Introduction
Head and neck cancer is the sixth most common type
of cancer in the world, accounting for more than 540,000 new cases
and 271,000 mortalities each year (1). These cancers occur in the lips, oral
cavity, nasal cavity, paranasal sinuses, pharynx and larynx, 90% of
which are squamous cell carcinomas. They significantly affect
long-term survival and the quality of life of patients. The
development of novel strategies is required for prevention and
early detection, to reduce cancer incidence and overcome problems
associated with treatment of late-stage tumors. Improved prediction
of outcome will lead to treatment decisions that prolong patients’
survival and quality of life.
Predictions concerning the outcome of head and neck
cancers are currently based mostly on clinicopathological features,
including tumor stage, differentiation, size and regional lymph
node or distant metastasis. However, increasing numbers of studies
utilize aberrant gene expression and genomic and epigenetic
alterations to predict prognosis.
It has been established that tobacco smoke and
alcohol consumption are the most significant risk factors for head
and neck cancers. They contribute to these cancers through multiple
genetic alterations, including the silencing of tumor suppressor
genes and oncogene activation (2).
A large body of knowledge has accumulated regarding gene
alterations that are associated with the development of this deadly
disease. However, greater understanding of the links between gene
alteration and head and neck cancer development and progression is
required.
DNA microarray profiling is an innovative technology
that facilitates analysis of a great number of genes
simultaneously. In this study, we performed an analysis of nearly
the entire human genome in order to detect altered gene expression
between primary laryngeal squamous cell carcinoma (LSCC) and
adjacent normal tissues. We identified 349 genes that are
differentially expressed between normal and malignant tissues, a
number of which have not previously been associated with LSCC.
Thus, we selected specific tumor-related genes from microarray
data, which have not been reported in LSCC before. We then verified
the differential expression of the seven genes in another set of
LSCC tissues. In the future, these genes may be further evaluated
as biomarkers or potential therapeutic targets for LSCC.
Materials and methods
Tissue samples
Between October 2007 and March 2010, tissue biopsy
specimens (tumor and matched adjacent normal tissues) were
collected from 36 patients (Table
I) with LSCC at the Department of Otorhinolaryngology Head and
Neck Surgery, Drum Tower Hospital Affiliated to Nanjing University
School of Medicine (Nanjing, China). Pathological analyses
confirmed the diagnosis of each patient. The tissues were
immediately frozen in liquid nitrogen and stored at −80°C until
use. Our institutional Human Ethics Committee approved the study.
Informed consent was obtained either from the patient or the
patient’s family.
 | Table I.Clinical characteristics of LSCC
cases studied.a |
Table I.
Clinical characteristics of LSCC
cases studied.a
| Case no. | Age (years) | Tobacco use | Alcohol use | Tumor
classification | Tumor
differentiation | TNM |
|---|
| LSCC-001 | 71 | Yes | No | Supra-GC | M | T3N0M0 |
| LSCC-002 | 62 | Yes | No | SGC | M | T3N0M0 |
| LSCC-003 | 61 | Yes | Yes | SGC | W | T3N0M0 |
| LSCC-004 | 63 | Yes | No | GC | W | T3N0M0 |
| LSCC-005 | 52 | Yes | Yes | Supra-GC | M | T4N0M0 |
| LSCC-006 | 75 | Yes | No | Supra-GC | P | T3N1M0 |
| LSCC-007 | 74 | Yes | No | GC | W | T4N0M0 |
| LSCC-008 | 59 | Yes | No | GC | W | T4N0M0 |
| LSCC-009 | 59 | Yes | Yes | GC | P | T3N0M0 |
| LSCC-010 | 68 | No | No | GC | M | T3N0M0 |
| LSCC-011 | 57 | Yes | Yes | Supra-GC | W | T2N0M0 |
| LSCC-012 | 84 | No | No | Supra-GC | P | T4N2M0 |
| LSCC-013 | 66 | Yes | No | Supra-GC | M | T3N0M0 |
| LSCC-014 | 74 | No | No | Supra-GC | M | T3N2M0 |
| LSCC-015 | 49 | Yes | No | Supra-GC | P | T3N1M0 |
| LSCC-016 | 54 | Yes | Yes | GC | W | T3N0M0 |
