Introduction
At present, prostate cancer is recognized as one of
the most important medical problems facing the male population. In
the USA and Europe, prostate cancer is currently the second most
common cause of cancer mortality in males (1,2). The
latest statistics reveal that among males in the USA, an estimated
240,890 new cancer cases and 33,720 mortalities due to prostate
cancer occurred in 2011 (1).
Prostate cancer-related mortality is largely due to its high
metastatic potential for bone and/or other organs (3,4).
Clinically, the prevention and treatment of prostate cancer
metastasis remains a significant challenge since the molecular
mechanisms of prostate cancer invasion and metastasis are not well
understood.
Metastasis is a complex and multi-step process in
the progression of malignant cancer (5). Cell migration results in the spreading
of cancer, which is the leading cause of cancer-related
mortalities. In the process of cancer progression, certain cell
adhesion molecules (CAMs) play a pivotal role in the development of
recurrent, invasive and distant metastasis (6). A loss or reduction in the expression
of CAMs, including cadherins, facilitates the detachment of single
cancer cells from the tumor bulk (7,8). One
of the key molecules critical for cell-to-cell adhesion is
E-cadherin, a membrane glycoprotein located at cell adherent
junctions (9,10). E-cadherin aids the assembly of
epithelial cells and maintains the quiescence of cells within
sheets by forming adherent junctions with adjacent epithelial cells
(11). A number of studies have
demonstrated that increased expression of E-cadherin is able to
inhibit invasion and metastasis, while a reduced expression
potentiates these phenotypes (11–14).
In order for epithelial cells to develop into cancer cells,
activation of the epithelial-mesenchymal transition (EMT) is
required (15). EMT causes the
epithelial cell layers to lose polarity and cell-cell contacts. It
therefore triggers the remodeling of the cellular skeleton
(16,17). Upregulation of E-cadherin is
implicated by the activation of EMT (14,18),
and E-cadherin is regarded as a main indicator of
epithelial/mesenchymal phenotype switching (19).
The metastasis-associated gene 1 (MTA1) was
originally identified by the differential screening of a cDNA
screening library using highly metastatic mammary adenocarcinoma
cell lines (20). MTA1 appears to
interact with, or may even be a member of, the histone deacetylase
(HDAC) complex, and acts as a co-activator of this complex
(21). Studies have demonstrated
that MTA1 overexpression is associated with the adhesion, invasion
and metastasis of certain cancer cells (22–24),
and with a higher tumor grade, the development, microvascular
invasion and poor prognosis in a number of malignant cancer types
(25). Through repression of the
estrogen receptor α (ERα), hypoxia-inducible factor-1α (HIF-1α) and
p53 protein, MTA1 converts cancer cells into a more aggressive
phenotype (26). Moreover, MTA1 has
been identified to determine EMT phenotypes mainly through
downregulating the expression of E-cadherin, which leads to EMT
(27,28). E-cadherin can be upregulated using
MTA1 small interfering RNA (siRNA) in melanoma cells, which was
also confirmed in our previous study in cervical cancer cells
(29,30). MTA1 and E-cadherin are involved in
the EMT process (28), since the
loss of E-cadherin expression has been demonstrated to increase
cancer metastasis progresses (13,14,31),
and tumor cells with increased expression of MTA1 indicate more
invasive phenotypes (32). Negative
feedback regulation is crucial for cells to determine their fate
and maintain function during gene regulation. Our other study
(unpublished data) provided information concerning the regulation
of E-cadherin expression by MTA1 when controlling malignant
phenotypes in prostate cancer. Whether E-cadherin has an effect on
MTA1 expression has not yet been elucidated.
In the present study, we examined whether the
expression of MTA1 is an important contributing factor to the
metastasis induced by E-cadherin loss in vitro. In addition,
we investigated the correlation between E-cadherin and MTA1
expression and location in prostate cancer and metastatic prostate
cancer tissue samples. We identified that loss of E-cadherin
expression changes the malignant phenotype of prostate cancer cells
through an MTA1-dependent pathway.
Materials and methods
Reagents and antibodies
All reagents were of analytical grade and
commercially purchased. Primary antibodies against MTA1 were from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). E-cadherin
was obtained from Epitomics Inc. (Burlingame, CA, USA). α-tublin,
β-actin and alkaline phosphatase-conjugated anti-rabbit/mouse/goat
IgGs were purchased from Sigma (Deisenhofen, Germany).
FITC/Cy3-conjugated IgG was obtained from Proteintech Group Inc.
(Chicago, IL, USA). 4,6-Diamino-2-pheylindoledi (DAPI), fibronectin
(FN) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) were purchased from Sigma. The SP histostain-plus kit
was obtained from ZhongShan Biotech Co. (Beijing, China). All other
chemicals were of analytical grade and obtained from standard
suppliers.
Cell lines
The human paired poorly-metastatic prostate
adenocarcinoma cell lines PC-3M-2B4 (2B4) and highly-metastatic
prostate adenocarcinoma cell lines PC-3M-1E8 (1E8) were kindly
provided by Dr Jie Zheng (Beijing University, Beijing, China)
(33). All cells were cultured in
RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
with 10% (v/v) fetal bovine serum (FBS; Sijiqing Co., Hangzhou,
China), 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C and
5% CO2 using a humidified incubator (Heraeus, Hanau,
Germany).
Western blot analysis
For western blot analysis, cells were lysed in RIPA
buffer containing 50 mM Tris/HCl (pH 7.2), 150 mM NaCl, 1% NP-40,
0.1% SDS and 0.5% (w/v) sodium deoxycholate. Equivalent amounts of
cell extracts (150 μg) were separated on 10% SDS-polyacrylamide gel
(SDS-PAGE) and transferred onto a polyvinylidene difluoride
membrane (PVDF). Filters were blocked in 25 mM Tris (pH 8.0)
containing 125 mM NaCl, 0.1% Tween 20 and 5% skimmed milk for 1 h,
and then incubated with the diluted primary antibody (MTA1, 1:200;
E-cadherin, 1:2000; β-actin, 1:1000) at 4°C overnight. After
incubation, the secondary antibodies were added (1:1000 dilution).
The immunoreactive bands were visualized with alkaline phosphatase
and 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium
(BCIP/NBT).
