Introduction
A dysembryoplastic neuroepithelial tumor (DNT) is
widely recognized as a benign lesion and is classified as a
neuronal and mixed neuronal-glial tumor, corresponding to WHO Grade
I (1). DNTs have been shown to
correlate with intractable epilepsy and are usually located in the
supratentorial cortex (2–4).
DNTs are considered curable with surgery alone,
without the use of adjuvant therapy (5–7).
However, studies have indicated certain instances of tumor
recurrence, with the majority of tumors recurring secondary to an
initial subtotal resection (STR) or partial resection surgery
(8–10). Certain cases have been shown to
exhibit recurrence following a gross total resection (GTR)
(11–14). Rare cases have reported tumors that
progressed to high-grade astrocytomas (8,9,14–17),
certain cases of which may have been the result of inappropriate
post-operative radiotherapy (14–15).
The present study reports a histological evolution DNT case. The
patient underwent a standard anterior temporal lobectomy in the
Department of Neurosurgery, Nan Fang Hospital (Guangzhou, China)
and a microscopic pathological evaluation demonstrated that the
lesion was a DNT. The patient was administered adjuvant
chemotherapy in another hospital at one month post-surgery. The
tumor recurred in situ five years after the initial surgery.
Microscopic pathological evaluation disclosed fibrillary
astrocytoma (WHO grade II) as the predominant component of the
recurrent tumor. Written informed consent was obtained from the
patient.
Case report
In October 2006, a 15-year old female with a
three-week history of partial complex seizures and an unusual
saline taste as the aura before seizure attack was admitted to the
Department of Neurosurgery, Nan Fang Hospital. Magnetic resonance
imaging (MRI) revealed a well-defined lesion in the right temporal
lobe that was 3.1×4.3×6.4 cm in size. No obvious peritumoral edema
or mass effect was observed. T1 weighted imaging (WI) revealed the
lesion to be hypointense (Fig. 1A),
while T2WI showed it to be hyperintense (Fig. 1B). Fluid-attenuated inversion
recovery (FLAIR) sequence imaging displayed the lesion as
heterogeneously hyperintense (Fig.
1C). Contrast-enhanced imaging revealed a patchy enhancement of
the tumor (Fig. 1D). During the
interictal period, a scalp EEG demonstrated α-waves with a
frequency of 9–10 Hz and an amplitude of 40–100 μV, mainly as the
background rhythm. A distribution of high amplitude sharp waves,
spikes and sharp-slow waves were observed in the lesion area.
The tumor was located in the cortex with a dim
appearance and moderate blood supply. A clear boundary and no
capsule was observed, as determined by pre-operative MRI. The
lesion, ipsilateral anterior temporal lobe, hippocampus and
amygdala were removed by a standard right anterior temporal
lobectomy and a GTR was achieved (Fig.
2).
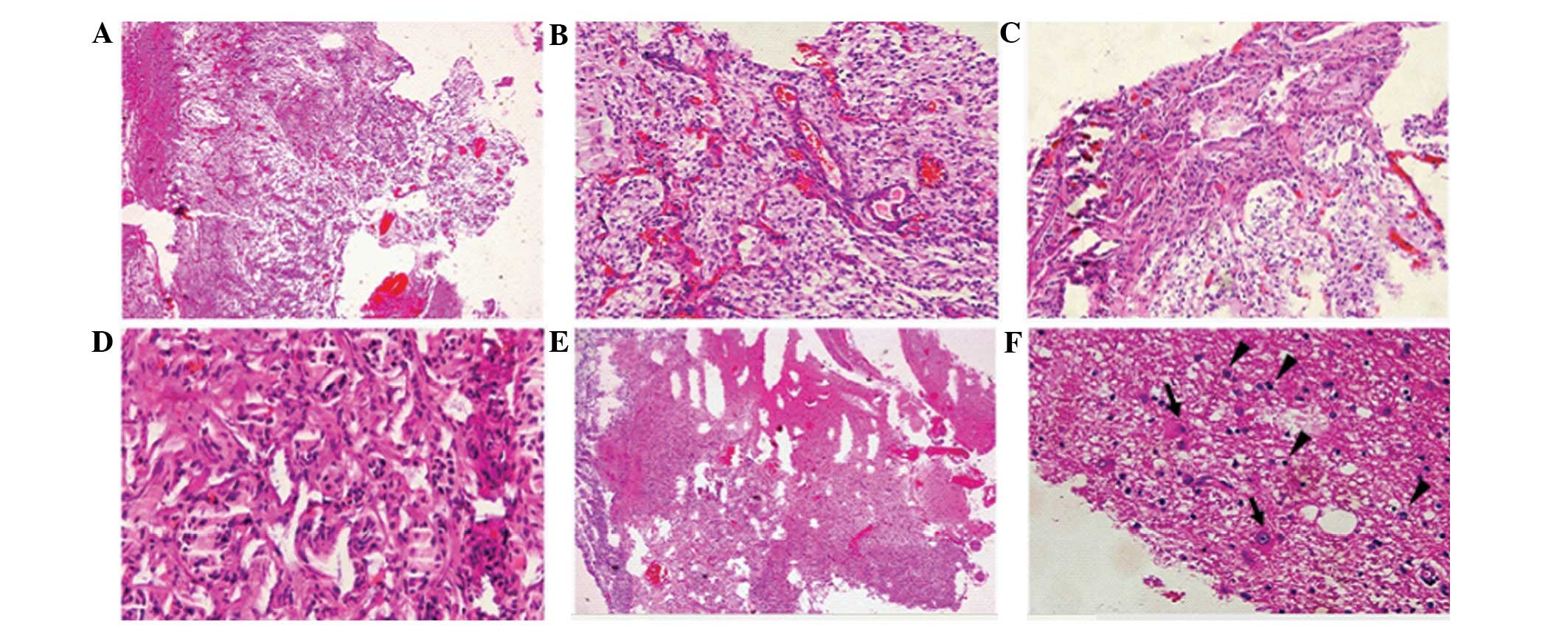
The microscopic evaluation revealed a typical form
of DNT (Fig. 3A and B), which was
composed of a specific glioneuronal element with floating neurons
within small mucoid lakes (Fig.
