Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of malignant tumors and the third leading cause of
cancer mortality worldwide (1).
Currently, surgery is the preferred treatment method for liver
cancer, but the five-year survival rate remains extremely low.
Autopsy studies confirm that nearly one-third of all HCC patients
have lymph node metastasis (LNM), which is the leading cause for
distant metastasis and mortality; however, the molecular mechanisms
of LNM from liver cancer remain unclear (2).
Hca-F and Hca-P cells are gynogenetic HCC cell lines
generated from mice. They are well-characterized with different
metastasis potentials exclusive to lymph nodes when inoculated
subcutaneously in 615 mice. Hca-F cells have a high metastatic
potential (LNM rate >75%), while Hca-P cells have low metastatic
potential (LNM rate <25%) (3,4).
Therefore, Hca-F and Hca-P cell lines were more appropriate for
investigating the mechanisms of LNM in comparison to clinical
samples, since high metastatic subclone cells exist in primary
cancers as a minority (5).
The critical role of the cancerous microenvironment
(cellular and non-cellular) is increasingly recognized as an
important factor markedly influencing cancer development and
metastasis (6,7). The tumor microenvironment plays a
decisive role in regulating the process of hepatocarcinogenesis,
epithelial-mesenchymal transition (EMT), tumor invasion and
metastasis (8). Additionally,
global gene expression profiling of HCC has revealed that the tumor
microenvironment is also an important factor in the biological and
prognostic classification of HCC. The tumor microenvironment can be
classified into cellular and non-cellular components. The major
cellular components include fibroblasts, hepatic stellate cells,
immune cells and endothelial cells. These cells produce the
non-cellular components including the extracellular matrix (ECM)
proteins, inflammatory cytokines, proteolytic enzymes and growth
factors, which modulate the biological behavior of HCC by their
effects on cancer signaling pathways in tumor cells and markedly
impact tumor invasion and metastasis (8). Certain investigations of the tumor
micro-environment have made significant advancements, but have
mainly focused on the effect(s) of individual cellular components
on tumor cells. However, the interactions between the tumor cells
and the surrounding components have not been comprehensively and
systematically demonstrated. Thus, the actual progress and
mechanism of metastasis is difficult to identify.
In current studies, certain candidate genes for LNM
were revealed by identifying genes with different expression levels
in the Hca-F and Hca-P cell lines (3–5,9).
However, LNM is a dynamic process and one limitation of such
studies (or the platform for such studies) is that the active
changes related to LNM were not revealed. It would be necessary in
the process of HCC cells encountering the lymphatic environment and
growing with lymph node components. Therefore, we used an in
vitro model where HCC cells with varying metastasis potential
were grown with lymph node components in order to gain an insight
into the possible and favorable LN niche condition(s) for
metastasis.
Materials and methods
Cell culture and animals
Mouse HCC cell lines, Hca-F and Hca-P (established
by the Department of Pathology, Dalian Medical University, Dalian,
China), were grown in the abdominal cavity of 8-10-week-old inbred
615 mice (males provided by the Animal Facility of Dalian Medical
University) for ∼seven days (5).
Cells were harvested and cultured in Rosewell Park Memorial
Institute-1640 medium (RPMI-1640; Gibco BRL, Gaithersburg, MD, USA)
supplemented with antibiotics (100 U/ml penicillin and 100 g/ml
streptomycin; Gibco BRL) and 10% heat-inactivated fetal bovine
serum (FBS; Gibco BRL), and incubated in a humidified incubator at
37°C with 5% CO2 for one day. The study was approved by
the ethics committee of Dalian Medical University, Dalian,
China.
Lymph node homogenates (LNHs)
The lymph nodes from inbred 615 mice were rinsed
thoroughly in PBS (pH 7.4) and homogenized using a homogenizer in
serum-free RPMI-1640. The homogenate was centrifuged at 2000 x g
for 10 min at room temperature, and the supernatant was quantified
using Bradford’s protein assay and immediately used for subsequent
experiments.
