Introduction
DNA damage response is essential for the maintenance
of genome integrity (1–5). As a complex, the DNA damage response
involves the recognition of DNA damage, activation of DNA
damage-responsive protein kinases, signal amplification by
downstream protein kinases and activation of the effector proteins
that trigger various cellular processes. At low DNA damage levels,
activation of the DNA damage response results in cell cycle arrest
and DNA repair. However, at higher levels or under severe
conditions, DNA damage response signaling frequently results in
cell death by apoptosis (1–5).
Phosphoinositide 3-kinase-related protein kinases,
ataxia telangiectasia mutated proteins (ATM) and Rad3-related
proteins (ATR), are the key regulators of the DNA damage response
(1–8). Once activated, ATM and ATR regulate an
array of substrates, including Chk1 and Chk2, which culminate in
DNA repair, cell cycle arrest and/or apoptosis. In the canonical
model, ATR activation involves the recruitment of the ATR-ATR
interacting protein (ATR-ATRIP) and Rad9-Hus1-Rad1 (9-1-1) protein
complexes to the DNA damage site via replication protein A (RPA).
As a result, the 9-1-1 complex brings topoisomerase-binding
protein-1 (TopBP1) (ATR activator) close to ATR for ATR activation
(1,4,8).
Mammalian TopBP1 functions at the DNA replication checkpoint
(9,10) and has multiple BRCA1 C-terminal
(BRCT) repeats, which usually function in tandem to bind
phosphoproteins (11,12). TopBP1 colocalizes with ATR-ATRIP at
the sites of DNA replication stress (9,10). The
N-terminus of TopBP1 is required for its recruitment and the
resulting activation of ATR via interaction with Rad9 in mammalian
cells (13).
Human MutY homolog (hMYH) is a base excision repair
DNA glycosylase that excises misincorporated adenine opposite
7,8-dihydro-8-oxoguanine (8-oxoG), a product of oxidative DNA
damage. Furthermore, in hMYH-disrupted cells, the phosphorylation
of ATR and Chk1 is decreased by hydroxyurea (HU) or ultraviolet
(UV) treatment (14). The hMYH is
known to interact with 9-1-1 (15),
and a recent study revealed that it also interacts with ATR and
MutS α via the human MutS homolog (hMSH) 6 subunit (14,16).
The mismatch repair protein hMSH2 may interact with
ATR and participate in ATR activation during DNA damage, leading to
apoptosis. Therefore, hMSH2-deficient cells are more resistant to
apoptosis (17). The mismatch
repair and MYH repair pathways share many common features. First,
both pathways function immediately following DNA replication to
distinguish newly synthesized DNA strands from their parental
counterparts (18,19). Second, hMYH and hMSH6 interact with
the replication proteins proliferating cell nuclear antigen and RPA
and colocalize at the same replication foci (20,21).
Finally, both pathways are involved in mutation avoidance following
DNA oxidation. Therefore, we suggest that the interaction between
hMYH and hATR is the same as hMSH2 and hATR.
Etoposide (ETP), a topoisomerase II inhibitor, is
known to induce apoptosis and activate ATR via TopBP1 (22,23);
however, it is unclear how hMYH is involved during the ETP
response. In this study, MYH, ATR and TopBP1 knockdown cells were
treated with ETP. In the absence of these proteins, the cells were
more resistant to ETP-induced apoptosis. We determined for the
first time that hMYH interacts with hTopBP1 or hATR in HEK293
cells, and that this interaction is increased following ETP
treatment. However, when hMYH is disrupted, the interaction between
hATR and hTopBP1 is decreased following ETP treatment. Since hATR
is inactive, the apoptosis signal cannot transduce to the
downstream proteins, specifically p-Chk2 and p-p53.