3C). Glial cell proliferation was notable, including numerous
oligodendrocyte-like cells and fewer astrocytes. The
oligodendrocyte-like cells shared the same appearance in a diffuse
manner with rare mitotic figures. In addition, focal cortical
dysplasia (FCD) was identified in the peritumoral cortex (Fig. 3D). The immunohistochemical results
demonstrated that glial fibrillary acidic protein (GFAP) and S-100
were positive in the astrocytes and oligodendrocyte-like cells
(Fig. 3E and F). Oligo-2 was
positive in the majority of the oligodendrocyte-like cells
(Fig. 3G). The immature neurons
were positive for synaptophysin (Syn; Fig. 3H) and neurofilament (NF; Fig. 3I). The pathological diagnosis of the
tumor was of a DNT, WHO grade I. The post-operative course was
uneventful and the seizures were controlled using an oral
antiepileptic. The dose of the drug was gradually reduced over two
years following the surgery. At one month post-surgery, the patient
was administered adjuvant chemotherapy with temozolomide in another
hospital. The regimen was recorded as 150 mg/m2/day,
orally, once a day on 5 consecutive days, for 28 days.
 | Figure 3Pathology data from the first surgery.
(A and B) A typical loose reticular degeneration with microcapsule
formation and matrix mucoid degeneration (HE staining;
magnification, ×4). (C) Hyperplastic oligodendrocyte-like cells
(black triangles) and immature neurons (black arrows) in the tumor
section (HE staining; magnification, ×40). (D) Evidence of FCD in
the peritumoral cortex (HE staining; magnification, ×20). (E)
Astrocytoma, (GFAP staining; magnification, ×40). (F) Hyperplastic
gliacyte component, including oligodendrocyte-like cells and
astrocytoma (S-100 staining; magnification, ×40). (G)
Oligodendrocyte-like cells, (Oligo-2 staining; magnification, ×40).
(H) Immature neurons (black arrows; Syn staining; magnification,
×40). (I) Immature neurons (black arrows; NF staining;
magnification, ×40). HE, hematoxylin and eosin; FCD, focal cortical
dysplasia; GFAP, glial fibrillary acidic protein; Syn,
synaptophysin; NF, neurofilament. |
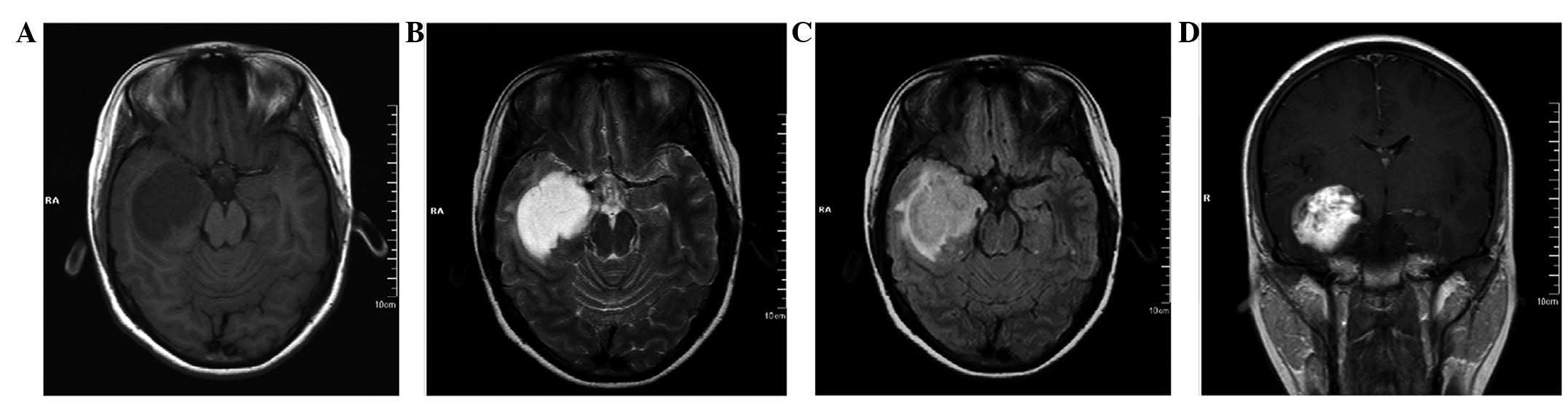
Five years after the initial surgery, the patient
reported an intermittent headache. A neurological examination did
not reveal any abnormalities. However, MRI revealed that a new
lesion had occurred at the base of the previous surgery, with a
size of 2.2×2.0×1.8 cm. The lesion was hypointense on T1WI
(Fig 4A) and hyperintense on T2WI
(Fig. 4B). No edema was observed on
the FLAIR sequence imaging (Fig.