Cell transfection
Cell lines were cultured in 6-well tissue culture
plates and flasks at 37°C and 5% CO2 using a humidified
incubator (Heraeus). Cells were transfected with 200 pmol/ml of
siRNA duplex using 5 μl Lipofectamine 2000 when cells were 20%
confluent following the manufacturer’s instructions of
Lipofectamine™ 2000 (Invitrogen Life Technologies) siRNA treatment.
The controls included untransfected cells and transfected cells
with a scrambled negative control siRNA (Ruibo Biotechnology Co.,
Guangzhou, China). For plasmid transfection, 4×105
cells/6-well plate were prepared using 4 μg of plasmid and 10 μl
Lipofectamine 2000 per well. The full length PCMV-SPORT6 E-cadherin
plasmid was purchased from Yrbio (Hunan, China). It was
reconstructed to pcDNA3.1 E-cadherin in our laboratory (Cancer
Biology Medical Center, Tongji Hospital) and the empty vector
pcDNA3.1 was preserved. Cells were harvested after 48 h and protein
levels were measured using western blotting. E-cadherin siRNA
sequences were as follows: forward, 5′-CAGACAAAGACCAGGACU-AdTdT-3′;
reverse, 3′-dTdT GUCUGUUUCUGGUCCUGAU-5′.
Wound healing assay
Exponentially growing cells were detached from
culture plates by trypsinization and seeded into 6-well plates
(1×105 cells/well in complete media) with 10% FBS in
DMEM. The plates were maintained at 37°C in 5% CO2 at
saturated humidity in an incubator overnight. The following day,
the confluent cell monolayers were converged and scrape-wounded
using a micropipette tip. Floating cells were removed after three
washes with serum-free medium, while scratched cells were observed
under an inverted phase microscope to identify the scratch widths.
Cells were cultured in serum-free culture media and images were
captured under phase contrast microscopy every 12 h after wounding.
For evaluation of ‘wound closure’, five randomly selected points
along each wound were marked, and the horizontal distance of the
migrating cells from the initial wound was measured. Each
experiment was performed twice or in triplicate. Data collected
were as the mean ± SD. The measurements were obtained by measuring
the area of the wound using Image J software (available from the
National Institute of Health website at http://imagej.nih.gov/ij/download/).
Invasion assay
Transwell chambers with polycarbonate membrane
filters with 24-well inserts, 6.5 mm diameter and 8 μm pore size
(Corning Life Sciences, Corning, NY, USA) were coated with 1.5
mg/ml Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA). The
lower chamber was filled with 600 μl RPMI-1640 containing 20% FBS
and mouse embryonic fibroblast cell line (NIH 3T3) supernatant,
acting as a chemotactic factor (CF), or just DMEM with 1% fetal
calf serum (FCS). A total of 1×104 cells/200 μl were
seeded into the upper compartment and incubated for 48 h at 37°C
and 5% CO2. Following removal of the non-migratory cells
from the upper surface of the filter, invasive cells that had
migrated through to the lower surface of the filter were fixed in
2.5% (v/v) glutaraldehyde for 15 min and stained with crystal
violet for 10 min. The number of invasive cells were calculated by
counting at least three randomly chosen visual fields at 100-fold
magnification (Leica, Solms, Germany). Three independent
experiments were conducted for each set.
Solid-phase adhesion assay
Adhesion was determined using an MTT assay.
Exponentially growing cells were detached from culture plates by
trypsinization. After washing, cells were resuspended in serum-free
medium. Equal numbers of cells (4×104 cells/well) were
seeded into 96-well plates precoated with 1 μg/ml FN (Sigma), which
proved to be the most evident in pre-experiments (data not shown).
All FN-coated wells were compared with cells seeded in 1% (w/v)
bovine serum albumin (BSA). After 1 h of incubation at 37°C, the
plate was immersed in PBS containing 1 mmol/l MgCl2 to
remove non-adherent cells. The adhered cells were then measured
using an MTT assay at 490 nm wavelength. Subsequently, 30 μl of MTT
(5 mg/ml) was added 4 h prior to the end of the incubation period.
Following incubation, the samples were centrifuged at 2200 × g for
5 min. Once the supernatant was removed, 150 μl of dimethyl
sulphoxide (DMSO) was added to resolve the crystals and the optical
density (OD) values of each well was measured at 490 nm with a
microplate reader after 15 min. The OD values reflect the
proportion of cells adhered to the FN-coated 96-well plate. The
rate of adhesion was calculated using the following equation: (the
OD value of test/the OD value of control) × 100. All experiments
were conducted four times and repeated twice.
Confocal microscopy imaging
For immunofluorescence cytoskeleton staining,
1×104 cells were seeded on glass coverslips (13 mm
diameter). Once the coverslips were fixated with 4%
paraformaldehyde (Merck, Haar, Germany) in PBS, cell membranes were
permeabilized with 0.2% (v/v) Triton X-100 in PBS, and non-specific
binding sites were blocked with 5% (w/v) BSA in PBS for 30 min at
37°C. Treatment with the first antibody (α-tublin, 1:50) was
conducted at 4°C overnight. Subsequently, the cells were washed
three times with cold PBS and incubated with Cy3-conjugated IgG
(1:50 dilution) in PBS for 30 min at room temperature. Nuclei were
stained for 5 min at room temperature with DAPI (2 μg/ml methanol).
Cells were then rinsed with PBS and were observed under confocal
microscopy (Olympus, Tokyo, Japan).
Immunohistochemistry
Tissues samples of normal prostate as well as
localized and metastatic prostate cancer were obtained from the
Department of Pathology at Tongji Hospital Affiliated to Huazhong
University of Science and Technology (Hubei, China).
Immunohistochemical staining for E-cadherin and MTA1 expression
were conducted following standard procedures. The 5 μm paraffin
sections cut on poly-L-lysine-coated microscopy slides were
deparaffinized and rehydrated in graded alcohol. The sections were
heated in 0.01 M citrate buffer (pH 6.0) in a 85–95°C microwave
oven (3 times for 5 min each) and the non-specific binding sites
were blocked with goat serum. Sections were incubated overnight at
4°C with rabbit monoclonal anti-human E-cadherin (1:500 dilution)
and goat polyclonal anti-human MTA1 (1:100 dilution) and then
washed with PBS. Biotinylated goat anti-rabbit immunoglobulin
(Dako, Kyoto, Japan) was then added to the sections for 30 min at
room temperature. Peroxidase-conjugated avidin (Dako) was applied
once the sections had been washed with PBS. Peroxidase activity was
detected by exposure of the sections to the solution of 0.05%
3,3′-diaminobenzidine (DAB) and 0.01% H2O2 in
Tris-HCl buffer (DAB solution) for 3–6 min at room temperature.