Materials and methods
Cell lines
Human embryonic kidney HEK293 cells were grown in
Dulbecco’s modified Eagle’s medium (Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(Invitrogen Life Technologies) and 1% penicillin-streptomycin
solution (Sigma, St. Louis, MO, USA) at 37°C in a 5% CO2
incubator. Prior to the experiments, HEK293 cells were seeded into
6-well plates at a density of 1×106 cells per well and
incubated for 24 h.
siRNA construction and transfection into
cells
The optimum siRNA sequences for the knockdown of
endogenous hMYH were designed and purchased from the Stealth™ RNAi
program of Invitrogen Life Technologies. siRNA corresponding to
nucleotides 415–439 of the green fluorescence protein (GFP) was
used as a negative control. The siMYH and siGFP sequences and
transfection method were conducted as previously described
(14). ATR siRNA (sc-29763) and
TopBP1 siRNA were purchased from Santa Cruz Biotechnology, Inc.
(sc-41068; Santa Cruz, CA, USA). ATR and TopBP1 knockdown were
conducted according to the manufacturer’s instructions.
Protein extraction and western blot
analysis
HEK293 cells were harvested, washed with
phosphate-buffered saline (PBS) and lysed with lysis buffer
containing 50 mM Tris-HCl phenylmethanesulfonylfluoride, protease
and a phosphatase inhibitor cocktail (Sigma), for 1 h at 4°C with
occasional vortexing. Protein extracts were collected following
centrifugation at 16,000 × g for 20 min, and protein concentration
was determined using a Bio-Rad DC protein assay kit (Bio-Rad,
Hercules, CA, USA). Protein extracts that were resolved on 8 or 12%
sodium dodecyl sulfate (SDS)-polyacrylamide gels were transferred
onto PVDF membranes (GE Healthcare Worldwide, Princeton, NJ, USA).
The membranes were blocked with 5% non-fat dried milk in
Tris-buffered saline with 0.05% Tween-20 and then incubated with
antibodies against ATR, Chk1, Chk2, phospho-Chk1 (Ser-345),
phospho-Chk2 (Thr-68), β-actin (all from Santa Cruz Biotechnology,
Inc.), phospho-ATR (Ser-428), caspase 9, caspase 7, p-p53 (Ser-15;
all from Cell Signaling Technology, Inc., Beverly, MA, USA), TopBP1
(Abcam, Cambridge, UK) and hMYH (Abnova, Teipei, Taiwan). Membranes
were then incubated with horseradish peroxidase-conjugated
secondary antibodies (Santa Cruz Biotechnology, Inc.). Protein
bands were detected using enhanced chemiluminescence (ECL) Pico
western blotting detection reagents (Pierce Biotechnology, Inc.,
Rockford, IL, USA).
Immunofluorescence microscopy
To determine the subcellular location of ATR, TopBP1
and hMYH, HEK293 cells were seeded onto polylysine-coated
coverslips and treated with 25 μM ETP for 48 h. At room
temperature, cells were fixed with 4% paraformaldehyde in PBS for
30 min and permeabilized with 0.25% Triton X-100 in PBS for 30 min.
After blocking with 1% bovine serum albumin in PBS containing 0.5%
Tween-20 (PBS-T) for 30 min, cells were incubated with ATR (1:100;
Santa Cruz Biotechnology, Inc.), TopBP1 (1:500; Abcam) and MYH
monoclonal antibodies (1:100; Abnova) for 2 h. The cells were
washed 3 times for 15 min each in PBS and incubated with Alexa
488-conjugated anti-mouse IgG (1:100; Sigma), fluorescein
isothiocyanate (FITC)-conjugated anti-rabbit IgG (1:100; Sigma) for
2 h. Cells were rinsed 3 times with 1 ml PBS and analyzed using a
confocal fluorescence microscope (Olympus FV-1000; software,
Olympus Fluoview; Olympus, Center Valley, PA, USA).