Two-dimensional gel electrophoresis
(2-DE) sample preparation, running and image analysis
Hca-F cells (2×106) were collected,
washed and cultured in 3 ml serum-free RPMI-1640 (control) and 3 ml
serum-free RPMI-1640 with 10 mg/ml LNH, at 37°C with 5%
CO2 for 24 h. Cells were then completely washed through
serum-free RPMI-1640. Cellular protein was extracted using strong
radio immunoprecipitation assay (RIPA) lysis buffer containing 50
mM TrisCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate and 0.1% sodium dodecyl sulfate (SDS) (Beyotime
Institute of Biotechnology, Haimen, Jiangsu, China) and purified
with ReadyPrep™ 2-D Cleanup Kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The purified proteins were solubilized in 2-D
rehydration buffer containing 7 M urea, 2 M thiourea, 4%
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
(CHAPS), 20 mM dithiothreitol (DTT) and 2% immobilized pH gradient
(IPG) buffer. The protein samples were then aliquoted and stored at
−80°C until use.
IPG gel strips (GE Healthcare, Piscataway, NJ, USA),
7 cm in size with a pH range of 3–10 (linear), were rehydrated in
125 μl rehydration buffer containing 600 μg of
proteins at 50 V for 12 h at 20°C. Isoelectric focusing (IEF) was
conducted using a Protean IEF Cell (Bio-Rad Laboratories, Inc.).
Proteins were focused at 200 V for 1 h, 500 V for 1 h and 800 V for
1 h. A gradient of 800–8,000 V was then applied for 30 min, and
focusing was continued at 8,000 V for 2.5 h.
Following IEF, IPG gel strips were equilibrated in
an equilibration buffer, containing 6 M urea, 50 mM Tris-HCl (pH
8.8), 2% SDS, 30% glycerol and a trace of bromophenol blue,
including 1% DTT, for 10 min whilst being agitated. Before being
transferred onto a 12% polyacrylamide gel, gels were transferred
into an equilibration solution containing 4.5% iodoacetamide and
equilibrated for 10 min. Separation of the second dimension was
carried out at a current of 5 mA/gel for 1 h and 10 mA/gel
thereafter. For each sample, the 2-DE was repeated three times.
Following SDS-polyacrylamide gel electrophoresis
(PAGE), gels were fixed in 30% ethanol and 10% acetic acid for 15
min at room temperature, rinsed three times in ultrapure water and
silver stained using Pierce Silver Stain for Mass Spectrometry kit
(Thermo Fisher Scientific, Inc., Fremont, CA, USA) according to the
manufacturer’s protocol. Images were captured using ChemiDoc XRS
image documentation system and analyzed using PDQust 8.0 (Bio-Rad
Laboratories, Inc.). Protein spots of interest, with an average
ratio of >2.0 or <−2.0 (P<0.05), were selected for protein
identification by matrix-assisted laser desorption/ionization
time-of-flight mass spectrometry (MALDI-TOF MS).
In-gel protein digestion and MALDI-TOF
MS
Protein spots of interest were manually excised from
gels and washed twice in ultra pure water in 96-cell boards for 10
min. The gel pieces were dehydrated completely with acetonitrile
(ACN) and dried in a vacuum centrifuge for 10 min. Subsequently,
the gel pieces were incubated in 10 mM DTT at 56°C for 1 h and in
55 mM IAM in a dark room for 45 min, then washed with 25 mM
NH4HCO3 for 2×10 min, 25 mM
NH4HCO3/50% ACN for 2×10 min and 100% ACN for
10 min. Gels were then dehydrated completely with ACN and dried in
a vacuum centrifuge for 10 min. Proteins in gel pieces were
rehydrated in 10 ng/ml trypsin (Roche Diagnostics GnbH, Mannheim,
Germany) in 25 mM NH4HCO3 and incubated
overnight at 37°C. Digestion was terminated using 0.1% TFA. Once
mixed and shocked, the mixtures were centrifuged. Supernatants (3
μl) were spotted onto the sample plates twice and dried in
air, followed by a spotting matrix (70% ACN/0.1% TFA with 4 mg/ml
a-cyano-4-hydroxycinnamic acid). Desalination was conducted twice
by spotting 1 μl 0.1% TFA on the sample plate and
aspirating. Peptide masses were determined using an Ultraflex
TOF/TOF MS (Bruker Daltonics, Inc., Billerica, MA, USA). The N2
laser (337 nm) was run at 100 μJ and 1 nsec pulse width.
Full scan MS were collected from 700-3500 m/z.
Database searching
The results of MALDI-TOF MS were analyzed by the
‘Calibrate Peptide Stanard-zk. FAMS Method’ using flexanalysis
software (Bruker Daltonics, Inc.). Peptide mass lists were searched
against online databases in the National Center for Biotechnology
Information (NCBI) using the MASCOT database search engine 2.1
(Matrix Science, London, UK). A MASCOT score >82 was considered
to indicate a statistically significant difference (P<0.05).
