Introduction
Neurofibromatosis type 1 (NF1), also referred to as
von Recklinghausen disease, is an autosomal dominant inherited
disease that affects approximately one in 3,000 individuals
(1). The disease is characterized
by the presence of multiple neurofibromas, café-au-lait spots, iris
hamartomas (Lisch nodules) and axillary and inguinal freckling
(1). It is well established that
the incidence of tumors in patients with NF1 is high compared with
the normal population, which is the main reason for the reduced
life span of NF1 patients (2). The
majority of tumors arising in NF1 patients are neurofibromas,
particularly plexiform neurofibromas, which is a hallmark of this
disease. Malignant peripheral nerve sheath tumors also affect these
patients. Furthermore, patients with NF1 have a greatly increased
risk of developing gliomas, leukemia, particularly juvenile
myelomonocytic leukemia, pheochromocytoma and rhabdomyosarcoma
(2,3). In addition, certain types of
carcinomas, including breast cancer, may also occur more frequently
in patients with NF1 (2,4,5).
However, the occurrence of cutaneous squamous cell carcinoma (SCC)
in patients with NF1 has been rarely documented (6). The present study describes a case of
an SCC adjacent to a neurofibroma of the forehead in a patient with
NF1. Written informed consent was obtained from the patient.
Case report
Patient
An 80-year-old female with NF1 presented with a
rapidly growing skin tumor of the forehead. A physical examination
revealed numerous cutaneous nodules across the entire body, which
were clinically diagnosed as neurofibromas. The forehead skin tumor
was well-circumscribed, dome-shaped with a central keratin plug and
adjacent to a neurofibroma. The lesion measured 2.5×2.4 cm in
diameter. Under the clinical diagnosis of keratoacanthoma, a total
resection of the forehead tumor with the adjacent neurofibroma was
performed.
Methods
The formalin-fixed, paraffin-embedded tissue blocks
of the resected skin specimen were cut into 3-μm thick sections,
deparaffinized and rehydrated. Each section was stained with
hematoxylin and eosin and used for immunostaining.
Immunohistochemical analyses were performed using an autostainer
(XT system BenchMark; Ventana Medical System, Inc., Tucson, AZ,
USA) according to the manufacturer’s instructions. A mouse
monoclonal antibody against Ki-67 (MM1; Novocastra Laboratories,
Ltd., Newcastle upon Tyne, UK) and a rabbit polyclonal antibody