Co-immunoprecipitation
Co-immunoprecipitation (IP) of endogenous proteins
using an ATR antibody was conducted using the ImmunoCruz™ IP/WB
Optima B System (Santa Cruz Biotechnology, Inc.) according to the
manufacturer’s instructions. IP was conducted using rabbit
anti-ATR, and immunoblot (IB) analysis was conducted using rabbit
anti-ATR, rabbit anti-TopBP1 and mouse anti-MYH. To determine the
effect of ETP on MYH-ATR and MYH-TopBP1 interaction, a similar
co-IP procedure was conducted using mouse anti-MYH for co-IP, and
IB analysis using rabbit anti-ATR, rabbit anti-TopBP1 and mouse
anti-MYH.
Statistical analysis
Experiments were conducted in triplicate and
statistical analyses were conducted using the Student’s t-test.
Data are expressed as the mean, and P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of ETP on HEK293 cell
proliferation
We examined the effect of ETP on cell viability by
treating cells with 3 different concentrations of ETP (10, 25 and
50 μM) for various periods of time. After 24, 48 and 72 h of
treatment, cell viability was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Fig. 1A shows that treatment
with ETP greatly reduced cell viability compared with the control
cells. ETP induced apoptosis in HEK293 cells in a time- and
dose-dependent manner. We determined that treatment with 25 μM ETP
was the best condition for our subsequent experiments.
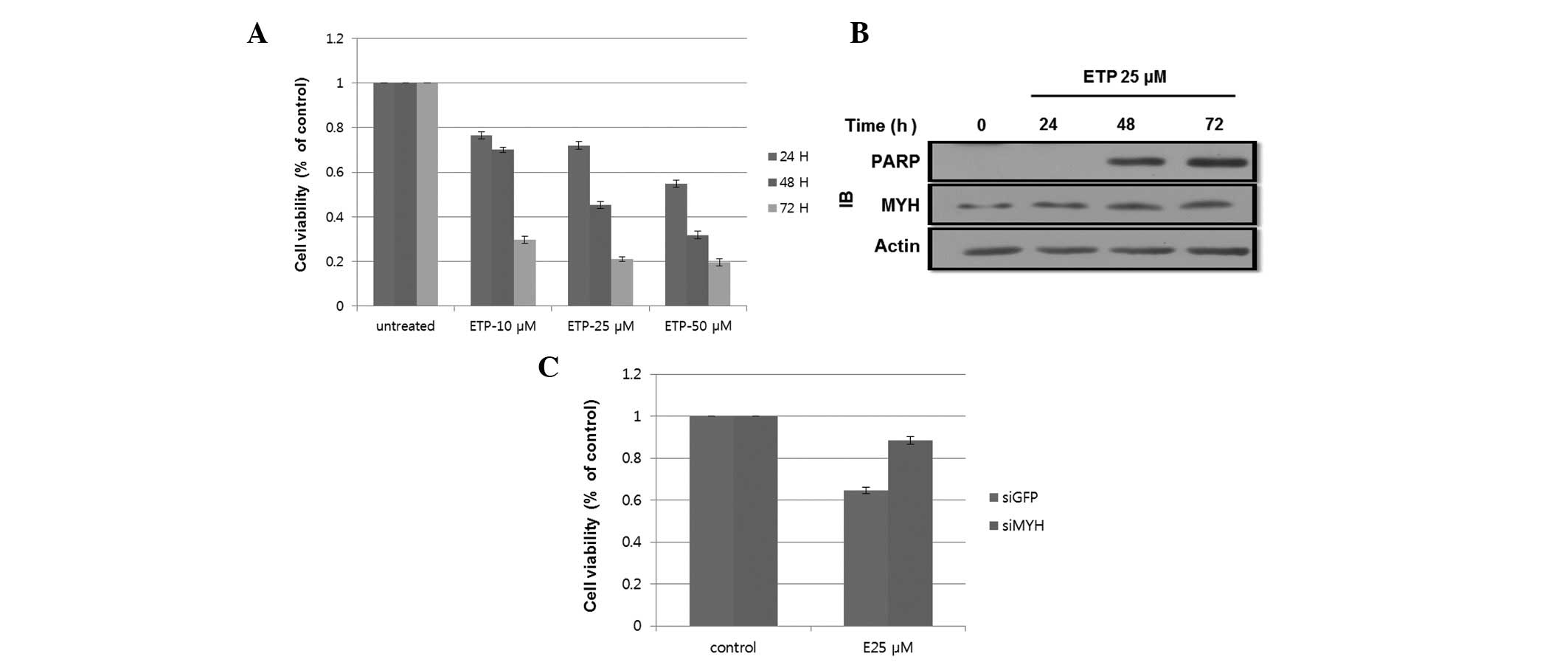 | Figure 1.ETP-induced apoptosis in HEK293
cells. (A) ETP treatment reduced cell viability. Cells were treated
with 0, 10, 25 and 50 μM ETP. After 24, 48 and 72 h, 20 μl MTT was
added to each well and incubated for an additional 4 h. The results
shown are the mean ± SD of 3 independent experiments. (B)
ETP-induced cleavage of PARP following 48 h of incubation. Cells
were incubated with 25 μM ETP for 24, 48 and 72 h. Cell lysates
were subjected to SDS-PAGE. PARP and MYH expression levels were
determined by western blotting using the indicated antibodies. (C)
hMYH knockdown reduced cell viability following ETP treatment.
Cells were transfected with siGFP or siMYH and incubated for 24 h.