Finally, the slides were counterstained with hematoxylin. Negative
control slides were processed similarly, but were incubated with
PBS instead of primary antibody.
Statistical analysis
Data were analyzed using the ANOVA test. Statistical
analysis was conducted using SPSS version 13.0 software. P<0.05
was considered to indicate a statistically significant difference.
The correlation between the indicators was analyzed using Spearman
correlation analysis.
Results
Silencing E-cadherin upregulates MTA1
expression
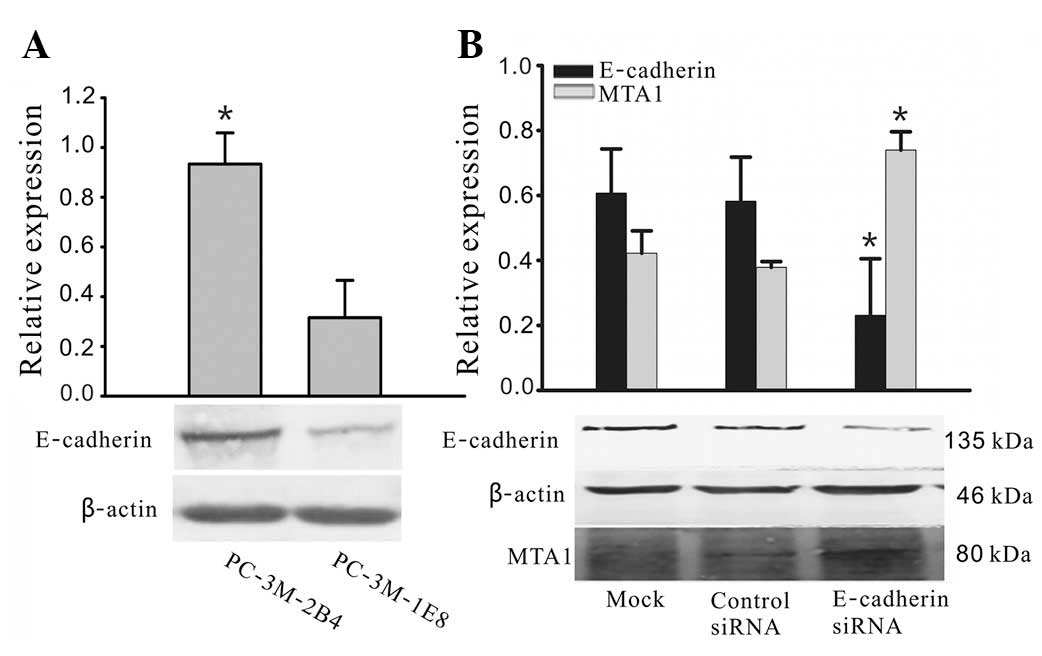
As shown in Fig. 1A,
E-cadherin was present in two prostate cancer cell lines, but at
different expression levels. The protein expression levels of
E-cadherin in 2B4 cells were over 2.5- to 3-fold higher compared
with those in 1E8 cells. Since the 2B4 cells expressed the highest
levels of endogenous E-cadherin, we selected this cell line to
investigate the effects of heterologous E-cadherin expression on
the cellular biological properties of prostate carcinoma cells
in vitro. Western blot analysis demonstrated that E-cadherin
siRNA is able to specifically downregulate E-cadherin expression
and significantly increase MTA1 expression (Fig. 1B).
Silencing E-cadherin induces migration
ability of 2B4 cells
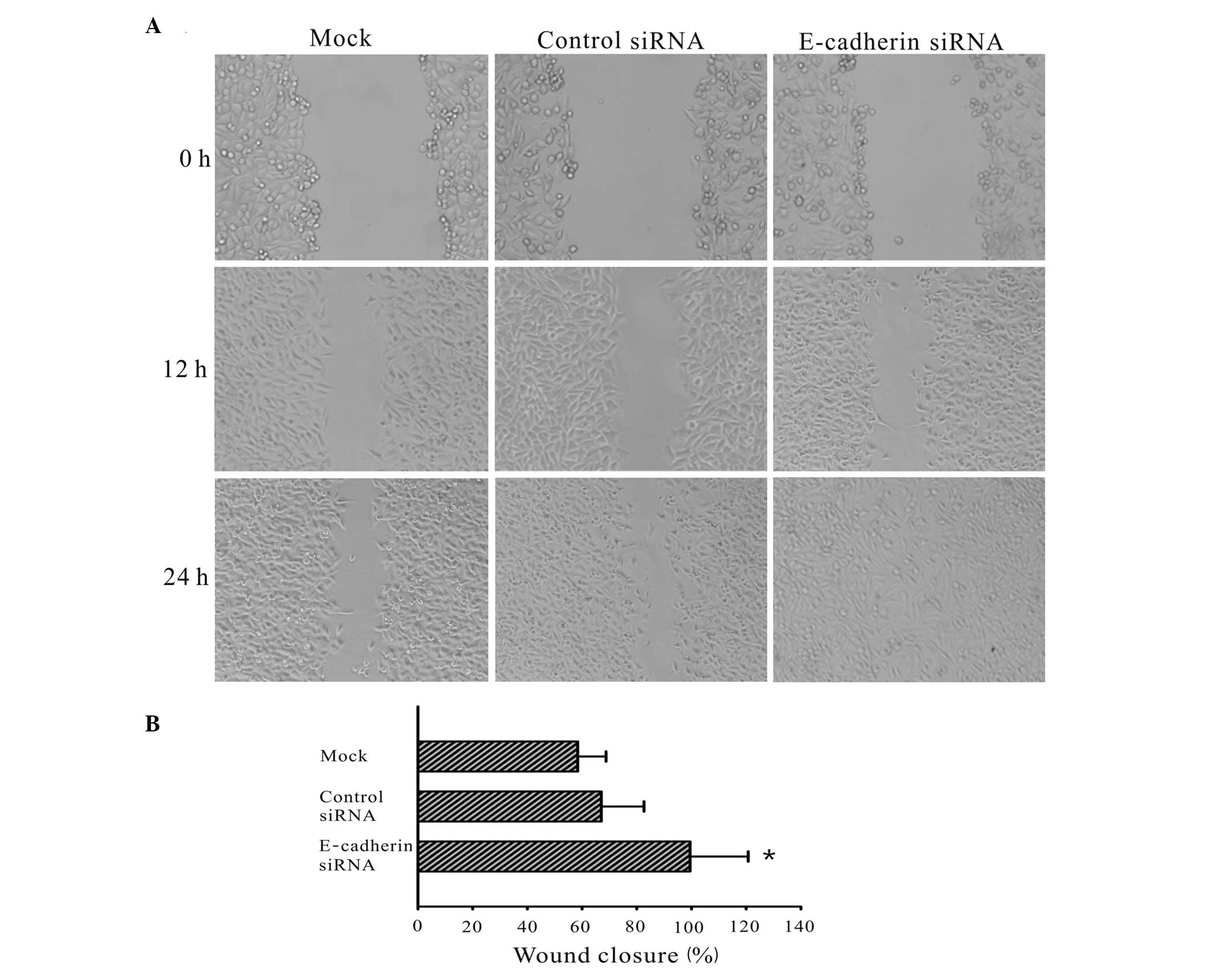
To determine whether E-cadherin affects the
migration ability of 2B4 cells, a wound healing assay was
conducted. The wound healing ability of cells reflects their
movement and migration on the surface on which they are anchored to
for growth. At 0, 12 and 24 h after wounding, transfected
E-cadherin siRNA cells demonstrated an increased level of wound
healing compared with the mock and control siRNA cells (Fig. 2). At 24 h after wounding, the wound
of the E-cadherin siRNA transfected group had healed, while that of
the cells from the control siRNA and mock groups had not closed
(P<0.01 compared with the control cells).