| LSCC-017 | 73 | No | No | Sub-GC | M | T3N0M0 |
| LSCC-018 | 53 | Yes | No | GC | W | T4N0M0 |
| LSCC-019 | 54 | Yes | No | Supra-GC | P | T4N1M0 |
| LSCC-020 | 70 | Yes | No | Supra-GC | P | T4N2M0 |
| LSCC-021 | 58 | Yes | Yes | Supra-GC | M | T4N2M0 |
| LSCC-022 | 63 | Yes | Yes | Supra-GC | M | T2N0M0 |
| LSCC-023 | 54 | Yes | Yes | Supra-GC | M | T1N0M0 |
| LSCC-024 | 60 | Yes | Yes | Supra-GC | P | T3N2M0 |
| LSCC-025 | 53 | Yes | No | Sub-GC | M | T3N0M0 |
| LSCC-026 | 59 | Yes | Yes | GC | W | T4N0M0 |
| LSCC-027 | 60 | Yes | Yes | GC | W | T3N0M0 |
| LSCC-028 | 75 | Yes | Yes | Supra-GC | M | T3N1M0 |
| LSCC-029 | 57 | Yes | Yes | GC | W | T1N0M0 |
| LSCC-030 | 63 | Yes | No | GC | W | T1N0M0 |
| LSCC-031 | 66 | Yes | Yes | Supra-GC | W | T2N0M0 |
| LSCC-032 | 52 | Yes | Yes | Supra-GC | M | T3N0M0 |
| LSCC-033 | 40 | Yes | No | GC | M | T3N0M0 |
| LSCC-034 | 75 | No | No | GC | P | T1N0M0 |
| LSCC-035 | 73 | Yes | Yes | GC | M | T1N0M0 |
| LSCC-036 | 70 | Yes | Yes | GC | M | T2N0M0 |
RNA isolation and microarray
analysis
To isolate RNA from the tissue specimens, both tumor
and normal mucosae were put into liquid nitrogen and ground into
powder in TRIzol reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA) using a rotor-stator homogenizer. Total RNA was then
isolated following the manufacturer’s instructions. The integrity
of the RNA was verified by visual inspection after 1% agarose gel
electrophoresis; the 28S ribosomal RNA band intensity was two times
that of the 18S ribosomal RNA band (3). Sample purity was ensured by an
OD260/OD280 ratio >1.8, measured with a
spectrophotometer.
For DNA microarray analysis, biotinylated probes
were prepared using 2 μg of total RNA. Briefly, the total
RNA obtained from tumor and normal tissues was mixed with 100 pmol
of T7-oligo(dT)24 primer and denatured at 70°C for 10
min, then chilled on ice. The first-strand cDNA synthesis was
performed using Superscript II reverse transcriptase (Life
Technologies, Carlsbad, CA, USA) and the second-strand with DNA
Polymerase I, E. coli DNA ligase and RNase H. The
biotinylated probes were then prepared from the entire cDNA
reaction using an ENZO Bioarray High Yield RNA Transcript Labeling
kit (ENZO Diagnostics, Toronto, Canada).
The purified probes were incubated with 1X
fragmentation buffer at 95°C for 35 min to reduce the average probe
length. Hybridization was performed at 45°C for 20 h with
biotinylated probes on the microarrays. The non-specific binding of
these probes was removed by low stringency washes (10 times) and
high stringency washes (4 times) using a GeneChip Fluidics Station
400 wash station (Agilent, San Diego, CA, USA). The positive signal
was detected by incubating the microar-rays with streptavidin
phycoerythrin (Molecular Probes, Camarillo, CA, USA) and scanned
with a GeneArray Scanner (Hewlett-Packard, San Diego, CA, USA). The
scanned data were analyzed with GeneChip Analysis Suite 3.3
(Agilent).
Semi-quantitative reverse transcription
polymerase chain reaction (RT-PCR)
To confirm the differential gene expression of
laryngeal cancer revealed during cDNA microarray analysis, we used
a 2-step method of semi-quantitative RT-PCR starting with tissues
from 32 cases of laryngeal cancer and matched normal adjacent
tissues. Briefly, total RNA was first reverse transcribed into cDNA
using Superscript II reverse transcriptase (Life Technologies) and
then amplified in a programmable Applied Biosystems 2720 thermal
cycler (Singapore). For each reaction, a 50-μl PCR mixture
containing 200 μM dNTPs, 1.25 units Taq polymerase in
10X Taq polymerase buffer (Takara Bio, Inc., Shiga, Japan),
and corresponding concentrations of primers (Table II) was set to an initial denaturing
at 95°C for 5 min and then appropriate PCR cycles for different
genes of 94°C for 1 min, annealing temperature (Table II) for 1 min, 72°C for 30 sec and a
final extension at 72°C for 10 min in a programmable 2720. The PCR
reactions were performed in triplicate.
| Table II.Primer sequences and PCR
conditions. |
Table II.
Primer sequences and PCR
conditions.