Cells were then treated with 25 μM ETP for 48 h. Cell viability was
determined by the MTT assay. The results shown are the mean ± SD of
3 independent experiments. ETP, etoposide; IB, immunoblot; PARP,
poly-ADP ribose polymerase; MYH, MutY homolog; GFP, green
fluorescence protein. MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel
electrophoresis; hMYH, human MYH. |
To assess the molecular mechanism of ETP-induced
apoptosis, we examined the expression of the 85-kDa cleaved form of
poly-ADP ribose polymerase (PARP), an apoptosis marker, and hMYH,
an apoptosis-related protein (25).
Cells were treated with 25 μM ETP for 24, 48 or 72 h. As shown in
Fig. 1B, cleavage of PARP was
observed after 48 and 72 h. The hMYH protein level increased in a
time-dependent manner, indicating the involvement of hMYH in
apoptosis. Taken together, treatment with 25 μM ETP for 48 h was
the best condition for the subsequent experiment.
To further determine the effect of hMYH on cell
viability, cells transfected with MYH-siRNA or control-siRNA were
treated with 25 μM ETP for 48 h. Fig.
1C shows that cell viability, as determined by the MTT assay,
decreased by ∼40% in the control cells, while only a 17% decrease
was observed in MYH-siRNA cells. These results suggested the
involvement of hMYH in apoptotic signaling during ETP
treatment.
hMYH, hTopBP1 and hATR deficient cells
are resistant to ETP-induced apoptosis and DNA-damage
signaling
Recently, it has been reported that ATR
phosphorylation upon HU or UV treatment was decreased in
hMYH-disrupted HEK293 and HaCaT cells (14). ATR is known to phosphorylate Chk1
and Chk2 (14,15), and here, we observed a
phosphorylation of Chk2 only in control-transfected cells, but not
in siMYH cells following ETP treatment (Fig. 2A). This result suggests that hMYH is
required for ATR-mediated Chk2 activation under these conditions.
The phosphorylation of Chk1 observed was similar to that of Chk2
(data not shown). Since transactivation of proapoptotic proteins
through the p53-dependent signaling pathway is an important
apoptotic mechanism, we focused on Chk2 because of its involvement
in p53 regulation and ETP-induced apoptosis (22,24).