Silencing E-cadherin alters the malignant
phenotype of 2B4 cells
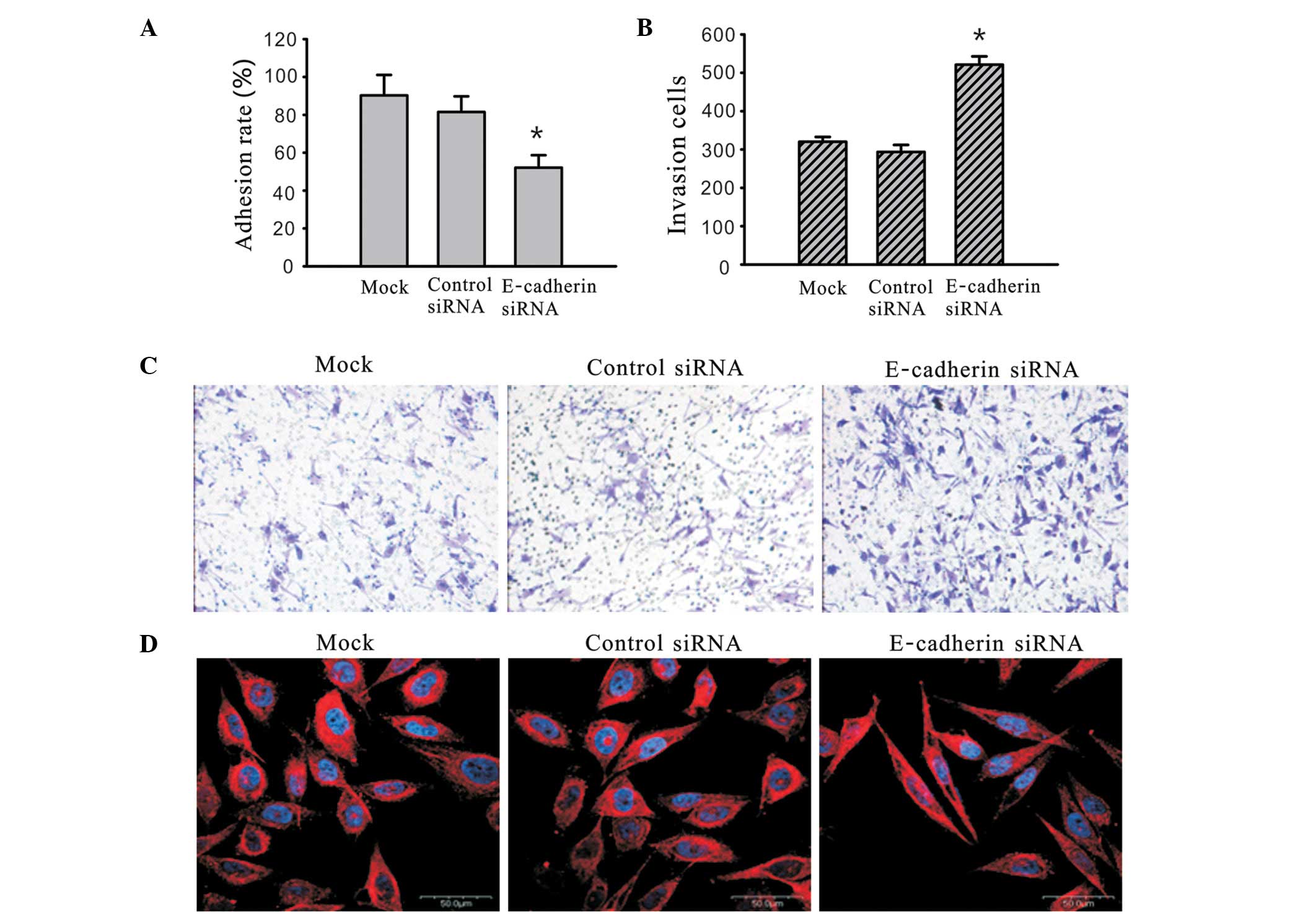
The 2B4 cells were transfected with siRNA against
E-cadherin for 48 h and a solid-phase adhesion assay was used to
detect the alteration in cell adhesion ability. For the cells with
a similar concentration of surface-coated FN, silencing of
E-cadherin by siRNA significantly inhibited the adhesion process
compared with the mock and control siRNA treated cells (Fig. 3A). 2B4 cells were transfected with
siRNA against E-cadherin for 48 h and a Matrigel-coated transwell
model was used to detect the alteration in cell invasion ability.
The number of cells at the lower side of the membrane reflects the
invasion ability of the cells being investigated. The number of
cells that invaded to the lower side of the membrane were
320.22±12.50, 293.56±18.32 and 521.13±21.67 for the mock, control
siRNA- treated and E-cadherin-siRNA-treated cells, respectively.