| Gene | Primer
sequences | Annealing
temperature (°C) | No. PCR cycles | Size (bp) |
|---|
| SENP1 |
5′-ACCCACCTCCTGCCACAAAC-3′ | 60 | 36 | 424 |
|
5′-TTCGACGACATGAACCACTCCA-3′ | | | |
| CD109 |
5′-AAGCCTTTGATTTAGATGTTGC-3′ | 60 | 36 | 445 |
|
5′-GAGTGATGATGGGAGCCTGA-3′ | | | |
| CKS2 |
5′-CAAGCAGATCTACTACTCGG-3′ | 56 | 36 | 222 |
|
5′-TGGAAGAGGTCGTCTAAAGA-3′ | | | |
| LAMA3 |
5′-TTCATGGGATACAGAGAGGT-3′ | 58 | 36 | 446 |
|
5′-TTGGAGAAACAAGGACAGAG-3′ | | | |
| LAMA2 |
5′-AATTTACCTCCGCTCGCTAT-3′ | 60 | 36 | 424 |
|
5′-CCTCCAATGTACTTTCCACG-3′ | | | |
| ITGAV |
5′-CTGGGATTGTGGAAGGAGGG-3′ | 60 | 36 | 462 |
|
5′-TGCTGTAAACATTGGGGTCG-3′ | | | |
| ITGB8 |
5′-TGGGCCAAGGTGAAGACAAT-3′ | 60 | 36 | 456 |
|
5′-ATGAGCCAAATCCAAGACGA-3′ | | | |
|
β-actin |
5′-TCGACAACGGCTCCGGCAT-3′ | 56 | 28 | 241 |
The PCR-amplified gene products were visualized in a
2% (w/v) agarose gel stained with ethidium bromide. Images of
resulting gels were captured with LabWorks45 (UVP, Upland, CA,
USA). The genes detected by PCR were SENP1, CD109,
CKS2, LAMA2, LAMA3, ITGAV, ITGB8
and β-actin (Table II).
β-actin was used as the loading control and normalizing
reference for each gene in these tissue samples. The primers were
designed according to their GenBank sequences using the Primer 3
online tool.
Protein extraction and western blot
analysis
Both LSCC and the matched adjacent normal tissues
were homogenized for total cellular protein extraction using a
commercial protein kit from Pierce Biotechnology (Rockford, IL,
USA). The protein concentration of the homogenates was determined
by a bicinchoninic acid protein assay kit (Shenergy Biocolor,
Shanghai, China).
Equal amounts of the protein samples (50 μg)
were separated via 10–15% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE), followed by electrophoretic transfer
onto polyvinylidene difluoride membranes (Roche Diagnostics,
Indianapolis, IN, USA). These membranes were incubated with 5%
non-fat milk in phosphate-buffered saline (PBS) for 2 h and then
with the primary antibody at 4°C overnight. The primary antibodies
CKS2 (#ab54658) and SENP1 (#ab3656) were purchased from Abcam
(Cambridge, MA, USA). CD109 (#SC33115) was obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). The next day, the
membranes were washed with PBS 3 times and then incubated with an
anti-goat or anti-rabbit horseradish peroxidase-conjugated
secondary antibody (Santa Cruz Biotechnology, Inc.). The
immunoreactive signals were visualized using an enhanced
chemiluminescence detection kit (Pierce Biotechnology) and
quantified with a densitometer (Kodak Digital Science 1D Analysis
Software, Rochester, NY, USA).
Statistical analysis
DNA microarray data were analyzed using the Agilent
GeneChip Analysis Suite 3.3 and summarized as fold changes. The
data from semi-quantitative RT-PCR and western blot analysis were
summarized as percentages of controls. The differential expression
levels of genes between the tumor and normal tissues were
statistically analyzed with paired-sample t-tests using SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA). The association of gene
expression levels with clinicopathological data was statistically
analyzed with an independent-samples t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Detection of differentially expressed
genes between the primary LSCC and corresponding normal
tissues
In this study, we first randomly selected 4 pairs of
primary laryngeal cancer and corresponding normal tissues for DNA
microarray analysis. We then isolated RNA from the frozen tissues
and performed DNA microarray analysis in Agilent chips. We
identified that 10,909 genes were differentially expressed between
laryngeal cancer and the matched normal tissues in case 1; 10,223
genes in case 2; 5,730 genes in case 3; and 14,665 genes in case 4.
Among these differentially expressed genes, there were 349 that
were identified in all four cases, of which 112 were significantly
upregulated with intensity ratios up 2.0, while 237 were
downregulated with ratios down 0.5 (Table III).
| Table III.Differentially expressed genes
between primary laryngeal cancer and corresponding normal
tissues. |
Table III.
Differentially expressed genes
between primary laryngeal cancer and corresponding normal
tissues.