As shown in Fig. 2A, ETP-induced
p53 phosphorylation was abrogated in siMYH-transfected cells.
 | Figure 2.MYH, ATR and TopBP1 knockdown
decreases ETP-induced apoptosis in HEK293 cells. (A) MYH knockdown
reduces the activation of apoptosis-related proteins. Control or
siMYH transfected cells were treated with 25 μM ETP for 48 h. Total
cell lysates were used for IB analysis of p-Chk2 (threonine-68),
p-p53 (threonine-15), caspase 9, caspase 7 and MYH. β-actin was
used as a loading control. (B and C) The same experiment as in A,
however, the cells were transfected with siRNA for either ATR or
TopBP1. IB, immunoblot; MYH, MutY homolog; PARP, poly-ADP ribose
polymerase; Cas, caspase; GFP, green fluorescence protein; ETP,
etoposide; ATR, Rad3-related protein; TopBP1, topoisomerase-binding
protein-1. |
Caspase is a well-known key molecule in DNA
damage-induced apoptosis (22). The
activation of caspase 9 and caspase 7 was examined via western blot
analysis in hMYH knockdown and control cells. ETP treatment induced
the cleavage of inactive 47-kDa procaspase 9 into smaller,
detectable, active 37-kDa fragments, and the cleavage of inactive
32-kDa procaspase 7 into the 20-kDa active form in control cells
(Fig. 2A). However, the active
forms of caspase 9 and 7 were not detected in hMYH knockdown cells
following ETP treatment.
To determine the role of ATR relative to Chk2 or p53
in ETP-induced apoptosis, cells were treated with either control or
ATR siRNA for 24 h, and then treated with ETP for 48 h. We observed
increased phosphorylation or activation of Chk2 and p53 in control
siRNA-transfected cells (Fig. 2B).
However, the active forms of caspase 9 and 7 were not observed in
hATR knockdown cells.
Since hTopBP1 is known to activate hATR, we observed
the apoptosis pattern in hTopBP1 knockdown cells. Fig. 2C shows that apoptosis was decreased
in hTopBP1 knockdown cells in comparison to the control cells, and
similar results were observed in hMYH and hATR knockdown cells.
hMYH associates with ATR or TopBP1 in
HEK293 cells during ETP treatment
ETP treatment activated hATR, hTopBP1 and hMYH
(A-T-M), which triggered a signaling cascade leading to apoptosis.
However, it is unclear how A-T-M is activated and regulates the
apoptosis pathway. To determine whether hMYH regulates hATR or
hTopBP1 in DNA damage models, we conducted a co-IP assay to confirm
that hMYH interacts with hATR or hTopBP1. Cells were treated with
25 μM ETP for 48 h, and whole cell extracts were used. As shown in
Fig. 3A, ETP treatment induced the
association of hMYH-hATR and hMYH-hTopBP1. Our results suggest that
hMYH interacts with hATR or hTopBP1 and participates in A-T-M
activation during ETP treatment, which leads to the DNA damage
response and signals apoptosis.
 | Figure 3.Interaction of MYH with ATR or TopBP1
increased following ETP treatment. (A) ATR or TopBP1 interact with
MYH increases following treatment with ETP. Cells were treated with
25 μM ETP for 48 h. Cells were lysed and total cell lysates were
used for co-IP with MYH antibody. IP samples were analyzed by
western blotting with ATR or TopBP1 and MYH antibodies. β-actin was
used as a loading control. (B) ATR or TopBP1 and MYH co-localized
following treatment with ETP. Cells were cultured overnight on
coverglass bottom dishes, then treated with 25 μM ETP for 48 h, and
fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton
X-100 in PBS. Cells were then stained with antibodies against ATR
or TopBP1 (FITC; yellow), MYH (Alexa®488/Green) and
To-pro®3 (Red). IB, immunoblot; ATR, Rad3-related
protein; TopBP1, topoisomerase-binding protein-1; MYH, MutY
homolog; IP, immunoprecipitation; Ab, antibody. ETP, etoposide;
PBS, phosphate-buffered saline; FITC, fluorescein
isothiocyanate. |
A critical function of hATR or hTopBP1 activation
during genotoxic stress is the accumulation of hATR or hTopBP1 in
the nucleus, where signaling proteins form multiple nuclei and
interact in response to DNA damage (6,11). To
examine the changes in hATR and hTopBP1 localization, we conducted
immunofluorescence (IF) experiments. We treated cells with 25 μM
ETP for 48 h, and conducted IF assays to determine whether hMYH and
hATR or hTopBP1 occupied the same subcellular locations during
genotoxic stress. In untreated control cells, light hMYH staining
was observed in the cytoplasm (Fig.