4C). Contrast-enhanced imaging revealed intense enhancement of
the tumor (Fig. 4D).
In October 2011, the patient underwent a second
surgery using the same route as for the previous procedure. The
tumor originated from the insular lobe and was a tenacious mass
with marked vascularity and a grey-colored appearance. GTR was
achieved (Fig. 5) and the
post-operative course was uneventful.
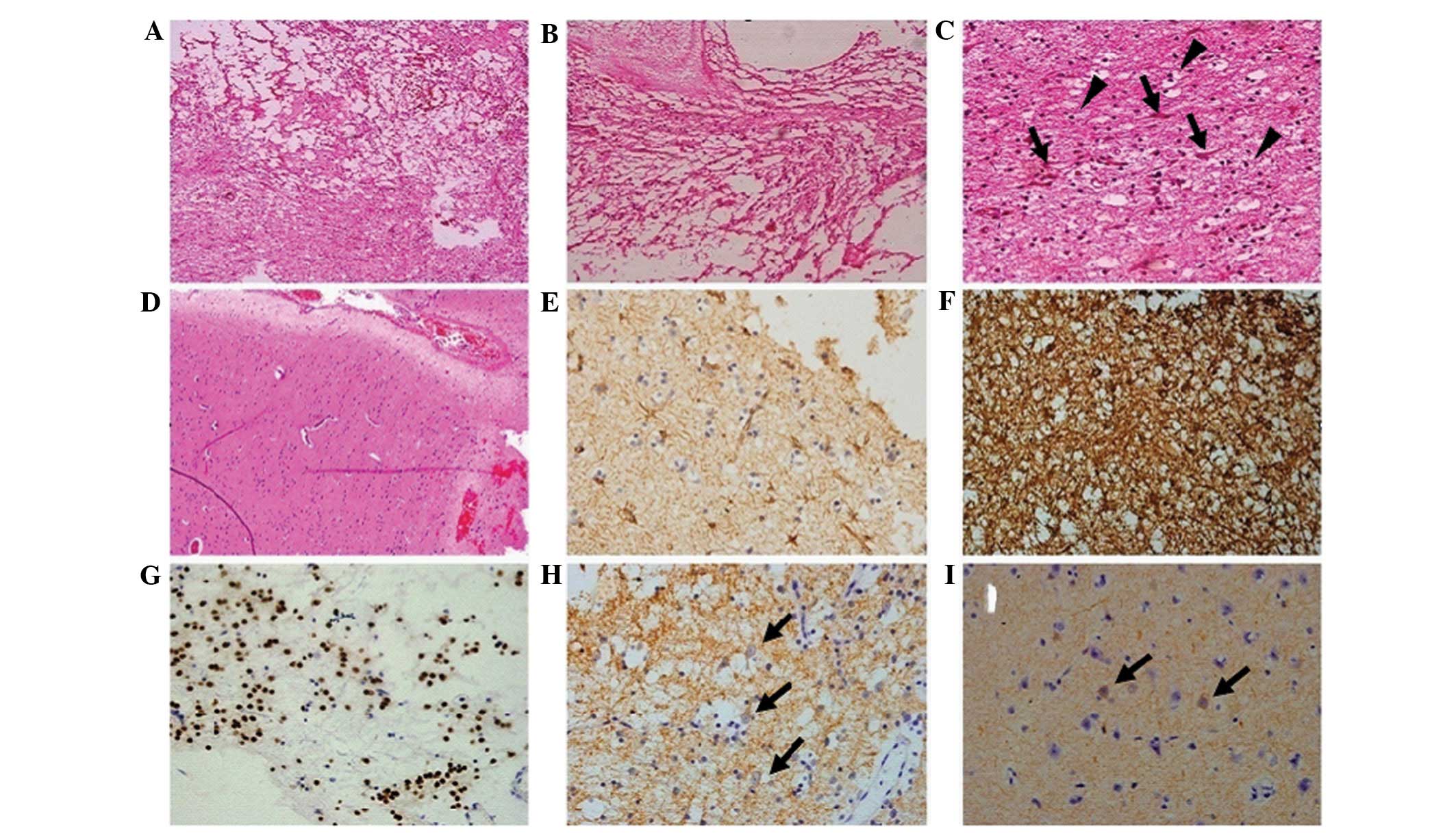
The microscopic evaluation of the recurrent lesion
disclosed two distinct morphological patterns. The prevailing area
revealed the typical appearance of a fibrillary astrocytoma
(Fig. 6A). The tumor cells were
diffusely distributed with a pale cytoplasm and polymorphous nuclei
(Fig. 6B) and mitotic activity was
absent. The tumor underwent small vessel proliferation (Fig. 6B), which may have accounted for the
results of the contrast-enhanced imaging. Certain tumor cells
exhibited a fibrous arrangement (Fig.