Western blot analysis
Cellular proteins were separately extracted from
2×107 Hca-F and Hca-P cells using RIPA lysis buffer
(Beyotime Institute of Biotechnology). The extracted proteins (40
μg of total protein) were subjected to 12% SDS-PAGE and
blotted onto nitrocellulose membranes (Invitrogen Life
Technologies, Carlsbad, CA, USA). Following incubation in 1% bovine
serum albumin (BSA) for 1 h, the blotted membranes were incubated
with primary antibodies (rabbit anti-GRP78 polyclonal antibody,
anti-GAL3 polyclonal antibody and mouse anti-beta actin monoclonal
antibody) overnight at 4°C. All primary antibodies were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Subsequently, membranes were washed, incubated with secondary
antibodies for 1 h and determined using Pierce ECL western blotting
kit (Thermo Fisher Scientific). The bands were analyzed using
Gel-Pro Analyzer 4.0 (Media Cybernetics, Bethesda, MD, USA).
Statistical analysis
Data are presented as mean ± SD and analyzed by the
Student’s t-test using SPSS version 19.0 software (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
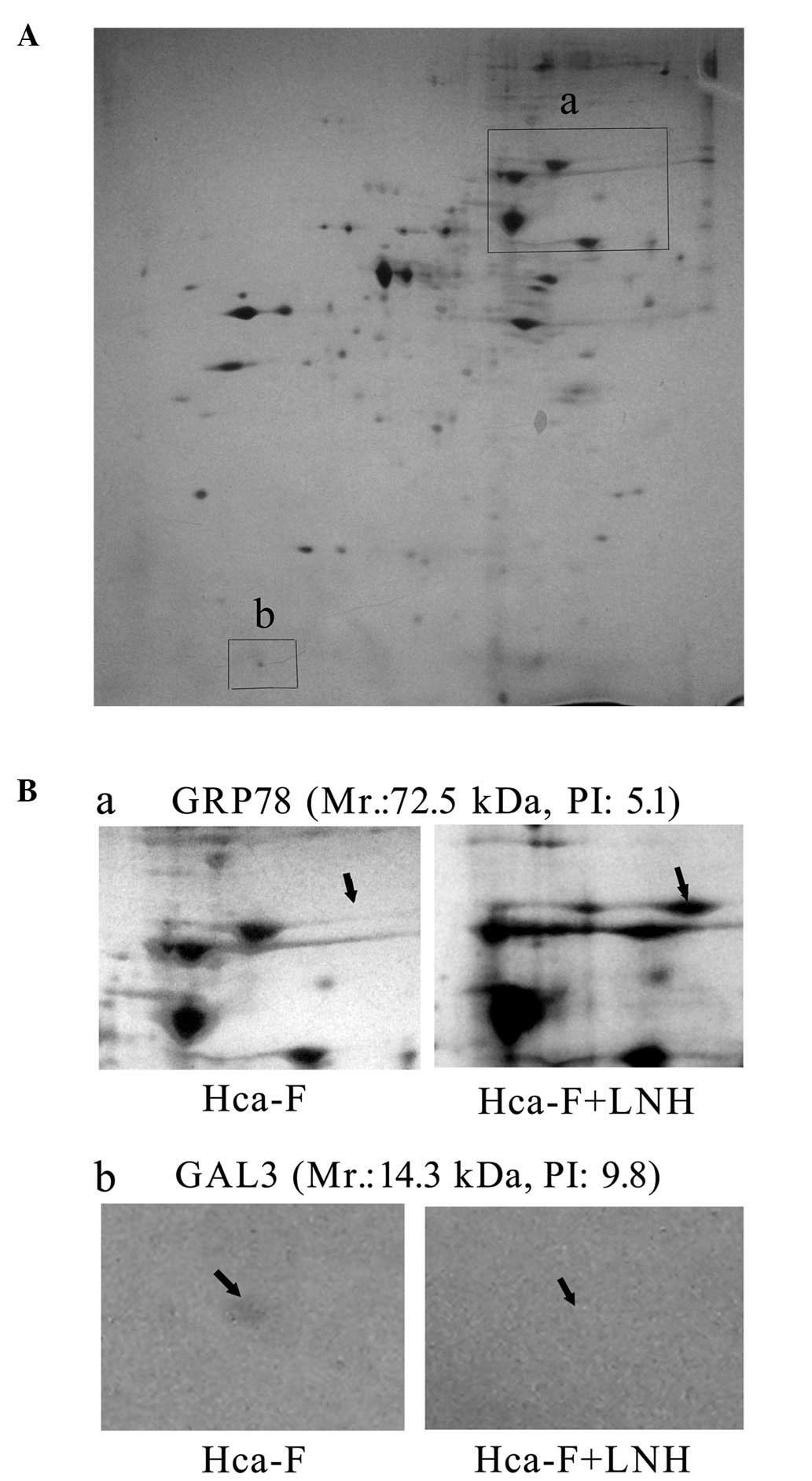
LNHs change the proteomic profiling in
Hca-F cells
The extracts of Hca-F cells incubated with and
without LNH were examined using 2-DE. Expression levels of 49
proteins were upregulated and 74 proteins were downregulated in
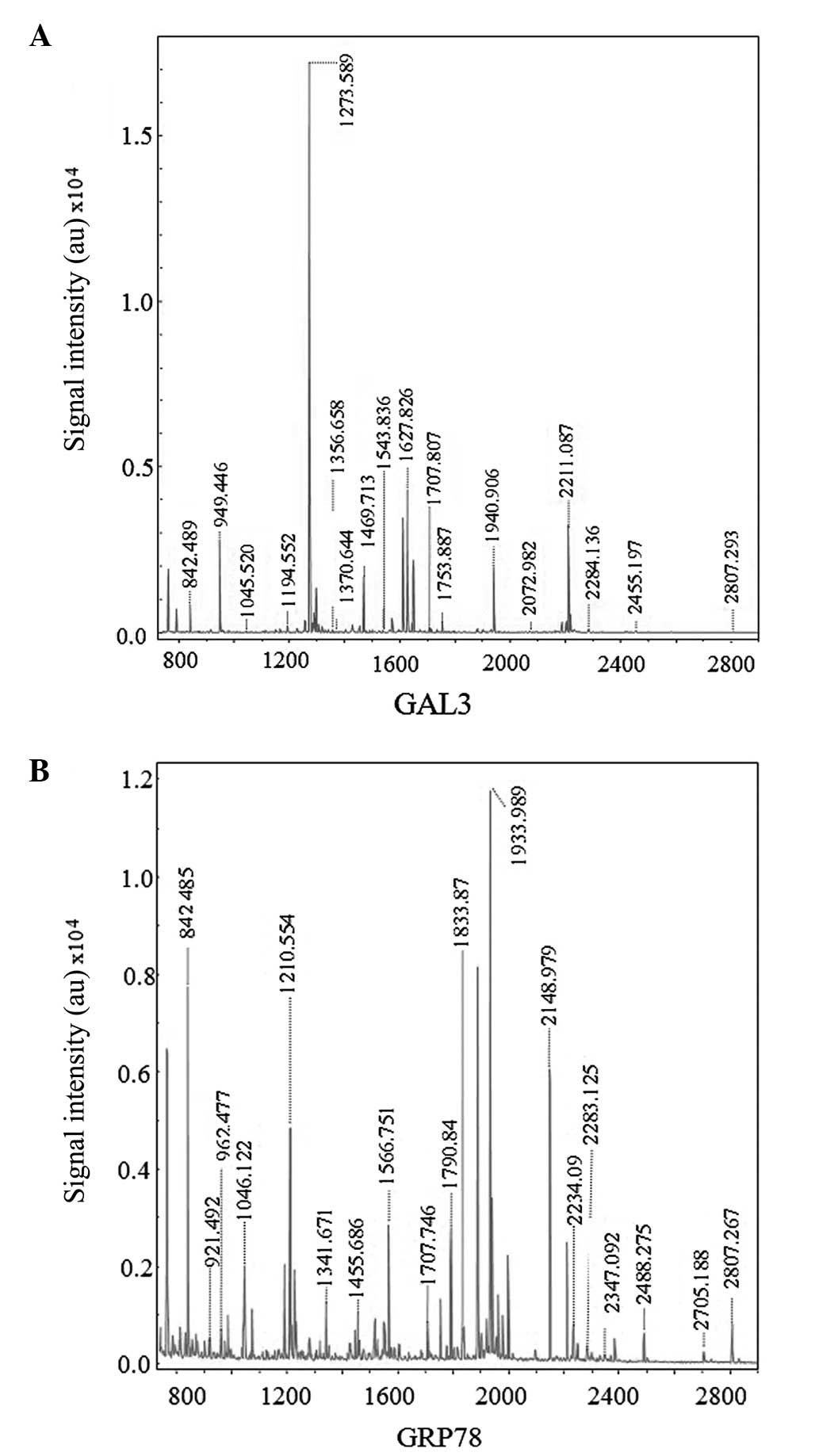
Hca-F cells following cell incubation in LNH for 24 h (Fig. 1). Once four upregulated and four
downregulated dots were detected by MALDI TOF/TOF MS (Fig. 2A and B), seven proteins were
identified (Table I). Among those
proteins was GRP78, an essential protein for pluripotent cell
survival and embryonic cell growth. It serves as a central
regulator of endoplasmic reticulum (ER) homeostasis due to its
multiple functional roles in protein folding and ER calcium
binding, and its control of transmembrane ER stress sensor
activation (10,11). GRP78 upregulation has been suggested
to correlate to metastases (12,13).