3B). However, following ETP treatment, we observed a higher
intensity hMYH staining that translocated to the nucleus,
indicating an increased expression. Staining of hATR and hTopBP1,
which are known to localize to the nucleus, also increased in
intensity following ETP treatment. Superimposition of
hMYH-dependent green fluorescence with hATR or hTopBP1-dependent
yellow fluorescence resulted in yellow-green images in cells.
Topro3 Red (red fluorescence) is a nuclear marker. These results
are consistent with the interaction of hMYH with hTopBP1 or hATR
and their colocalization in cells.
ATR and TopBP1 interaction decreased in
hMYH-deficient cells during ETP treatment
The decrease in hMYH expression results in decreased
apoptotic signaling following ETP treatment (Fig. 2A). Furthermore, hMYH interacts with
hATR or hTopBP1 and this interaction increased following treatment
with ETP (Fig. 3A). To observe the
implication of hATR, hTopBP1 and hMYH interaction in this pathway,
we conducted co-IP assays using ATR antibody. Cells were
transfected with siGFP, as a control, or siMYH. Then, cells were
treated with or without ETP. Total cell lysates were used in co-IP,
and the presence of A-T-M was observed using western blot
analysis.
A decreased hMYH expression was observed in MYH
knockdown cells (Fig. 4A), while
changes in ATR and TopBP1 expression levels were not observed in
MYH knockdown cells. Following treatment with ETP, the expression
of A-T-M increased. Furthermore, the co-IP result suggests that the
interaction between hATR and hTopBP1 was decreased in MYH knockdown
cells following treatment with ETP.
 | Figure 4.Knockdown of hMYH abrogated the
interaction between ATR and TopBP1. (A) Co-IP and western blot
analysis were conducted using lysates of cells transfected with
siGFP or siMYH for 24 h. Cells were treated with 25 μM ETP for 48 h
and co-IP was conducted using an ATR antibody. IP samples were
further analyzed by western blotting with ATR, MYH and TopBP1
antibodies. Rabbit IgG was used as a control for co-IP. (B) A model
of hMYH-mediated ATR activation in DNA damage signaling. DNA damage
signaling involving ATR pathway activation is initiated by hMYH.
For ATR activation, hMYH interacts with ATR and TopBP1 at damaged
sites, leading to the activation of Chk2 and p53, culminating in
cell cycle arrest and apoptosis. IB, immunoblotting; ATR,
Rad3-related protein; TopBP1, topoisomerase-binding protein-1;
hMYH, human MutY homolog; ETP, etoposide; GFP, green fluorescence
protein; IP, immunoprecipitation; Ab, antibody. |
Discussion
hMYH interacts not only with hATR, but also with the
human MutS homologs (hMSH2/hMSH6) via the hMSH6 subunit (14,16).
hMSH2 directly participates in ATR recruitment and activation,
leading to DNA damage signaling and subsequent apoptosis.
Therefore, hMSH2 deficiency increases the resistance of cells to
apoptosis (17). Another study
suggests that the nuclear isoforms of hMYH initiate cell death by
sensing adenine opposite 8-oxoG during nuclear DNA replication,
thus suppressing tumorigenesis. In addition, accumulation of 8-oxoG
in mitochondrial DNA and initiation cell death by MYH may also
contribute to the tumor suppression (25).