| GenBank accession
no. | Gene name | Gene symbol | Potential
functions | Fold changes |
|---|
| AK091217 | Amine oxidase
(flavin containing) domain 1 | AOF1 | Transcription | 4.157 |
| AB037807 | Ankyrin repeat and
IBR domain containing 1 | ANKIB1 | Signaling | 3.332 |
| NM_019862 | ATP-binding
cassette, sub-family C (CFTR/MRP), member 1 | ABCC1 | Signaling | 3.012 |
| AL834478 | CD109 antigen (Gov
platelet alloantigens) | CD109 | Signaling | 3.448 |
| NM_001274 | CHK1 checkpoint
homolog (S. pombe) | CHEK1 | Cell cycle | 4.564 |
| NM_001827 | CDC28 protein
kinase regulatory subunit 2 | CKS2 | Cell cycle | 3.336 |
| NM_018098 | Epithelial cell
transforming sequence 2 oncogene | ECT2 | Signaling | 5.918 |
| NM_000165 | Gap junction
protein, α 1, 43 kDa (connexin 43) | GJA1 | Signaling | 5.305 |
| NM_005329 | Hyaluronan synthase
3 | HAS3 | Metabolism | 5.493 |
| NM_002210 | Integrin, α V
(vitronectin receptor) | ITGAV | Adhesion | 3.778 |
| BC002630 | Integrin, β 8 | ITGB8 | Adhesion | 5.953 |
| X85108 | Laminin, α 3 | LAMA3 | Cell structure | 2.707 |
| NM_022045 | Mdm2, transformed
3T3 cell double minute 2 | Mdm2 | Apoptosis | 4.994 |
| BC004887 | LanC lantibiotic
synthetase component C-like 2 | LANCL2 | Transcription | 2.667 |
| NM_014554 | SUMO1/sentrin
specific protease 1 | SENP1 | Transcription | 2.688 |
| AF061512 | Tumor protein
p73-like |
TP73L/P63 | Cell cycle | 5.089 |
| NM_000667 | Alcohol
dehydrogenase 1A (class I) | ADH1A | Metabolism | 0.109 |
| NM_000669 | Alcohol
dehydrogenase 1C (class I) | ADH1C | Metabolism | 0.045 |
| NM_032827 | Atonal homolog 8
(Drosophila) | ATOH8 | Transcription | 0.216 |
| NM_006763 | BTG family, member
2 | BTG2 | Transcription | 0.274 |
| NM_175709 | Chromobox homolog
7 | CBX7 | Transcription | 0.305 |
| NM_005064 | Chemokine (C-C
motif) ligand 23 | CCL23 | Signaling | 0.219 |
| NM_006274 | Chemokine (C-C
motif) ligand 19 | CCL19 | Signaling | 0.215 |
| NM_005756 | G protein-coupled
receptor 64 | GPR64 | Signaling | 0.074 |
| M65062 | Insulin-like growth
factor binding protein 5 | IGFBP5 | Signaling | 0.253 |
| L36531 | Integrin, α 8 | ITGA8 | Adhesion | 0.395 |
| NM_138284 | Interleukin
17D | IL17D | Signaling | 0.123 |
| NM_000426 | Laminin, α 2 | LAMA2 | Cell structure | 0.293 |
| NM_005924 | Mesenchyme homeo
box 2 | MEOX2 | Transcription | 0.136 |
| AK090729 | Sodium channel,
voltage-gated, type II, β | SCN2B | Signaling | 0.163 |
| NM_003256 | Tissue inhibitor of
metalloproteinase 4 | TIMP4 | Growth factors | 0.2 |
Validation of microarray data using
semi-quantitative RT-PCR or western blot analysis
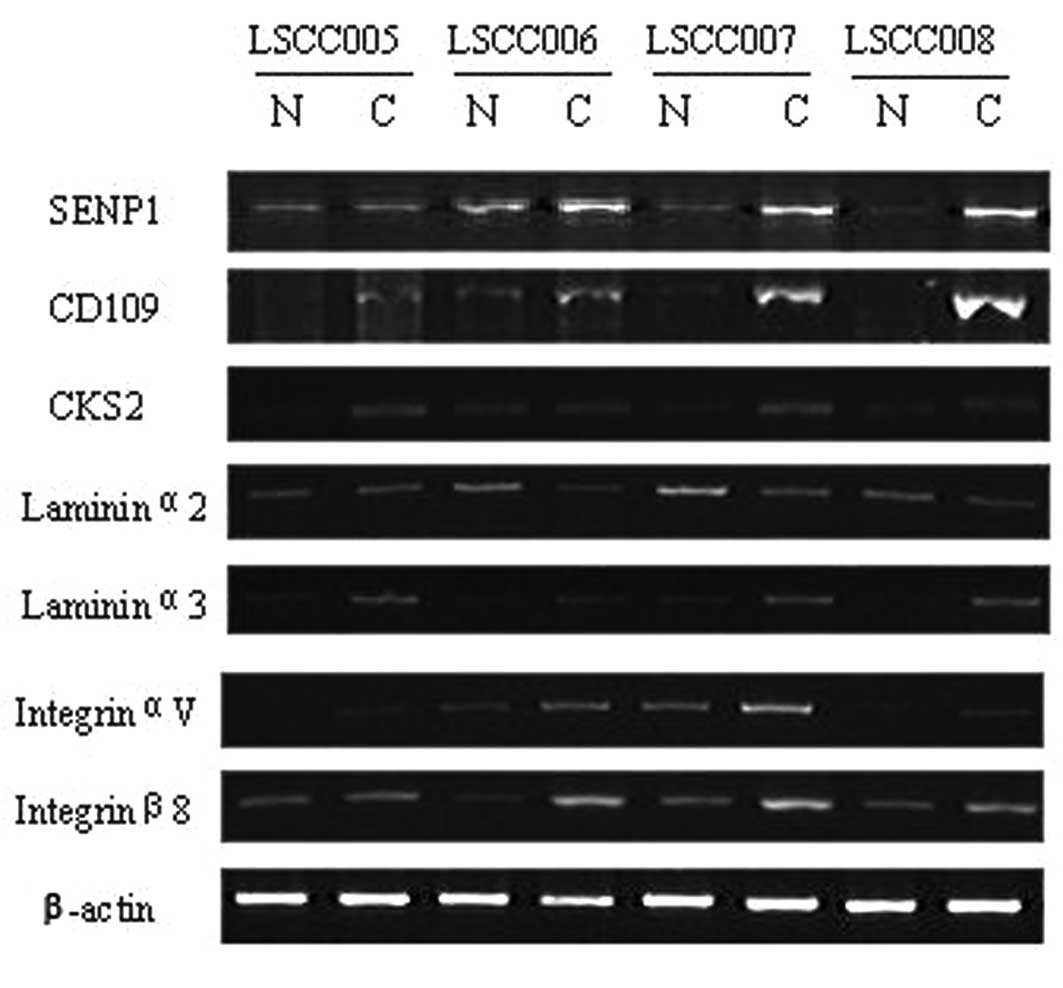
From the microarray data we chose 7 genes whose
differential status was validated with semi-quantitative RT-PCR or
western blot analysis, using source material from 32 cases of