Compared with the mock and control-siRNA treated group, the
invasion ability of the cells treated with E-cadherin siRNA was
significantly greater (Fig. 3B and =
C). Each assay was repeated three times. The decreased adhesion
ability as well as an increased invasion and migration ability,
influences a change in the architecture and composition of the
cytoskeleton. To examine whether the inhibition of E-cadherin
expression affects the subcellular distribution and organization of
the cytokeratin filaments, immunofluorescence analyses were
conducted using E-cadherin siRNA-transfected, control-transfected
siRNA and mock cells. In the E-cadherin siRNA-transfected cells we
observed an increased elongated spindle-shape morphology with
extended pseudopodia between adjacent cells and an increase in
cellular polarity strength compared with the control siRNA and mock
cells (Fig. 3D). These results
suggest that E-cadherin plays a key role in controling the
malignant phenotypes of 2B4 cells, including the adhesion ability,
invasion ability and cytoskeleton organization.
E-cadherin regulates MTA1 expression
The 2B4 cells were treated with E-cadherin siRNA and
full length E-cadherin plasmid for various time periods between 24
and 48 h, and western blot analyis was used to detect E-cadherin
and MTA1 protein expression. As demonstrated in Fig. 4A, 48 h after treatment with
E-cadherin siRNA, E-cadherin expression was significantly
downregulated, while MTA1 expression was significantly upregulated.
Additionally, following empty vector pCMV-SPORT6 transfection, MTA1
expression was significantly downregulated and E-cadherin was
significantly upregulated 48 h after treatment (Fig. 4B). All experiments were repeated
three times.
Orientation of E-cadherin and MTA1
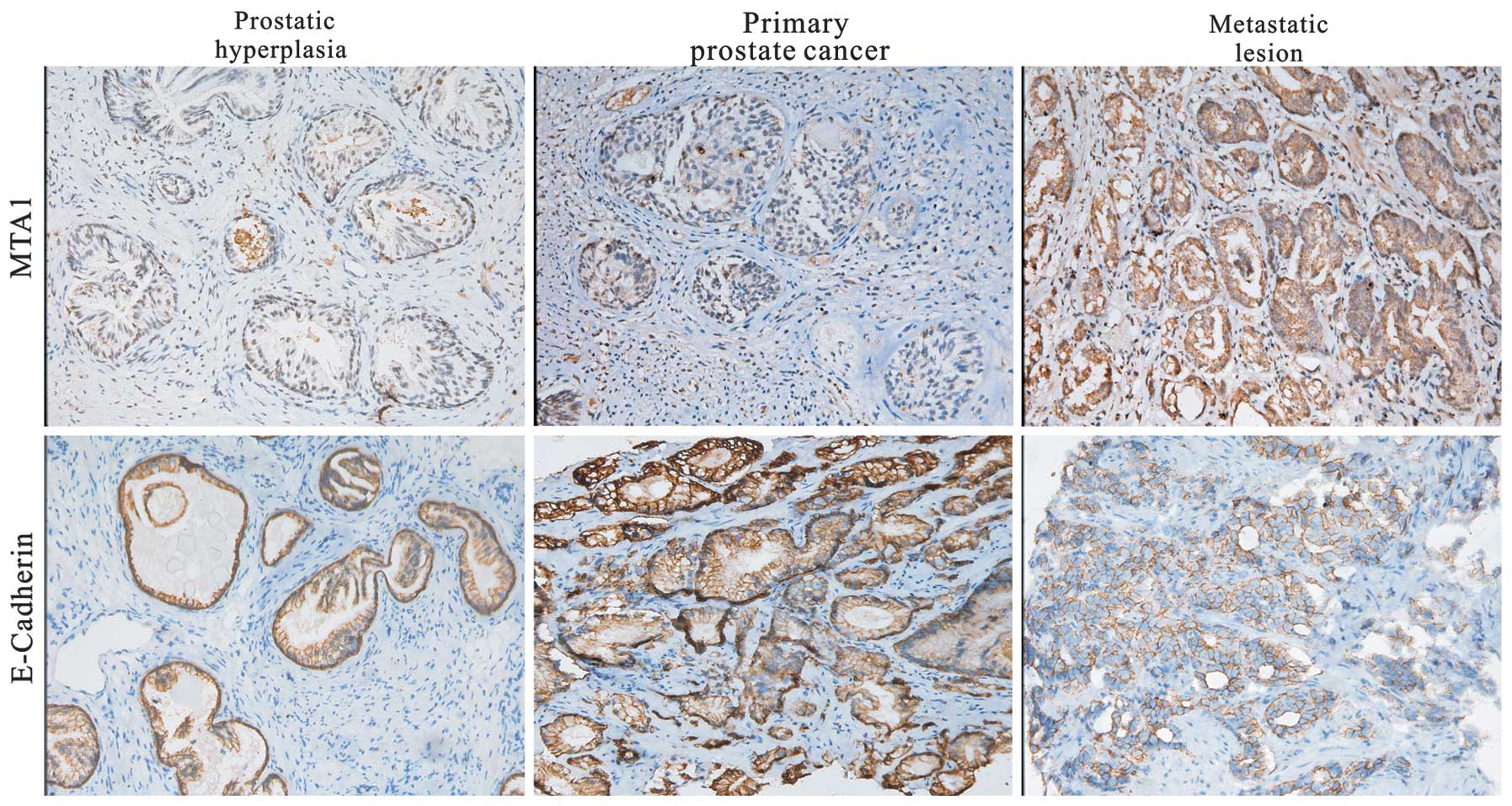
protein in prostate tissue
E-cadherin expression was detected predominantly in
the cytoplasm and membrane of the prostate cancer tissues. MTA1
protein positive signaling was detected in the cytoplasm or nucleus
of cancer cells. A representative result of the
immunohistochemistry for MTA1 and E-cadherin in prostate cancer is
shown in Fig. 5. The samples probed
with E-cadherin antibody demonstrated 2+ or 3+ membrane staining of
the epithelial cells. Cytoplasmic staining was 0− or 1+. The
positive staining in the prostate lesions had distinct
circumferential E-cadherin immunoreactivity of high intensity,
which revealed that E-cadherin is a cell membrane protein.