In this study, we implicated the DNA glycosylase,
hMYH, in ETP-induced apoptosis, and revealed that hMYH knockdown
cells reduce Chk2 (T-68) and p53 (T-15) phosphorylation. We also
observed similar results in ATR and TopBP1 knockdown cells
(Fig. 2). This result suggests the
possibility that hATR, hTopBP1 and hMYH (A-T-M) function in the
same pathway. This was accompanied by the suppression of
proapoptotic protein expression and decreased apoptosis. The
interaction between hMYH with ATR and TopBP1 is dependent on ETP
treatment (Fig. 3).
In hMYH knockdown cells, ATR and TopBP1 interaction
decreases following ETP treatment (Fig.
4A). We suggest that hMYH functions as a sensor in ETP induced
apoptosis. In the absence of hMYH, cells cannot recognize the
damage signal, and thus the ATR pathway is not activated, which
results in tumor development (14,25).
Based on these observations, we suggest new pathways
for A-T-M sensor activation (Fig.
4B). This pathway is initiated by MYH, which recruits and
activates ATR-related proteins in ETP-induced apoptosis. In
summary, binding of MYH directly participates in ATR and TopBP1
activation in DNA damage signaling, leading to apoptosis. An MYH
protein deficiency increases the resistance of cells to
apoptosis.
Acknowledgements
This study was supported by the
project from the Advanced Technology Center (ATC) Support Program
of the Ministry of Knowledge Economy (Republic of Korea; Grant No.
10033024), the Basic Science Research Program through the National
Research Foundation of Korea (NRF; Grant No. 2012R1A1A3005889) and
the World Class University (WCU; Grant No. R33-2008-000-1071)
program through the Korea Science and Engineering Foundation funded
by the Ministry of Education, Science and Technology.
References
|
1.
|
JW HarperSJ ElledgeThe DNA damage
response: ten years afterMol Cell28739745200718082599
|
|
2.
|
J BarketJ BartkovaJ LukasDNA damage
signaling guards against activated oncogenes and tumor
progressionOncogene2677737779200710.1038/sj.onc.121088118066090
|
|
3.
|
BB ZhouSJ ElledgeThe DNA damage response:
putting checkpoint in
perspectiveNature408433439200010.1038/3504400511100718
|
|
4.
|
SP JacksonJ BartekThe DNA-damage response
in human biology and
diseaseNature46110711078200910.1038/nature0846719847258
|
|
5.
|
A SancarLA Lindsey-BoltzK Unsal-KaçmazS
LinnMolecular mechanisms of mammalian DNA repair and the DNA damage
checkpointsAnnu Rev
Biochem733985200410.1146/annurev.biochem.73.011303.07372315189136
|
|
6.
|
RT AbrahamPI 3-kinase related kinases:
‘big’ players in stress-induced signaling pathwaysDNA
repair38838872004
|
|
7.
|
PJ HurleyF BunzATM and ATR: components of
an integrated circuitCell
cycle6414417200710.4161/cc.6.4.388617312392
|
|
8.
|
KA CimprichD CortezATR: an essential
regulator of genome integrityNat Rev Mol Cell
Biol9616628200810.1038/nrm245018594563
|
|
9.
|
M MäkiniemiT HillukkalaJ TuusaK ReiniM
VaaraD HuangH PospiechI MajuriT WesterlingTP MäkeläJE SyväojaBRCT
domain-containing protein TopBP1 Functions in DNA replication and
damage responseJ Biol Chem2763039930406200111395493
|
|
10.
|
V GarciaK FuruyaAM CarrIdentification and
functional analysis of TopBP1 and its homologsDNA
Repair412271239200510.1016/j.dnarep.2005.04.00115897014
|
|
11.
|
IA MankeDM LoweryA NguyenMB YaffeBRCT
repeats as phosphopeptide-binding modules involved in protein
targetingScience302636639200310.1126/science.108887714576432
|
|
12.