against S-100 protein (Nichirei Bioscience, Tokyo, Japan) were
used.
Results
Macroscopically, the cut section of the tumor
revealed two tumorous lesions. One was a marked hyperkeratotic
tumor invading the upper subcutis and the other was a
well-circumscribed nodule in the dermis and subcutis (Fig. 1). Although the two lesions were
adjacent, no continuity was noted.
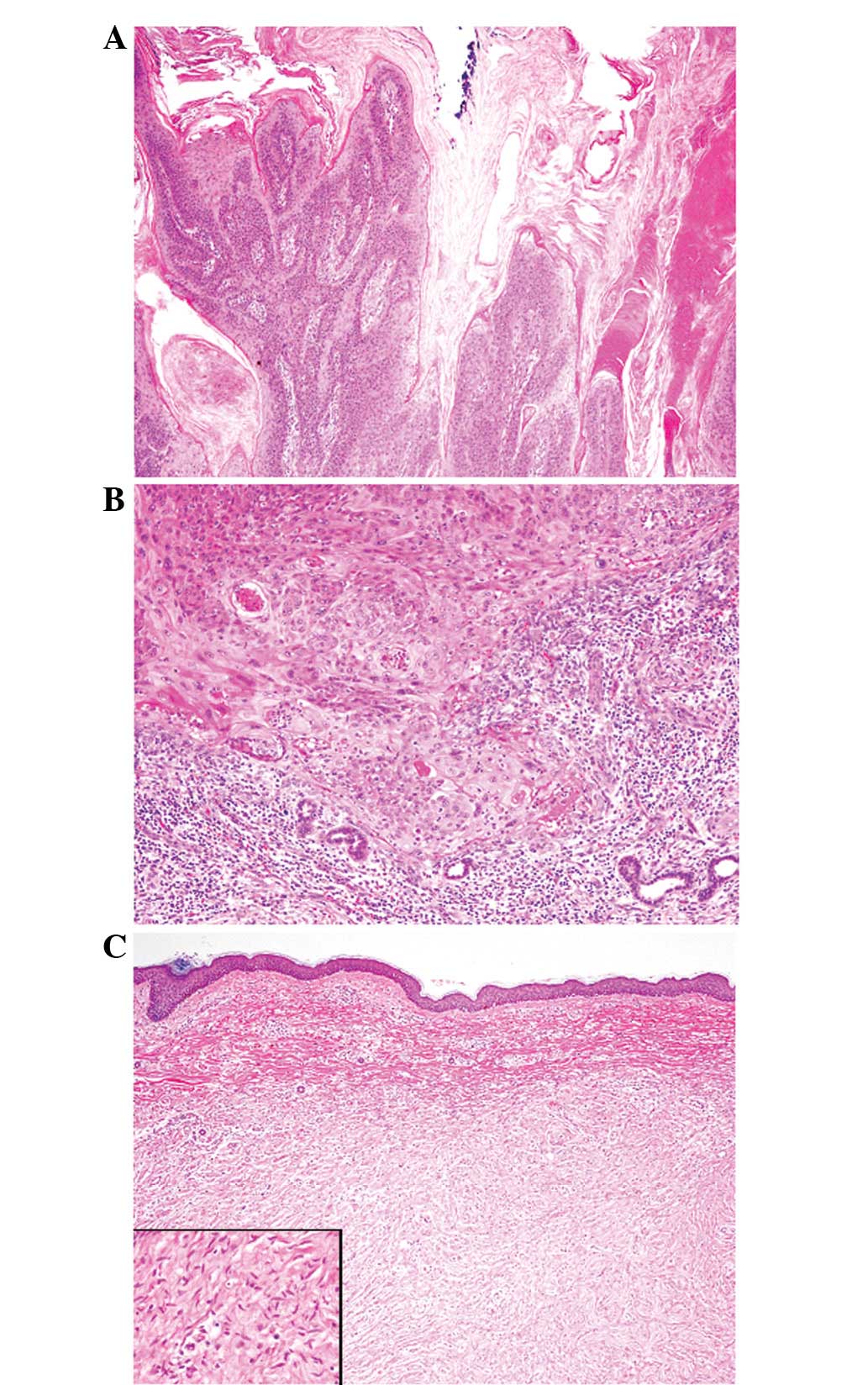
Microscopically, the former tumor revealed papillary
proliferation of atypical squamous cells with marked
hyperparakeratosis (Fig. 2A). These
atypical squamous cells contained large nuclei with coarse
chromatin, conspicuous nucleoli and a rich eosinophilic cytoplasm
(Fig. 2B). Mitotic figures were
frequently observed. The tumor had invaded into the upper subcutis
and peritumoral lymphoplasmacytic infiltration was also noted
(Fig. 2B). These histopathological
features were typical for an invasive SCC. The latter component was
a neurofibroma, which was composed of proliferating spindle cells
containing bland cigar-shaped nuclei with inconspicuous nucleoli
and an eosinophilic cytoplasm (Fig.
2C). No mitotic figures were observed. Immunohistochemically,
the spindle cells were diffusely positive for S-100 protein and the
Ki-67 labeling index was <1%. Therefore, this component was
diagnosed as a neurofibroma.
Discussion
Patients with NF1 are at an increased risk of
non-epithelial neoplasms of several types, including neurofibromas,
malignant peripheral nerve sheath tumors, gliomas, leukemia,
pheochromocytoma and rhabdomyosarcoma (2,3).
Furthermore, studies have also suggested an increased risk of
certain types of carcinomas in patients with NF1 (2,4,5).
Seminog and Goldacre (2) recently
analyzed 697 cases of carcinomas in 6,739 patients with NF1. The
study revealed that patients with NF1 have a significantly high
risk of developing carcinomas of the esophagus, stomach, colon,
liver, lung, thyroid, breast and ovary (2). Furthermore, a slightly increased risk
of non-melanoma skin cancers was reported, though the
histopathological subtypes of the skin cancers were not available
(2). However, SCC of the skin
accounts for only a small number of malignancies in NF1 patients
and to the best of our knowledge, only one case of an invasive SCC
of the sole of the foot, with analyses of the histopathological
features, has been reported in an NF1 patient (6). The present study is the second
documented case of an invasive SCC with histopathological analyses
in a patient with NF1.