Correlation between expression of
E-cadherin and MTA1 in prostate carcinoma tissue
To further assess whether E-cadherin expression
levels correlate with the expression of MTA1 in prostate cancer
metastasis, we analyzed the results for E-cadherin and MTA1
immunoreactivity. Intact E-cadherin immunostaining was observed in
70% (14/20) of benign prostatic hyperplasia (BPH), 53.6% (15/28) of
primary cancer and 16.7% (2/12) of metastatic lesion tissues. MTA1
expression levels in BPH, primary cancer and metastatic lesion
tissues were 30 (6/20), 75 (21/28) and 25% (3/12), respectively.
E-cadherin expression was decreased in the prostate cancer tissues
compared with the BPH tissues (P<0.001), while MTA expression
was increased in the prostate cancer tissues compared with the BPH
tissues (P<0.05; Table I).
Furthermore, we analyzed the correlation between the expression
levels of E-cadherin and MTA1 in the tumor group. As shown in
Table II, we observed a
statistically significant correlation between E-cadherin and MTA1
immunoreactivity in the prostate cancer group (P<0.05;
rs=−0.434, respectively).
 | Table I.Expression of E-cadherin and MTA1 in
prostate carcinoma tissue. |
Table I.
Expression of E-cadherin and MTA1 in
prostate carcinoma tissue.
| | E-cadherin
| | MTA1
| |
|---|
| Group | No. of cases | − | + | P-value | − | + | P-value |
|---|
| BPH | 20 | 6 | 14 | 0.044 | 14 | 6 | 0.028 |
| Tumor | 40 | 23 | 17 | | 16 | 24 | |
| Primary cancer | 28 | 13 | 15 | 0.030 | 7 | 21 | 0.003 |
| Metastatic
lesion | 12 | 10 | 2 | | 9 | 3 | |
| Table II.Correlation between
immunohistochemically detected expression of MTA1 and E-cadherin in
prostate cancer. |
Table II.
Correlation between
immunohistochemically detected expression of MTA1 and E-cadherin in
prostate cancer.
| MTA1
| | | |
|---|
| E-cadherin | − | + | Total | P-value | rs |
|---|
| − | 5 | 18 | 23 | 0.005 | −0.434 |
| + | 11 | 6 | 17 | | |
| Total | 16 | 24 | 40 | | |
Discussion
Cancer metastasis is a complex and multi-step
process that commonly leads to the mortality of cancer patients.
Cell adhesion molecules play an important role in the development
of recurrent, invasive and distant metastasis in the process of
cancer progression (6). E-cadherin
is a key cell-to-cell adhesion molecule, which plays a significant
role in the invasion and metastasis of tumor cells (10). In the majority of, if not all,
cancers of epithelial origin, the loss of cell-to-cell adhesion
mediated by E-cadherin may occur concomitantly with the progression
towards tumor malignancy (10).
The majority of previous studies indicate that
E-cadherin has a close correlation with metastasis and invasion of
a number of tumors, including ovarian (13), breast (34,35),
pancreatic (36), gastric (37) and prostate cancer (38,39).
With regard to prostate cancer, E-cadherin expression in the cancer
cells at metastasis in lymph node sites is lower than in the
primary prostate cancer (38).
Studies have suggested that prostate cancer cells with low
expression of E-cadherin are more invasive (40); therefore, the absence of E-cadherin
expression in prostate cancer predicts the potential of metastasis
to the bone compared with prostate cancer that does express
E-cadherin (41). A recent study
suggested that upregulating E-cadherin expression by valproic acid
inhibits prostate cancer cell migration (42). The effects of E-cadherin on the
malignant phenotype of prostate cancer cells and possible molecular
mechanisms are not entirely understood.
MTA1 is a highly expressed with high potential to
metastasize in a number of cancer types (22–24).
Earlier studies have demonstrated an inverse correlation between
the levels of MTA1 and E-cadherin, and identified that increased
MTA1 expression is associated with increased invasiveness and
reduced expression of E-cadherin in tumor cells (23,43).
However, studies with regard to the expression and correlation of
E-cadherin and MTA1 in prostate cancer have rarely been reported.
In this study, we demonstrate that the loss of E-cadherin is able
to induce metastasis of prostate cancer cells through upregulated
MTA1 expression.
Downregulated expression of E-cadherin in prostate
cancer and upregulated expression of MTA1 is consistent with
previous studies (38,41,44).
Therefore, it was hypothesized that there is a certain inherent
connection between E-cadherin and MTA1, and their coordination may
lead to tumor cell invasion and metastasis. E-cadherin may be
upregulated by MTA1 siRNA in melanoma cells, which was also
confirmed in our previous study in cervical cancer cells (29,30).
MTA1 overexpression resulted in the downregulation of E-cadherin
expression in ovarian cancer (23).
In the present study, E-cadherin protein was
markedly expressed in 2B4 (poorly-metastatic prostate
adenocarcinoma cell lines) cells and weakly expressed in 1E8
(highly-metastatic prostate adenocarcinoma cell lines) cells. To
clarify the molecular characteristics of E-cadherin, E-cadherin
siRNA was transfected into 2B4 cells for 48 h. Our results revealed
that MTA1 expression was increased when E-cadherin expression was
decreased. E-cadherin downregulation is one of the principal events
during EMT and a frequently reported phenomena in embryonic
development and cancer progression (45,46).
MTA1 has been suggested to determine EMT phenotype, mainly through
the downregulated expression of E-cadherin, which leads to EMT
(28). Developing cancer cells
acquire migratory features concomitant with a loss of E-cadherin
expression during carcinogenesis (13,47).
We identified that downregulation of E-cadherin expression resulted
in the promotion of 2B4 cell migration and invasion, and the
inhibition of adhesion capability via upregulated MTA1 in
vitro.
The increased adhesion ability and increased
invasion and migration ability is accompanied by a change in the
architecture and composition of the cytoskeleton. EMT is a key step
toward cancer metastasis, and E-cadherin is regarded as a main
indicator of the epithelial/mesenchymal phenotype switching
(19). E-cadherin loss is
suggestive of EMT, and tumor cell invasion and metastasis are
associated with EMT (10,48). Cell changes, including morphological
and gene expression alterations, are necessary for EMT (49,50).
In the present study, cells acquired an elongated spindle-shape
morphology with extended pseudopodia between adjacent cells due to
a decrease in E-cadherin and an increase in cellular polarity
strength. Our results indicate that E-cadherin may play an
important role in the cellular polarity of prostate cancer cells.
We identified that the protein level of MTA1 was upregulated when
E-cadherin was decreased. We also identified that expression of
E-cadherin regulates MTA1 expression in 2B4 cells treated with
E-cadherin siRNA or full length E-cadherin plasmid at different
times (24–48 h). Our data suggest that E-cadherin regulates MTA1 in
a time-dependent manner.