6C) or epithelioid-like proliferation (Fig. 6D), in which marked hyperchromatism
and pleomorphism were observed. The secondary component was
identified only in a small section of the tumor (<20% of the
total tumor bulk) and shared similar pathological features with the
initial tumor that was identified in 2006 (Fig. 6E and F). However, the
oligodendrocyte-like cells in this area were pleomorphic and
binucleated and multinucleated cells were visible (Fig. 6F). The prevailing section of the
recurrent tumor revealed the strong expression of GFAP and S-100
(Fig. 7A and B). The endothelium of
the hyperplastic vessel was positive for CD-34 (Fig. 7C) and the Ki-67 index was <3%
(Fig. 7D). In the secondary section
of the tumor, only the oligodendrocyte-like cells were positive for
Oligo-2 (Fig. 7E), while the
astrocytes and the oligodendrocyte-like cells were positive for
GFAP (Fig. 7F). The immature
neurons expressed NeuN, Syn and NF (Fig. 7G and I). The final pathological
diagnosis was fibrillary astrocytoma, WHO grade II.
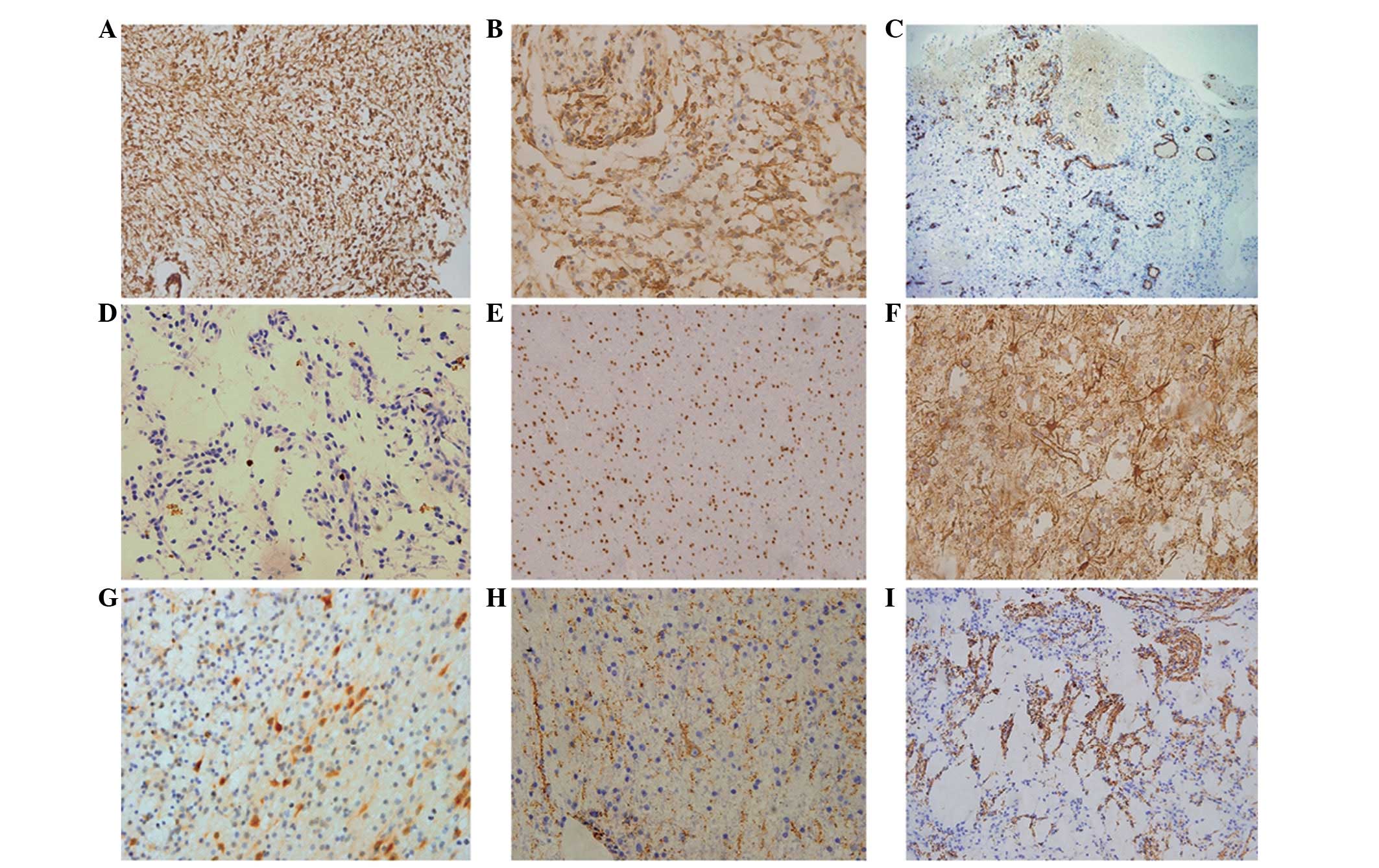 | Figure 7Pathology data following the second
surgery. (A) Hyperplastic gliacyte component (GFAP staining;
magnification, ×20). (B) Hyperplastic gliacyte component (S-100
staining; magnification, ×40). (C) Hyperplasia of the capillaries
(CD34 staining; magnification, ×4). (D) Rare mitotic figures of the
tumor (Ki-67 staining; magnification, ×40). (E)
Oligodendrocyte-like cells in the region of the microcapsules
(Oligo-2 staining; magnification, ×20). (F) Astrocytoma in the
region of microcapsules (GFAP staining; magnification, ×40). (G)
Immature neurons (NeuN staining; magnification, ×40). (H) Immature
neurons (Syn staining; magnification, ×40). (I) The focal positive
reaction of the tumor (NF staining; magnification, ×20). GFAP,
glial fibrillary acidic protein; Syn, synaptophysin; NF,
neurofilament. |
Adjuvant radiotherapy was administered
post-operatively. The radiation dose in the tumor region was 54
Gy/27 F and 48 Gy/27 F in the edema region. During an 11-month
follow-up period, the patient was in good condition without any
neurological disorder. No recurrence or residual tumor was
identified using MRI.