GAL3, one of the β-galactoside-binding proteins, has been
associated with cell proliferation, recognition, adhesion,
differentiation, immunomodulation, apoptosis and angiogenesis.
Subsequently, reduced GAL3 expression has been associated with
lymph node metastasis in gastric cancer (14).
 | Table I.Identification of differentially
expressed proteins in Hca-F cells upon LNH treatment. |
Table I.
Identification of differentially
expressed proteins in Hca-F cells upon LNH treatment.
| No. | Protein
description | NCBI GI | Protein score | Matched
peptides | Cover age (%) | Fold changea |
|---|
| 1 | TPI | 1864018 | 165 | 15 | 66 | 19.83 |
| 2 | mCG15924 | 148694806 | 67 | 21 | 19 | 3.29 |
| 3 | HP1BP3 | 18043549 | 50 | 12 | 23 | 3.27 |
| 4 | GRP78 | 254540166 | 143 | 24 | 42 | 2.16 |
| 5 | DLD | 74223108 | 82 | 11 | 30 | −21.27 |
| 6 | CCDC157 isoform 2
precursor | 114145528 | 86 | 14 | 26 | −3.11 |
| 7 | GAL3 | 148688796 | 108 | 14 | 87 | −2.09 |
LNH differentially influences GRP78 and
GAL3 expression in Hca-F and Hca-P cells on western blotting
The expression levels of GRP78 and GAL3 varied in
Hca-F and Hca-P cells after culturing with LNH for 24 h (Fig. 3A and B). GRP78 was increased
2.4-fold (P<0.05) in Hca-F cells but decreased almost to half
(P<0.05) in Hca-P cells (Fig.
3B). GAL3 decreased by half in Hca-F cells and only slightly
increased in Hca-P cells (Fig. 3B).
These lines of evidence suggest that following incubation with LNH
the high metastatic potential cell line was induced to regulate
protein expression in favor of metastases, but the low metastatic
potential cell line was induced in an opposite trend against
metastasis. This molecular evidence may also explain their known
opposite phenotytpic metastatic characteristics in animal
models.
Discussion
Organ microenvironments influence the biological
behaviors of tumor cells, including cancer cell survival,
proliferation, angiogenesis, invasion and metastases (15,16).
These studies provide important supplementary evidence for the
hypothesis of metastatic niche, the modern version of ‘seed and
soil’ hypothesis (17), which
suggests that the intrinsic properties of the metastatic cells and
the host microenvironment are important determinants in metastatic
spread (7). Our results reveal that
the ‘non-cellular’ components in LNs may regulate the cells with
high metastatic potential to engage in appropriate changes for
metastases. This phenomenon suggests that HCC cells with high
metastatic potential demonstrate dynamic and variable superiorities
compared to their low metastastic potential counterpart when they
meet plausible metastatic microenvironments.