laryngeal cancer and the corresponding normal tissues. The results
demonstrated that expression of these 7 genes were in accordance
with the microarray data (Figs. 1
and 2). Expression levels of
SENP1, CD109, CKS2, LAMA3, ITGAV
and ITGB8 mRNA were all increased compared with the normal
tissues, while LAMA2 mRNA was significant decreased in tumor
tissues compared with normal tissues. As shown in Table IV, of the 32 laryngeal cancers,
compared with normal epithelial tissues mRNA expression of
SENP1 was significantly elevated in 22 cases (68.8%),
LAMA3 in 23 (71.9%), CD109 in 26 (81.3%), CKS2
in 25 (78.1%), ITGAV in 22 (68.8%) and ITGB8 in 20
(62.5%), while LAMA2 was significantly less in 18 (56.3%).
Western blot data showed that of these 32 laryngeal cancer tissues,
compared with the corresponding normal tissues, SENP1 protein
levels were markedly higher in 21 cases (65.6%), CD109 in 24 (75%)
and CKS2 in 23 (71.9%; Table
V).
| Table IV.Semi-quantitative RT-PCR analysis of
gene expression. |
Table IV.
Semi-quantitative RT-PCR analysis of
gene expression.
| Gene | LSCC tissues | Related adjacent
normal tissues | P-value |
|---|
| SENP1 | 0.540±0.248 | 0.395±0.327 | 0.013 |
| CD109 | 0.941±0.452 | 0.293±0.294 | 0.000 |
| CKS2 | 13.895±4.787 | 10.351±4.297 | 0.000 |
| LAMA2 | 7.085±4.382 | 11.967±7.298 | 0.000 |
| LAMA3 | 6.276±3.922 | 2.849±3.723 | 0.002 |
| ITGAV | 1.013±0.478 | 0.759±0.468 | 0.019 |
| ITGB8 | 1.736±1.385 | 1.227±0.936 | 0.033 |
| Table V.Western blot analysis of SENP1, CD109
and CKS2 protein expression in laryngeal cancer and the
corresponding adjacent normal tissues. |
Table V.
Western blot analysis of SENP1, CD109
and CKS2 protein expression in laryngeal cancer and the
corresponding adjacent normal tissues.
| Protein | LSCC tissues | Related adjacent
normal tissues | P-value |
|---|
| SENP1 | 0.987±0.257 | 0.775±0.237 | 0.003 |
| CD109 | 1.827±0.676 | 1.606±0.746 | 0.021 |
| CKS2 | 0.827±0.389 | 0.628±0.252 | 0.013 |
Association of the expression of three
genes with patient clinicopathological data
Statistical analysis revealed that protein levels of
the three genes SENP1, CD109 and CKS2 were
significantly different between tumor and corresponding normal
tissues (P≤0.05). We examined their expression levels for
associations with clinicopathological data, including age and tumor
classification, stage, differentiation and lymph node metastasis
(Table VI). SENP1 expression
differed between stage I+II and III+IV tumors. CKS2 expression
differed with tumor classification, tumor differentiation and lymph
node metastasis. CD109 expression differed between glottic
carcinoma and subglottic carcinoma.
| Table VI.Table
IV. Association of SENP1, CD109 and CKS2 expression levels with
patient clinicopathological data. |
Table VI.