Discussion
First reported in 1988 (18), DNT was classified as a neuronal and
mixed neuronal-glial tumor in 2000 (1). The correlation between DNT and
intractable epilepsy has been widely recognized and epilepsy caused
by DNT may account for 0.8–6.8% of all intractable epilepsy cases
(1,16,19–21).
In patients with DNT, seizures are not controlled by an oral
antiepileptic drug and surgical treatment is the most effective
method of management. Usually, post-operative adjuvant treatment is
unnecessary, as the tumor seldom recurs following a GTR. According
to the literature, the majority of STR cases share a similar
prognosis to GTR cases (6,11,5,16).
Since the initial study of a recurrent case in 2000
(9), 36 similar cases have been
reported, including the present study. All the reported cases of
recurrent DNT are listed in Table
I, in which three cases of radiographic progression are
included. These cases have shown that DNT has a wider spectrum of
clinical behaviors than those that were initially reported by
Daumas-Duport et al(18). As
a brain tumor corresponding to WHO Grade I, DNT retains the
potential for recurrence and malignant transformation.
 | Table ISummary of the previous and current
reports of recurrent or progressive DNTs. |
Table I
Summary of the previous and current
reports of recurrent or progressive DNTs.
| First author, year
(ref) | Case no. | Age at first
resection (years)/gender | Initial pathological
diagnosis | Location | Resection | Adjuvant therapy | Interval between
first and second resection |
Recurrence/progression |
|---|
| Hammond et al,
2000 (9) and Duggal et al,
2008 (8) | 1 | 29/M | Fibrillary
astrocytoma (1984), rediagnosed as DNT (1995) | Left frontal
lobe | STR | None | 11 years | Astrocytoma, WHO
grade IV |
| Rushing et al,
2003 (15) | 2 | 14/M | Mixed low-grade
oligoastrocytoma (1974), rediagnosed as DNT (2003) | Right temporo-
paritetal lobe | STR | Radiotherapy,
chemotherapy | 3 years | DNT plus anaplastic
astrocytoma plus radiation changes |
| Fernandez et
al, 2003 (11) | 3 | 6/M | DNT | Frontal lobe | GTR | None | 125 months | Pathological
diagnosis unknown |
| Nolan et al,
2004 (5) | 4 | Unknown | DNT | Unknown | STR | None | ≤12 months | Radiographic
progression |
| 5 | Unknown | DNT | Unknown | STR | None | ≤12 months | Radiographic
progression |
| 6 | Unknown | DNT | Unknown | STR | None | ≤12 months | Radiographic
progression |
| Sakuta et
al, 2005 (7) | 7 | 8/M | DNT | Left parietal
lobe | STR | None | 6 years | Recurrence |
| 8 | 4/F | DNT | Left parietal
lobe | STR | None | 1 year | Recurrence |
| 9 | 10/M | DNT | Right temporal
lobe | GTR | None | 6.9 years | Recurrence |
| Jensen et
al, 2006 (12) | 10 | 29 (MRI)/F | - | Temporal lobe | No initial
resection radiographic progression | None | 15 years | DNT (first MRI to
but surgery) |
| Josan et al,
2007 (19) | 11 | 3
(MRI)/unknown | - | Parietal lobe | STR | None | 11 years | Radiographic
progression without initial pathological diagnosis |
| Gonzales et
al, 2007 (23) | 12 | 34/M | DNT | Left temporal
lobe | Not stated | Unknown | 125 months | Recurrence |
| 13 | 32/F | DNT | Right frontal
lobe | Not stated | Unknown | 98 months | Recurrence |
| 14 | 33/F | DNT | Right amygdala | Not stated | Unknown | 64 months | Recurrence |
| 15 | 47/F | DNT | Left frontal
lobe | STR | None | 40 months | DNT plus
oligoastrocytoma, WHO grade II |
| Schittenhelm et
al, 2007 (10) | 16 | 7/F | DNT | Mutifocal
lesions | STR | None | 7 years | DNT with atypia and
Ki-67 index up to 10% |
| Maher et al,
2008 (13) | 17 | 6/M | DNT | Temporo-occipital
lobe | GTR | None | 6 years | Recurrence |
| Minkin et
al, 2008 (24) | 18 | Unknown | DNT and pilocytic
astrocytomas | Unknown | GTR | None | Unknown | No sign of
progression as stated |
| Sung and Suh, 2009
(25) | 19 | 16/M | DNT | Occipital lobe | Not stated | Not stated | Not stated | Not stated |
| 20 | 64/F | DNT | Occipital lobe | Not stated | Not stated | Not stated | Not stated |
| Lee et al,
2009 (26) | 21 | 16/M | DNT | Occipital lobe | Lesionectomy | None | 5 years | No sign of
progression as stated |
| Ray et al,
2009 (14) | 22 | 20/F | DNT | Right frontal
lobe | GTR | None | 72 months | Recurrence |
| 23 | 37/M | DNT | Left temporal