To further investigate the expression levels of and
correlation between E-cadherin and MTA1 in prostate cancer, we
examined BPH, carcinoma in situ and metastatic carcinoma
tissues using immunohistochemical staining. Our results
demonstrated that there is an inverse correlation between protein
expression of E-cadherin and MTA1 in prostate cancer. Loss of
E-cadherin expression in 57.5% of prostate cancer tissues in
association with the increase of MTA expression in 60% of tissues,
suggests that the two proteins are closely related to prostate
cancer progression. This result suggests the possibility that
E-cadherin and MTA1 act together as prognosis predictors of
metastasis and progression of prostate cancer. A combined testing
strategy for detecting MTA1 and E-cadherin may be sufficient for
selecting high-risk patients with metastasis.
We revealed that loss of E-cadherin-induced MTA1
expression in prostate cancer 2B4 cells promotes the migration and
invasion of 2B4 cells. Our results provide a new insight into the
mechanisms of E-cadherin regulation of MTA1 in prostate cancer, and
suggests that E-cadherin may combine with MTA1 and alter the
malignant phenotype of prostate cancer. A combined testing strategy
for detecting MTA1 and E-cadherin may be sufficient for selecting
high-risk patients with metastasis. E-cadherin and MTA1 may be
potential powerful targets for the treatment of various types of
cancer.
Acknowledgements
This study was supported by grants
from the Major State Basic Research Development Program of China
(973 Program, 2009; No. CB521808) and the National Science
Foundation of China (Nos. 30700895, 30770913, 81001006, 30901587
and 81101971).
References
|
1.
|
R SiegelE WardO BrawleyA JemalCancer
statistics, 2011: the impact of eliminating socioeconomic and
racial disparities on premature cancer deathsCA Cancer J
Clin61212236201110.3322/caac.2012121685461
|
|
2.
|
P BoyleJ FerlayCancer incidence and
mortality in Europe, 2004Ann
Oncol16481488200510.1093/annonc/mdi09815718248
|
|
3.
|
GR MundyMetastasis to bone: causes,
consequences and therapeutic opportunitiesNat Rev
Cancer2584593200210.1038/nrc86712154351
|
|
4.
|
CJ LogothetisSH LinOsteoblasts in prostate
cancer metastasis to boneNat Rev
Cancer52128200510.1038/nrc152815630412
|
|
5.
|
SA EcclesDR WelchMetastasis: recent
discoveries and novel treatment
strategiesLancet36917421757200710.1016/S0140-6736(07)60781-817512859
|
|
6.
|
T OkegawaRC PongY LiJT HsiehThe role of
cell adhesion molecule in cancer progression and its application in
cancer therapyActa Biochim Pol51445457200415218541
|
|
7.
|
T BogenriederM HerlynAxis of evil:
molecular mechanisms of cancer
metastasisOncogene2265246536200310.1038/sj.onc.120675714528277
|
|
8.
|
U CavallaroE DejanaAdhesion molecule
signalling: not always a sticky businessNat Rev Mol Cell
Biol12189197201110.1038/nrm306821346732
|
|
9.
|
MA HuberN KrautH BeugMolecular
requirements for epithelial-mesenchymal transition during tumor
progressionCurr Opin Cell
Biol17548558200510.1016/j.ceb.2005.08.00116098727
|
|
10.
|
U CavallaroG ChristoforiCell adhesion and
signalling by cadherins and Ig-CAMs in cancerNat Rev
Cancer4118132200410.1038/nrc127614964308
|
|
11.
|
D HanahanRA WeinbergHallmarks of cancer:
the next
generationCell144646674201110.1016/j.cell.2011.02.01321376230
|
|
12.
|
G BerxF van RoyInvolvement of members of
the cadherin superfamily in cancerCold Spring Harb Perspect
Biol1a003129200910.1101/cshperspect.a00312920457567
|
|
13.
|
K SawadaAK MitraAR RadjabiLoss of
E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin,
which is a therapeutic targetCancer
Res6823292339200810.1158/0008-5472.CAN-07-516718381440
|
|
14.
|
TT OnderPB GuptaSA ManiJ YangES LanderRA
WeinbergLoss of E-cadherin promotes metastasis via multiple
downstream transcriptional pathwaysCancer
Res6836453654200810.1158/0008-5472.CAN-07-293818483246
|
|
15.
|
A SinghJ SettlemanEMT, cancer stem cells
and drug resistance: an emerging axis of evil in the war on
cancerOncogene2947414751
|
|
16.
|
M YilmazG ChristoforiEMT, the
cytoskeleton, and cancer cell invasionCancer Metastasis
Rev281533200910.1007/s10555-008-9169-019169796
|
|
17.
|
G Moreno-BuenoF PortilloA
CanoTranscriptional regulation of cell polarity in EMT and
cancerOncogene2769586969200810.1038/onc.2008.34619029937
|
|
18.
|
K GravdalOJ HalvorsenSA HaukaasLA AkslenA
switch from E-cadherin to N-cadherin expression indicates
epithelial to mesenchymal transition and is of strong and
independent importance for the progress of prostate cancerClin
Cancer Res1370037011200710.1158/1078-0432.CCR-07-1263
|
|
19.
|
YN LiuY LiuHJ LeeYH HsuJH ChenActivated
androgen receptor downregulates E-cadherin gene expression and
promotes tumor metastasisMol Cell
Biol2870967108200810.1128/MCB.00449-0818794357
|
|
20.
|
Y TohSD PencilGL NicolsonA novel candidate
metastasis-associated gene, mta1, differentially expressed in
highly metastatic mammary adenocarcinoma cell lines. cDNA cloning,
expression, and protein analysesJ Biol Chem26922958229631994
|
|
21.
|
B ManavathiS PengSK RayalaRepression of
Six3 by a corepressor regulates rhodopsin expressionProc Natl Acad
Sci USA1041312813133200710.1073/pnas.070587810417666527
|
|
22.
|
H QianN LuL XueReduced MTA1 expression by
RNAi inhibits in vitro invasion and migration of esophageal
squamous cell carcinoma cell lineClin Exp
Metastasis22653662200510.1007/s10585-006-9005-216703414
|
|
23.