Table
IV. Association of SENP1, CD109 and CKS2 expression levels with
patient clinicopathological data.
| | SENP1
| CKS2
| CD109
|
|---|
| Parameter | No. | Mean ± SD | P-value | Mean ± SD | P-value | Mean ± SD | P-value |
|---|
| Age (years) | | | | | | | |
| ≤62 | 17 | 1.000±0.246 | 0.793 | 0.862±0.325 | 0.472 | 1.868±0.619 | 0.612 |
| >62 | 15 | 1.020±0.156 | | 0.770±0.392 | | 1.968±0.450 | |
| Tumor
classification | | | | | | | |
| Supra-GC | 16 | 1.012±0.154 | 0.989a | 0.938±0.441 | 0.040a | 1.906±0.573 | 0.523a |
| GC | 14 | 1.013±0.263 | 0.792b | 0.674±0.177 | 0.158b | 2.029±0.447 | 0.028b |
| Sub-GC | 2 | 0.959±0.290 | 0.678c | 0.882±0.260 | 0.866c | 1.187±0.524 | 0.112c |
| Tumor
differentiation | | | | | | | |
| Well | 10 | 1.030±0.257 | 0.857d | 0.626±0.256 | 0.068d | 1.906±0.671 | 0.679d |
| Moderate | 14 | 1.020±0.148 | 0.690e | 0.900±0.394 | 0.906e | 2.004±0.353 | 0.378e |
| Poor | 8 | 0.980±0.244 | 0.656f | 0.919±0.329 | 0.049f | 1.770±0.665 | 0.674f |
| Tumor stage | | | | | | | |
| I+II | 9 | 0.920±0.102 | 0.047 | 0.689±0.493 | 0.212 | 2.063±0.457 | 0.339 |
| III+IV | 23 | 1.050±0.227 | | 0.870±0.282 | | 1.857±0.568 | |
| Lymph node
metastasis | | | | | | | |
| Yes | 9 | 1.030±0.193 | 0.750 | 1.039±0.358 | 0.026 | 1.935±0.504 | 0.897 |
| No | 23 | 1.000±0.214 | | 0.733±0.322 | | 1.907±0.564 | |
Discussion
A profile of the genes that were differentially
expressed between laryngeal cancers and corresponding normal
mucosae was created using cDNA microarray analysis. A total of 349
differentially expressed genes were identified in four patients, of
which 112 were significantly upregulated and 237 were
downregulated. We also identified certain genes that were altered
in LSCC, including P63 and Mdm2. We also found other genes that
were altered in human cancers, but which have not been identified
before in LSCC. Thus, we selected 7 genes to study the differential
mRNA and protein expression in LSCC using semi-quantitative RT-PCR
and western blot analysis, respectively. The data demonstrated
that, compared with the normal mucosae, 6 of the 7 genes were
upregulated in laryngeal cancer and 1 was downregulated.
Associations between these genes and
clinicopathological data from the patients were identified. For
example, the expression of SENP1 was associated with tumor stage,
while CKS2 was associated with tumor classification,
differentiation and lymph node metastasis. These results imply that
the detection of elevated levels of expression of SENP1 and CKS2
should be further evaluated as tumor markers for early detection or
prognosis of laryngeal cancer.
The identification of genes differentially expressed
between normal and malignant tissues is the first step to
understanding how altered expression may contribute to
tumorigenesis. These genes are likely to represent critical points
of alteration in pathways that regulate the cell cycle, cell-cell
adhesion and cell motility. Our current data identified a large
number of genes differentially expressed in tumor tissues compared
with normal tissues of the larynx. Specifically, in 4 cancer cases
there were 10,909, 10,223, 5,730 and 14,665 differentially
expressed genes. Only 349 genes were identified to be
differentially expressed in all 4 cases. These data indicate that
each patient has an individualized profile with genes that may be
targeted for personalized medical treatment in future studies.
However, there are also genes that are commonly altered in
laryngeal cancer that may be evaluated as biomarkers for early
detection and prediction of prognosis of laryngeal cancer.
Of the genes whose expression is commonly elevated
in laryngeal cancer, the small ubiquitin-like modifier (SUMO) is
involved in numerous cellular processes, including
nuclear-cytosolic transport, transcriptional regulation, apoptosis,
protein stability, response to stress and progression through the
cell cycle (4). The family of
sentrin/SUMO-specific proteases (SENPs) is one of a group of
enzymes that process newly synthesized SUMO1s into the conjugate
form and catalyze the deconjugation of SUMO-containing species to
regulate the function of the SUMO protein. SENP1, a member
of the SENP family, has been reported to be overexpressed in
colon cancer tissues (5) and has
been demonstrated to regulate androgen receptor transactivation by
targeting histone deacetylase I, and induce c-Jun activity through
de-SUMOylation of p300 (6).
Previous studies have demonstrated that SENP1 was able to
transform normal prostate epithelium into a dysplasia, and also
directly modulate several oncogenic pathways in prostate cells
(7,8). However, it remains unknown whether and
how the expression of SENP1 plays a role in laryngeal
cancer.
In the present study, we identified that
SENP1 mRNA and protein were highly expressed in laryngeal
cancer tissues. SENP1 expression was also statistically different
between stage I+II and III+IV tumors. Together, these findings
indicate that SENP1 may play a role in tumorigenesis and
progression of laryngeal cancer.