lobe | STR | None | 83 months | Recurrence |
| 24 | 16/M | DNT | Left temporo-
occipital lobe | GTR | None | 88 months | Pilocytic
astrocytoma, WHO grade I |
| 25 | 18/M | DNT | Right parietal
lobe | GTR | None | 23 months | Recurrence |
| 26 | 12/M | Protoplasmic
astrocytoma, rediagnosed as DNT | Right
fronto-parietal lobe | STR | Radiotherapy | 80 months | Astrocytoma, WHO
grade III |
| Zakrzewski et
al, 2009 (17) | 27 | 7/F | DNT | Right temporal
lobe | STR | None | 4 years | Pilocytic
astrocyoma, WHO grade I |
| Kawataki et
al, 2010 (27) | 28 | 1 (MRI)/M | - | Left frontal
lobe | No initial
resection, but radiographic progression | None | 7 years (first CT
to surgery) | DNT |
| Qaddoumi et
al, 2010 (28) | 29 | 6/unknown | DNT | Temporal lobe | STR | None | 12 years | No sign of
progression as stated |
| 30 | 9/unknown | DNT | Temporal lobe | GTR | None | Unknown | Unknown |
| 31 | 9/unknown | DNT | Temporal lobe | GTR | None | Unknown | Unknown |
| 32 | 13/unknown | DNT | Temporal-parietal-
occipital lobe | GTR | None | Unknown | Unknown |
| 33 | 11/unknown | DNT | Temporal lobe | GTR | None | Unknown | Unknown |
| Thom et al,
2011 (16) | 34 | 56/unknown | DNT | Left temporal
lobe | STR | None | 2 years | Anaplastic mixed
glioneuronal tumor, WHO grade III |
| Wagner et
al, 2012 (29) | 35 | 9/unknown | DNT | Unknown | Unknown | Unknown | 10 months | Recurrence |
| Present case
study | 36 | 15/F | DNT | Right temporal
lobe | GTR | Chemotherapy | 5 years | Astrocyoma, WHO
grade II |
The excessive growth of any component of DNT,
including immature neurons, oligodendrocyte-like cells and
astrocytes, may lead to tumor recurrence or post-operative
malignant transformation. The present case of recurrence may be
attributed to the administration of inappropriate adjuvant
chemotherapy following surgery. The patient is also the first
histological evolution case that may have been caused by
chemotherapy alone. Based on the current treatment guidelines for a
low-grade brain tumor, adjuvant therapy is unnecessary for a WHO
grade I tumor. However, the patient was administered chemotherapy
with temozolomide in another hospital and the tumor recurrence was
considered to be directly associated with this inappropriate
chemotherapy. A further two similar cases have been identified in
previous studies, cases 2 (15) and
26 (14), in which the patients
underwent an STR of the tumor prior to 1988, when little was known
about DNT. Case two was diagnosed as a fibrillary astrocytoma at
the initial surgery, after which, the patient was administered
adjuvant radiotherapy and chemotherapy. The patient from case 26
was administered adjuvant radiotherapy for the initial diagnosis of
a protoplasmic astrocytoma. At that time, adjuvant therapy for
residual tumors of WHO grade II was acceptable. In the follow-up
period, the tumors recurred and progressed into astrocytoma, WHO
grade III, at 72 months (case 2) and 80 months (case 26).
The present case had a typical imaging manifestation
shown by T1WI, T2WI and FLAIR sequence imaging, as described in
previous studies of DNT. However, contrast-enhanced imaging
revealed the tumor with rare, patchy enhancement. Although the
tumor was initially diagnosed as a pilocytic astrocytoma or
pleomorphic xanthoastrocytoma based on the manifestation on the
neuroimaging, microscopic pathology revealed the tumor to be a
typical DNT. From a review of the previous literature, similar
presentations have been identified using enhanced imaging in a
number of cases (11,12,22,30,31).
In the 36 reported cases, including the present
study, 20 cases exhibited clear pathological evidence that
demonstrated the tumor recurrence or malignant transformation. The
average tumor-free survival time was 65.3 months, (range, 10–132
months). This survival time was similar to that reported by Ray
et al(14). The extent of
the resection following the initial surgery was available in 16
cases, of which, 10 resulted in an STR compared with six GTRs. The
average tumor-free survival time of the STR and GTR groups was 61.1
months (range, 12–132 months) and 66.3 months (range, 23–88
months), respectively. No statistical difference was observed
between the two groups based on the independent-samples t-test
(P=0.757).