|
X YuCC ChiniM HeG MerJ ChenThe BRCT domain
is a phosphor-protein binding
domainScience302639642200310.1126/science.108875314576433
|
|
13.
|
S DelacroixJM WagnerM KobayashiK
YamamotoLM KarnitzThe Rad9-Hus1-Rad1 (9-1-1) clamp activates
checkpoints signaling via TopBP1Genes
Dev2114721477200710.1101/gad.154700717575048
|
|
14.
|
SH HahmJH ParkSI KoYR LeeIS ChungJH
ChungLW KangYS HanKnock-down of human MutY homolog (hMYH) decreases
phosphorylation of checkpoint kinase 1 (Chk1) induced by
hydroxyurea and UV treatmentBMB
Rep44352357201110.5483/BMBRep.2011.44.5.35221615992
|
|
15.
|
PJ LuncsfordDY ChangG ShiK BernsteinA
MadabushiDN PattersonAL LuEA TothA structural hinge in eukaryotic
MutY homologues mediates catalytic activity and Rad9-Rad1-Hus1
checkpoint complex interactionsJ Mol
Biol403351370201010.1016/j.jmb.2010.08.045
|
|
16.
|
Y GuA ParkerTM WilsonH BaiDY ChangAL
LuHuman MutY homolog, a DNA glycosylase involved in base excision
repair, physically and functionally interacts with mismatch repair
proteins human MutS homolog 2/Human MutS homolog 6J Biol
Chem2771113511142200210.1074/jbc.M108618200
|
|
17.
|
N PablaZ MaMA McllhattonR FishelZ
DonghMSH2 recruits ATR to DNA damage sites for activation during
DNA damage-induced apoptosisJ Biol
Chem2861041110428201110.1074/jbc.M110.21098921285353
|
|
18.
|
RD KolodnerGT MarsischkyEukaryotic DNA
mismatch repairCurr Opin Genet
Dev98996199910.1016/S0959-437X(99)80013-6
|
|
19.
|
P ModrichR LahueMismatch repair in
replication fidelity, genetic recombination, and cancer biologyAnnu
Rev
Biochem65101133199610.1146/annurev.bi.65.070196.0005338811176
|
|
20.
|
AB ClarkF ValleK DrotschmannRK GaryTA
KunkelFunctional interaction of proliferating cell nuclear antigen
with MSH2-MSH6 and MSH2-MSH3 complexesJ Biol
Chem2753649836501200010.1074/jbc.C00051320011005803
|
|
21.
|
H Flores-RozasD ClarkRD
KolodnerProliferation cell nuclear antigen and Msh2p-Msh6p interact
to form an active mispair recognition complexNat
Genet26375378200010.1038/8170811062484
|
|
22.
|
NO KarpinichM TafaniRJ RothmanMA RussoJL
FarberThe course of Etoposide-induced apoptosis from damage to DNA
and p53 activation tomitochondrial release of cytochrome cJ Biol
Chem2771654716552200210.1074/jbc.M11062920011864976
|
|
23.
|
R RossiMR LidonniciS SozaG BiamontiA
MontecuccoThe dispersal of replication proteins after Etoposide
treatment requires the cooperation of Nbs1 with ataxia
telangiectasis Rad3-related/Chk1 pathwayCancer
Res6616751683200610.1158/0008-5472.CAN-05-2741
|
|
24.
|
Y XiaP OngusahaSW LeeYC LiouLoss of Wip1
sensitizes cells to stress- and DNA damage-induced apoptosisJ Biol
Chem2841742817437200910.1074/jbc.M109.00782319395378
|
|
25.
|
S OkaY NakabeppuDNA glycosylase encoded by
MUTYH functions as a molecular switch for programmed cell death
under oxidative stress to suppress tumorigenesisCancer
Sci102677682201110.1111/j.1349-7006.2011.01869.x21235684
|














