficulties in recognizing families with
Hereditary Non-polyposis Colorectal Carcinoma. Presentation of 4
families with proven mutationEur J Surg
Oncol3413221327200810.1016/j.ejso.2008.01.00618289827
|
|
14.
|
HT LynchT SmyrkHereditary nonpolyposis
colorectal cancer (Lynch syndrome). An updated
reviewCancer7811491167199610.1002/(SICI)1097-0142(19960915)78:6%3C1149::AID-CNCR1%3E3.0.CO;2-58826936
|
|
15.
|
A WagnerC TopsJT WijnenK ZwindermanC van
der MeerM KetsMF NiermeijerGenetic testing in hereditary
non-polyposis colorectal cancer families with a MSH2, MLH1, or MSH6
mutationJ Med Genet39833837200210.1136/jmg.39.11.83312414824
|
|
16.
|
AS WarthinHeredity with reference to
carcinoma as shown by the study of the cases examined in the
pathological laboratory of the University of Michigan 1895–1913Arch
Intern Med1254655519133931868
|
|
17.
|
HT LynchGJ VoorheesSJ LanspaPS McGreevyJF
LynchPancreatic carcinoma and hereditary nonpolyposis colorectal
cancer: a family studyBr J
Cancer52271273198510.1038/bjc.1985.1874027169
|