Of the 17 cases in which the exact pathological
diagnosis of the recurrent tumor was available (the three patients
who were administered adjuvant radiotherapy and chemotherapy were
excluded from this comparison), DNT recurrence without malignant
transformation was demonstrated in 11 cases and the remaining six
cases represented malignant transformation or histological
evolution. The average tumor-free survival time of the recurrence
group was 64.9 months (range, 10–125 months) compared with an
average tumor-free survival time of 69.3 months (range, 24–132
months) in the malignant-transformation group. No statistical
difference was observed between the two groups based on the results
of the independent-samples t-test (P=0.818).
In all recurrent cases with a pathological
diagnosis, the average recurrence time of those who were
administered adjuvant therapy was 58.7 months (range, 36–80 months)
compared with 69.3 months (range, 24–132 months) for the patients
who only underwent surgery. No statistical difference was observed
between the two groups based on the independent-samples t-test
(P=0.684). Therefore, this may be evidence that post-operative
adjuvant therapy for DNT is not able to prolong the tumor-free
survival time and benefit patients. The reason for tumor recurrence
in the three patients who were administered adjuvant therapy is
unknown. Hammond et al(9)
and Ray et al(14)
hypothesized that radiotherapy was a risk factor for tumor
recurrence, and the present study may indicate that chemotherapy is
another risk factor. Notably, malignant tumor transformation
without any adjuvant therapy has been reported in six cases and
remains unexplained. The recurrence and malignant transformation of
DNT may have occurred due to the innate potential of the tumor
itself, and the adjuvant radiotherapy or chemotherapy may have
initiated the key steps towards malignant transformation.
To date, no treatment guidelines for recurrent DNT
have been available. Usually, a second surgery is performed in
cases of tumor recurrence without malignant transformation
(7,12–14).
For malignant transformation or histological evolution cases, there
are no fixed views on treatment. We prefer a comprehensive
treatment, including surgery and post-operative adjuvant therapy.
Ray et al(14) reported a
patient whose tumor recurred and progressed into an astrocytoma
(WHO grade III). The patient was administered adjuvant chemotherapy
with temozolomide and showed a favorable post-operative prognosis.
A similar case has been reported in which the patient underwent a
GTR of the recurrent tumor without any adjuvant therapy (16). In the majority of patients in whom
the recurrent tumor has progressed into a WHO Grade II tumor, a
favorable prognosis may be achieved through a GTR of the recurrent
lesion. The case of a patient who remained tumor-free five years
following the second surgery has also been reported (10,17,22,23).
In the present case, the patient underwent focal radiotherapy and
the tumor did not recur within a follow-up period of 11 months.
In conclusion, as a WHO Grade I tumor, DNT retains
the potential for recurrence and malignant transformation following
GTR. Post-operative adjuvant therapy is not able to prolong the
tumor-free survival time and may be a risk factor for tumor
recurrence. For recurrent cases, the prognosis is favorable if a
GTR of the recurrent lesion is achieved. Based on the previous
evidence, adjuvant therapy is not recommended for a definitive
diagnosis of DNT. The use of regular imaging examinations and the
maintenance of a long-term follow-up is of importance following a
tumor resection.
Abbreviations:
|
DNT
|
dysembryoplastic neuroepithelial
tumors
|
|
WI
|
weighted imaging
|
|
FCD
|
focal cortical dysplasia
|
|
Syn
|
synaptophysin
|
|
GTR
|
gross total resection
|
|
STR
|
subtotal resection
|
References
|
1
|
Daumas-Duport C, Pietsch T and Lantos PL:
Dysembryoplastic neuroepithelial tumour. Pathology and Genetics of
Tumors of the Nervous System (IARC WHO Classification of Tumors).
Kleihues P and Cavenee WK: 2nd edition. IARC Press; Lyon, France:
pp. 103–106. 2000
|
|
2
|
Fellah S, Callot V, Viout P, et al:
Epileptogenic brain lesions in children: the added-value of
combined diffusion imaging and proton MR spectroscopy to the
presurgical differential diagnosis. Childs Nerv Syst. 28:273–282.
2012. View Article : Google Scholar
|
|
3
|
Prayson RA and Napekoski KM: Composite
ganglioglioma/dysembryoplastic neuroepithelial tumor: a
clinicopathologic study of 8 cases. Hum Pathol. 43:1113–1118. 2012.
View Article : Google Scholar
|
|
4
|
Thom M, Blümcke I and Aronica E: Long-term
epilepsy-associated tumors. Brain Pathol. 22:350–379. 2012.
View Article : Google Scholar
|
|
5
|
Nolan MA, Sakuta R, Chuang N, et al:
Dysembryoplastic neuroepithelial tumors in childhood: long-term
outcome and prognostic features. Neurology. 62:2270–2276. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Piao YS, Lu DH, Chen L, et al:
Neuropathological findings in intractable epilepsy: 435 Chinese
cases. Brain Pathol. 20:902–908. 2010.PubMed/NCBI
|
|
7
|
Sakuta R, Otsubo H, Nolan MA, et al:
Recurrent intractable seizures in children with cortical dysplasia
adjacent to dysembryoplastic neumepithelial tumor. J Child Neurol.