CD109 is a
glycosylphosphatidylinositol-linked glycoprotein which belongs to
the α2 macroglobulin/C3/C4/C5 family of thioester-containing
proteins. A previous study revealed that CD109 was a useful
diagnostic marker for basal-like breast carcinoma (9). Another study identified that
CD109 may be involved in bladder tumorigenesis and might be
a potential target for cancer immunotherapy (10). Zhang et al (11) detected CD109 expression in
half of lung squamous cell carcinomas cases investigated, but not
in lung adenocarcinomas, large cell carcinomas or small cell
carcinoma. In addition, CD109 expression was found to be
upregulated in approximately 50% of esophageal squamous cell
carcinoma cases. In the present study, we identified that
CD109 was highly expressed in laryngeal cancer tissues, and
CD109 expression differed between glottic carcinoma and subglottic
carcinoma. However, the number of cases of subglottic carcinoma in
this study were too few to make a definitive statement; these data
require confirmation with additional studies with larger sample
sizes.
CKS2, an essential component of
cyclin/cyclin-dependent kinase complexes, contributes to cell cycle
progression. Earlier studies identified CKS2 as a
transcriptional target that was downregulated by the tumor
suppressor p53 (12). Other studies
demonstrated that expression of CKS1 and CKS2 were
elevated in prostate cancer, while knockdown of CKS2
expression induced programmed cell death and inhibited
tumorigenicity (13). Another study
demonstrated that CKS2 was significantly expressed in
metastasized tumors (14). Using
oligomicroarray analysis and qRT-PCR, Uchikado et al
(15) identified CKS2 as a
gene associated with the lymph node metastasis of esophageal
squamous cell carcinoma. The present study also revealed that CKS2
was highly expressed in laryngeal cancer and associated with tumor
classification, tumor differentiation and lymph node
metastasis.
Laminin, a basement membrane protein consisting of
α, β and γ chains, plays a critical role in the maintenance of
tissue structures (16). Laminin
expression is a prerequisite for normal embryonic development
(17). Abnormal expression of
LAMA332 and its integrin receptors is a hallmark of certain
types of tumor and is considered to promote the invasion of colon,
breast and skin cancer cells (18).
Our present study confirmed these previous findings, although its
role in laryngeal cancer requires further study.
Integrins are a group of cell adhesion molecules
that regulate a wide variety of dynamic cellular processes,
including cell migration, phagocytosis, growth and embryonic
development. The interaction of integrins with extracellular
ligands is regulated from inside the cell through the short
cytoplasmic α- and β-integrin tails, and transmits biochemical and
mechanical signals to the cytoskeleton to change cell shape,
behavior and fate (19).
ITGAVB6 has a role in the inhibition of colon cancer cell
apoptosis through targeting the mitochondrial pathway (20). Another study (21) demonstrated that antisense
ITGAV and ITGB3 inhibited tumor vascularization and
growth, but enhanced the apoptosis of tumor cells. Antisense
ITGAV suppressed tumor growth more markedly than antisense
ITGB3. Loss of the ITGB8 subunit resulted in abnormal
blood vessel development in the yolk sac, placenta and brain
(22); animals lacking the
ITGB8 gene die either at mid-gestation (due to insufficient
vascularization of the placenta and yolk sac) or shortly after
birth with severe intra-cerebral hemorrhage (22). Our present study also demonstrated
altered expression of these integrins in laryngeal cancer.
In conclusion, our study provides the first evidence
that SENP1, CD109, CKS2, LAMA2,
LAMA3, ITGAV and ITGB8 are differentially
expressed in laryngeal cancer tissue specimens. Further study of
these seven genes may aid the understanding of the multistep
process of laryngeal tumorigenesis, and evaluate them as tumor
biomarkers for early detection or prediction of prognosis of
laryngeal cancer.
Acknowledgements
We would like to thank Dr J.D. Wu of
Nanjing Medical University for his helpful advice and Mr. J.Y. Shen
and Mr. L. Zhang for their technical assistance. This study was
supported in part by grants from the Nanjing Technology Development
Project Fund (No. 200702075), the Medical Science and Technology
Development Foundation, Nanjing Department of Health (No.
YKK10175), and the BenQ Medical Center Research Fund (No.
SRD20100001).
References
|
1.
|
BW StewartP KleihuesWorld Cancer
ReportInternational Agency for Research on
CancerGeneva2322362003
|
|
2.
|
R MehrotraS YadavOral squamous cell
carcinoma: etiology, pathogenesis and prognostic value of genomic
alterationsIndian J
Cancer436066200610.4103/0019-509X.2588616790942
|
|
3.
|
S TaylorM WakemG DijkmanM AlsarrajM
NguyenA practical approach to RT-qPCR-publishing data that conform
to the MIQE
guidelinesMethods50S1S5201010.1016/j.ymeth.2010.01.00520215014
|
|
4.
|
RT HaySUMO-specific proteases: a twist in
the tailTrends Cell
Biol17370376200710.1016/j.tcb.2007.08.00217768054
|
|
5.
|
Y XuJ LiY ZuoJ DengLS WangGQ
ChenSUMO-specific protease 1 regulates the in vitro and in vivo
growth of colon cancer cells with the upregulated expression of CDK
inhibitorsCancer
Lett3097884201110.1016/j.canlet.2011.05.01921669491
|
|
6.