|
C DannenmannN ShabaniK FrieseU JeschkeI
MylonasA BruningThe metastasis-associated gene MTA1 is upregulated
in advanced ovarian cancer, represses ERbeta, and enhances
expression of oncogenic cytokine GROCancer Biol
Ther714601467200810.4161/cbt.7.9.642718719363
|
|
24.
|
SH RyuYH ChungH LeeMetastatic tumor
antigen 1 is closely associated with frequent postoperative
recurrence and poor survival in patients with hepatocellular
carcinomaHepatology47929936200810.1002/hep.2212418306220
|
|
25.
|
GL NicolsonA NawaY TohS TaniguchiK
NishimoriA MoustafaTumor metastasis-associated human MTA1 gene and
its MTA1 protein product: role in epithelial cancer cell invasion,
proliferation and nuclear regulationClin Exp
Metastasis201924200310.1023/A:1022534217769
|
|
26.
|
Y TohGL NicolsonThe role of the MTA family
and their encoded proteins in human cancers: molecular functions
and clinical implicationsClin Exp
Metastasis26215227200910.1007/s10585-008-9233-819116762
|
|
27.
|
SB PakalaK SinghSD ReddyTGF-β1 signaling
targets metastasis-associated protein 1, a new effector in
epithelial cellsOncogene3022302241
|
|
28.
|
E RadaelliP DamonteRD
CardiffEpithelial-mesenchymal transition in mouse mammary
tumorigenesisFuture Oncol511131127200910.2217/fon.09.9319852725
|
|
29.
|
H QianJ YuY LiRNA interference of
metastasis-associated gene 1 inhibits metastasis of B16F10 melanoma
cells in a C57BL/6 mouse modelBiol
Cell99573581200710.1042/BC2006013017868030
|
|
30.
|
Y RaoH WangL FanG ChenSilencing MTA1 by
RNAi reverses adhesion, migration and invasiveness of cervical
cancer cells (SiHa) via altered expression of p53, and
E-cadherin/β-catenin complexJ Huazhong Univ Sci Technolog Med
Sci3119201121336715
|
|
31.
|
NC HuntAG Douglas-JonesB JasaniJM MorganM
PignatelliLoss of E-cadherin expression associated with lymph node
metastases in small breast carcinomasVirchows
Arch430285289199710.1007/BF010927519134039
|
|
32.
|
B ManavathiR KumarMetastasis tumor
antigens, an emerging family of multifaceted master coregulatorsJ
Biol Chem28215291533200710.1074/jbc.R60002920017142453
|
|
33.
|
Y LiuJ ZhengW FangIsolation and
characterization of human prostate cancer cell subclones with
different metastatic potentialZhonghua Bing Li Xue Za
Zhi283613641999(In Chinese)
|
|
34.
|
YL ChaoCR ShepardA WellsBreast carcinoma
cells re-express E-cadherin during mesenchymal to epithelial
reverting transitionMol
Cancer9179201010.1186/1476-4598-9-17920609236
|
|
35.
|
PJ KowalskiMA RubinCG KleerE-cadherin
expression in primary carcinomas of the breast and its distant
metastasesBreast Cancer Res5R217R222200310.1186/bcr65114580257
|
|
36.
|
J von BurstinS EserMC PaulE-cadherin
regulates metastasis of pancreatic cancer in vivo and is suppressed
by a SNAIL/HDAC1/HDAC2 repressor complexGastroent
erology137361371200919362090
|
|
37.
|
B TangZH PengPW YuG YuF QianExpression and
significance of Cx43 and E-cadherin in gastric cancer and
metastatic lymph nodesMed
Oncol28502508201110.1007/s12032-010-9492-520373058
|
|
38.
|
L ChengM NagabhushanTP PretlowSB AminiTG
PretlowExpression of E-cadherin in primary and metastatic prostate
cancerAm J Pathol1481375138019968623909
|
|
39.
|
MA RubinNR MucciJ FigurskiA FeckoKJ
PientaML DayE-cadherin expression in prostate cancer: a broad
survey using high-density tissue microarray technologyHum
Pathol32690697200110.1053/hupa.2001.25902
|
|
40.
|
Q MaoX ZhengK YangSuppression of migration
and invasion of PC3 prostate cancer cell line via activating
E-cadherin expression by small activating RNACancer
Invest2810131018201010.3109/0735790080262084420690797
|
|
41.
|
J Pontes JrM SrougiPM BorraMF Dall’
OglioLA Ribeiro-FilhoKR LeiteE-cadherin and beta-catenin loss of
expression related to bone metastasis in prostate cancerAppl
Immunohistochem Mol
Morphol18179184201010.1097/PAI.0b013e3181640bca18685493
|
|
42.
|
L ZhangG WangL WangC SongX WangJ
KangValproic acid inhibits prostate cancer cell migration by
up-regulating E-cadherin
expressionPharmazie66614618201121901986
|
|
43.
|
H ZhangLC StephensR KumarMetastasis tumor
antigen family proteins during breast cancer progression and
metastasis in a reliable mouse model for human breast cancerClin
Cancer Res1214791486200610.1158/1078-0432.CCR-05-1519
|
|
44.
|
MD HoferR KueferS VaramballyThe role of
metastasis-associated protein 1 in prostate cancer
progressionCancer
Res64825829200410.1158/0008-5472.CAN-03-275514871807
|
|
45.
|
JP ThieryEpithelial-mesenchymal
transitions in tumour progressionNat Rev
Cancer2442454200210.1038/nrc82212189386
|
|
46.
|
K PolyakRA WeinbergTransitions between
epithelial and mesenchymal states: acquisition of malignant and
stem cell traitsNat Rev
Cancer9265273200910.1038/nrc262019262571
|
|
47.
|
PJ RichmondAJ KarayiannakisA NagafuchiAV
KaisaryM PignatelliAberrant E-cadherin and alpha-catenin expression
in prostate cancer: correlation with patient survivalCancer
Res573189319319979242448
|
|
48.
|
L Di CrocePG PelicciTumour-associated
hypermethylation: silencing E-cadherin expression enhances invasion
and metastasisEur J Cancer39413414200312751369
|
|
49.
|
M MaedaKR JohnsonMJ WheelockCadherin
switching: essential for behavioral but not morphological changes
during an epithelium-to-mesenchyme transitionJ Cell
Sci118873887200510.1242/jcs.0163415713751
|
|
50.
|
G ChristoforiNew signals from the invasive
frontNature441444450200610.1038/nature0487216724056
|