20:377–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Duggal N, Taylor R, Zou GY and Hammond RR:
Dysembryoplastic neuroepithelial tumors: clinical, proliferative
and apoptotic features. J Clin Pathol. 61:127–131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hammond RR, Duggal N, Woulfe JM and Girvin
JP: Malignant transformation of a dysembryoplastic neuroepithelial
tumor. Case report. J Neurosurg. 92:722–725. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schittenhelm J, Mittelbronn M, Wolff M, et
al: Multifocal dysembryoplastic neuroepithelial tumor with signs of
atypia after regrowth. Neuropathology. 27:383–389. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fernandez C, Girard N, Paz Paredes A, et
al: The usefulness of MR imaging in the diagnosis of
dysembryoplastic neuroepithelial tumor in children: a study of 14
cases. AJNR Am J Neuroradiol. 24:829–834. 2003.PubMed/NCBI
|
|
12
|
Jensen RL, Caamano E, Jensen EM and
Couldwell WT: Development of contrast enhancement after long-term
observation of a dysembryoplastic neuroepithelial tumor. J
Neurooncol. 78:59–62. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maher CO, White JB, Scheithauer BW and
Raffel C: Recurrence of dysembryoplastic neuroepithelial tumor
following resection. Pediatr Neurosurg. 44:333–336. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ray WZ, Blackburn SL, Casavilca-Zambrano
S, et al: Clinicopathologic features of recurrent dysembryoplastic
neuroepithelial tumor and rare malignant transformation: a report
of 5 cases and review of the literature. J Neurooncol. 94:283–292.
2009. View Article : Google Scholar
|
|
15
|
Rushing EJ, Thompson LD and Mena H:
Malignant transformation of a dysembryoplastic neuroepithelial
tumor after radiation and chemotherapy. Ann Diagn Pathol.
7:240–244. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thom M, Toma A, An S, et al: One hundred
and one dysembryoplastic neuroepithelial tumors: an adult epilepsy
series with immunohistochemical, molecular genetic, and clinical
correlations and a review of the literature. J Neuropathol Exp
Neurol. 70:859–878. 2011. View Article : Google Scholar
|
|
17
|
Zakrzewski K, Biernat W, Liberski PP,
Polis L and Nowoslawska E: Pilocytic astrocytoma as a predominant
component of a recurrent complex type DNT. Folia Neuropathol.
47:284–288. 2009.PubMed/NCBI
|
|
18
|
Daumas-Duport C, Scheithauer BW,
Chodkiewicz JP, et al: Dysembryoplastic neuroepithelial tumor: a
surgically curable tumor of young patients with intractable partial
seizures. Report of thirty-nine cases. Neurosurgery. 23:545–556.
1988. View Article : Google Scholar
|
|
19
|
Josan V, Smith P, Kornberg A, et al:
Development of a pilocytic astrocytoma in a dysembryoplastic
neuroepithelial tumor. Case report. J Neurosurg. 106:509–512.
2007.PubMed/NCBI
|
|
20
|
Prayson RA: Tumors arising in the setting
of pediatric chronic epilepsy. Pathol. 42:426–431. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prayson RA, Fong J and Najm I: Coexistent
pathology in chronic epilepsy patients with neoplasms. Modern
Pathol. 23:1097–1103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dozza DC, Rodrigues FF and Chimelli L:
Dysembryoplastic neuroepithelial tumor originally diagnosed as
astrocytoma and oligodendroglioma. Arq Neuropsiquiatr. 70:710–714.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gonzales M, Dale S, Susman M, et al:
Dysembryoplastic neuroepithelial tumor (DNT)-like
oligodendrogliomas or Dnts evolving into oligodendrogliomas: two
illustrative cases. Neuropathology. 27:324–330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Minkin K, Klein O, Mancini J and Lena G:
Surgical strategies and seizure control in pediatric patients with
dysembryoplastic neuroepithelial tumors: a single-institution
experience. J Neurosurg Pediatr. 1:206–210. 2008. View Article : Google Scholar
|
|
25
|
Sung CO and Suh YL: Different pattern of
expression of nestin in the non-specific form of dysembryoplastic
neuroepithelial tumors compared to the simple and complex forms. J
Neurooncol. 92:7–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee J, Lee BL, Joo EY, et al:
Dysembryoplastic neuroepithelial tumors in pediatric patients.
Brain Dev. 31:671–681. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawataki T, Sato E, Kato T, et al: A
cortical dysembryoplastic neuroepithelial tumor initially occurring
in the periventricular white matter. J Neurosurg Pediatr.
6:600–603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qaddoumi I, Ellison DW, Morris EB, et al:
Dysembryoplastic neuroepithelial tumors and cognitive outcome: cure
at a price? Cancer. 116:5461–5469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wagner AS, Yin NS, Tung S, et al: Intimal
thickening of meningeal arteries after serial corticectomies for
Rasmussen encephalitis. Hum Pathol. 43:1308–1313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ostertun B, Wolf HK, Campos MG, et al:
Dysembryoplastic neuroepithelial tumors: MR and CT evaluation. AJNR
Am J Neuroradiol. 17:419–430. 1996.PubMed/NCBI
|
|
31
|
Yu AH, Chen L, Li YJ, et al:
Dysembryoplastic neuroepithelial tumors: magnetic resonance imaging
and magnetic resonance spectroscopy evaluation. Chin Med J (Engl).
122:2433–2437. 2009.PubMed/NCBI
|