|
J ChengX KangS ZhangET YehSUMO-specific
protease1 is essential for stabilization of HIF1a during
hypoxiaCell131584595200710.1016/j.cell.2007.08.04517981124
|
|
7.
|
T Bawa-KhalfeET YehSUMO losing balance:
SUMO proteases disrupt SUMO homeostasis to facilitate cancer
development and progressionGenes
Cancer1748752201010.1177/194760191038255521152235
|
|
8.
|
T Bawa-KhalfeJ ChengZ WangET YehInduction
of the SUMO-specific protease 1 transcription by the androgen
receptor in prostate cancer cellsJ Biol
Chem2823734137349200710.1074/jbc.M70697820017932034
|
|
9.
|
M HasegawaS MoritaniY MurakumoT SatoS
HagiwaraC SuzukiS MiiM JijiwaA EnomotoN AsaiS IchiharaM
TakahashiCD109 expression in basal-like breast carcinomaPathol
Int58288294200810.1111/j.1440-1827.2008.02225.x
|
|
10.
|
M HagikuraY MurakumoM HasegawaM JijiwaS
HagiwaraS MiiS HagikuraY MatsukawaY YoshinoR HattoriK WakaiS
NakamuraM GotohM TakahashiCorrelation of pathological grade and
tumor stage of urothelial carcinomas with CD109 expressionPathol
Int60735743201010.1111/j.1440-1827.2010.02592.x20946523
|
|
11.
|
JM ZhangM HashimotoK KawaiY MurakumoT
SatoM IchiharaS NakamuraM TakahashiCD109 expression in squamous
cell carcinoma of the uterine cervixPathol
Int55165169200510.1111/j.1440-1827.2005.01807.x15826242
|
|
12.
|
K RotherM DenglJ LorenzK TschöpR
KirschnerJ MössnerK EngelandGene expression of cyclin-dependent
kinase subunit Cks2 is repressed by the tumor suppressor p53 but
not by the related proteins p63 or p73FEBS
Lett58111661172200710.1016/j.febslet.2007.02.028
|
|
13.
|
Y LanY ZhangJ WangC LinMM IttmannF
WangAberrant expression of Cks1 and Cks2 contributes to prostate
tumorigenesis by promoting proliferation and inhibiting programmed
cell deathInt J Cancer123543551200810.1002/ijc.2354818498131
|
|
14.
|
M LiYM LinS HasegawaT ShimokawaK MurataM
KameyamaO IshikawaT KatagiriT TsunodaY NakamuraY FurukawaGenes
associated with liver metastasis of colon cancer, identified by
genome-wide cDNA microarrayInt J Oncol24305312200414719106
|
|
15.
|
Y UchikadoH InoueN HaraguchiK MimoriS
NatsugoeH OkumuraT AikouM MoriGene expression profiling of lymph
node metastasis by oligomicroarray analysis using laser
microdissection in esophageal squamous cell carcinomaInt J
Oncol2913371347200617088971
|
|
16.
|
Y KariyaT MoriC YasudaN WatanabeY KanekoY
NakashimaT OgawaK MiyazakiLocalization of laminin alpha3B chain in
vascular and epithelial basement membranes of normal human tissues
and its down-regulation in skin cancersJ Mol
Histol39435446200810.1007/s10735-008-9183-0
|
|
17.
|
D MalanM ReppelR DobrowolskiW RoellN
SmythJ HeschelerM PaulssonW BlochBK FleischmannLack of laminin
gamma1 in embryonic stem cell-derived cardiomyocytes causes
inhomogeneous electrical spreading despite intact differentiation
and functionStem Cells278899200910.1634/stemcells.2008-0335
|
|
18.
|
D TsurutaH KobayashiH ImanishiK SugawaraM
IshiiJC JonesLaminin-332-integrin interaction: a target for cancer
therapy?Curr Med
Chem1519681975200810.2174/09298670878513283418691052
|
|
19.
|
MA ArnaoutSL GoodmanJP XiongStructure and
mechanics of integrin-based cell adhesionCurr Opin Cell
Biol19495507200710.1016/j.ceb.2007.08.00217928215
|
|
20.
|
Z Zhao-YangX Ke-SenH Qing-SiN Wei-BoW
Jia-YongM Yue-TangW Jin-ShenW Guo-QiangY Guang-YunN JunSignaling
and regulatory mechanisms of integrin alphavbeta6 on the apoptosis
of colon cancer cellsCancer
Lett266209215200810.1016/j.canlet.2008.02.05418381232
|
|
21.
|
J LiH TanX DongZ XuC ShiX HanH JiangGW
KrissansenX SunAntisense integrin alphaV and beta3 gene therapy
suppresses subcutaneously implanted hepatocellular carcinomasDig
Liver Dis39557565200710.1016/j.dld.2007.01.025
|
|
22.
|
JM ProctorK ZangD WangR WangLF
ReichardtVascular development of the brain requires beta8 integrin
expression in the neuroepitheliumJ
Neurosci2599409948200510.1523/JNEUROSCI.3467-05.200516251442
|