In our new model, we have used whole non-cellular
components of LNs to simulate a relatively non-cellular metastatic
LN microenvironment. These components, with high LNM potential,
regulate the HCC cell line to express appropriate proteins
necessary for LNM. Profiling/fractionation of LNH may serve to
standardize our experimental conditions. We propose that within the
experimental limitations of the current study, GRP78 and GAL3 may
be potential biomarkers for LNM diagnosis in HCC.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition;
|
|
FBS
|
fetal bovine serum;
|
|
GRP78
|
78-kDa glucose-regulated protein;
|
|
GAL3
|
galectin-3;
|
|
HCC
|
hepatocellular carcinoma;
|
|
LN
|
lymph node;
|
|
LNH
|
lymph node homogenate;
|
|
RPMI-1640
|
Rosewell Park Memorial Institute-1640
medium
|
References
|
1.
|
DM ParkinGlobal cancer statistics in the
year 2000Lancet Oncol2533543200111905707
|
|
2.
|
VO MelnikovaM Bar-EliInflammation and
melanoma metastasisPigm Cell Melanoma
Res22257267200910.1111/j.1755-148X.2009.00570.x19368690
|
|
3.
|
H ChuH ZhouY LiuY HuJ ZhangFunctional
expression of CXC chemokine recepter-4 mediates the secretion of
matrix metalloproteinases from mouse hepatocarcinoma cell lines
with different lymphatic metastasis abilityInt J Biochem Cell
Biol39197205200710.1016/j.biocel.2006.07.008
|
|
4.
|
H ZhouL JiaS WangH WangH ChuY HuJ CaoJ
ZhangDivergent expression and roles for caveolin-1 in mouse
hepatocarcinoma cell lines with varying invasive abilityBiochem
Biophys Res
Commun345486494200610.1016/j.bbrc.2006.03.24616684506
|
|
5.
|
B SongJW TangB WangXN CuiL HouL SunLM
MaoIdentify lymphatic metastasis-associated genes in mouse
hepatocarcinoma cell lines using gene chipWorld J
Gastroenterol1114631472200510.3748/wjg.v11.i10.146315770722
|
|
6.
|
RR LangleyIJ FidlerThe seed and soil
hypothesis revisited - the role of tumor-stroma interactions in
metastasis to different organsInt J
Cancer12825272535201110.1002/ijc.2603121365651
|
|
7.
|
RG BagleyThe Tumor
MicroenvironmentSpringer Science + Business MediaNew
York201010.1007/978-1-4419-6615-5
|
|
8.
|
L JiaS WangH ZhouJ CaoY HuJ
ZhangCaveolin-1 upregulates CD147 glycosylation and the invasive
capability of murine hepatocarcinoma cell linesInt J Biochem Cell
Biol3815841593200610.1016/j.biocel.2006.03.01916702020
|
|
9.
|
JD YangI NakamuraLR RobertsThe tumor
microenvironment in hepatocellular carcinoma: current status and
therapeutic targetsSemin Cancer
Biol213543201110.1016/j.semcancer.2010.10.00720946957
|
|
10.
|
C JollyRI MorimotoRole of the heat shock
response and molecular chaperones in oncogenesis and cell deathJ
Natl Cancer Inst9215641572200010.1093/jnci/92.19.156411018092
|
|
11.
|
S LuoC MaoB LeeAS LeeGRP78/BiP is required
for cell proliferation and protecting the inner cell mass from
apoptosis during early mouse embryonic developmentMol Cell
Biol2656885697200610.1128/MCB.00779-0616847323
|
|
12.
|
JM LukCT LamAFM SiuBY LamIO NgM HuCM CheST
FanProteomic profiling of hepatocellular carcinoma in Chinese
cohort reveals heat-shock proteins (Hsp27, Hsp70, GRP78)
up-regulation and their associated prognostic
valuesProteomics610491057200610.1002/pmic.20050030616400691
|
|
13.
|
D DongC StapletonB LuoA critical role for
GRP78/BiP in the tumor microenvironment for neovascularization
during tumor growth and metastasisCancer
Res7128482857201110.1158/0008-5472.CAN-10-315121467168
|
|
14.
|
K OkadaT ShimuraT SuehiroE MochikiH
KuwanoReduced galectin-3 expression is an indicator of unfavorable
prognosis in gastric cancerAnticancer Res2613691376200616619546
|
|
15.
|
IJ FidlerSJ KimRR LangleyThe role of the
organ microenvironment in the biology and therapy of cancer
metastasisJ Cell Biochem101927936200710.1002/jcb.2114817177290
|
|
16.
|
IJ FidlerThe organ microenvironment and
cancer
metastasisDifferentiation70498505200210.1046/j.1432-0436.2002.700904.x12492492
|
|
17.
|
S PagetThe distribution of secondary
growths in cancer of the breast 1889Cancer Metastasis
Rev89810119892673568
|