NF1 is caused by inactivating mutations in the
NF1 gene, which is located on chromosome 17q11.2. This gene
has a tumor suppressor function as the gene product of NF1,
neurofibromin, is a major negative regulator of the
RAS/mitogen-activated protein kinase (MAPK) pathway, which
transmits mitogenic signals to the nucleus (7). Consistent with Knudson’s two-hit
hypothesis, NF1 patients with a heterozygous germline NF1
mutation develop a somatic mutation in the second wild-type
NF1 allele, resulting in the development of neurofibromas.
The loss of heterozygosity of the NF1 gene is observed in
certain types of non-epithelial tumors in patients with NF1.
Biallelic NF1 inactivation is observed in neurofibromas and
malignant peripheral nerve sheath tumors (8–10). A
high proportion of astrocytomas from patients with NF1 also
demonstrate a loss of neurofibromin expression and a loss of
heterozygosity of the NF1 gene (11). Furthermore, the loss of
heterozygosity of the NF1 region has been observed in the
majority of pheochromocytoma cases in patients with NF1 (12).
The genetic mechanism of carcinoma development in
patients with NF1 is not well understood. However, Güran and Safali
(13) reported a case of breast
carcinoma in an NF1 patient and loss of heterozyosity of the
NF1 gene in the carcinoma tissue. BRCA1 or
BRCA2 mutations were not observed in this case. Furthermore,
Ceccaroni et al(14)
reported the cases of five individuals in a family with NF1 who
presented with neurofibromas and breast, ovary, peritoneal or
rectal carcinomas. The study clearly demonstrated that three
individuals shared a common haplotype, including the NF1 and
BRCA1 loci on chromosome 17 and speculated that the
occurrence of NF1 and breast carcinoma in this family was due to
the presence of two linked mutations at the NF1 and
BRCA1 foci (14).
In conclusion, the present study is the second
documented case of cutaneous SCC with an analysis of the
histopathological features in a patient with NF1. The increased
risk of various types of carcinomas, including non-melanoma skin
carcinoma, in patients with NF1 is recognized, however, the
molecular mechanism of carcinoma development in patients with NF1
is not well understood. Additional studies are required to clarify
this mechanism
References
|
1
|
Brems H, Beert E, de Ravel T and Legius E:
Mechanisms in the pathogenesis of malignant tumours in
neurofibromatosis type 1. Lancet Oncol. 10:508–515. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seminog OO and Goldacre MJ: Risk of benign
tumours of nervous system, and of malignant neoplasms, in people
with neurofibromatosis: population-based record-linkage study. Br J
Cancer. 108:193–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zöller ME, Rembeck B, Odén A, Samuelsson M
and Angervall L: Malignant and benign tumors in patients with
neurofibromatosis type 1 in a defined Swedish population. Cancer.
79:2125–2131. 1997.PubMed/NCBI
|
|
4
|
Salemis NS, Nakos G, Sambaziotis D and
Gourgiotis S: Breast cancer associated with type 1
neurofibromatosis. Breast Cancer. 17:306–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharif S, Moran A, Houson SM, et al: Women
with neurofibromatosis 1 are at a moderately increased risk of
developing breast cancer and should be considered for early
screening. J Med Genet. 44:481–484. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Friedrich RE, Al-Dam A and Hagel C:
Squamous cell carcinoma of the sole of the foot in
neurofibromatosis type 1. Anticancer Res. 32:2165–2168.
2012.PubMed/NCBI
|
|
7
|
Seizinger BR: NF1: a prevalent cause of
tumorigenesis in human cancers? Nat Genet. 3:97–99. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Laycock-van Spyk S, Thomas N, Cooper DN
and Upadhyaya M: Neurofibromatosis type 1-associtated tumours:
their somatic mutational spectrum and pathogenesis. Human Genomics.
5:623–690. 2011.PubMed/NCBI
|
|
9
|
Garcia-Linares C, Fernández-Rodriguez J,
Terribas E, et al: Dissecting loss of heterozygosity (LOH) in
neurofibromatosis type 1-associated neurofibromas: Importance of
copy neutral LOH. Hum Mutat. 32:78–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Upadhyaya M, Kluwe L, Spurlock G, et al:
Germline and somatic NF1 gene mutation spectrum in NF1-associated
malignant peripheral nerve sheath tumors (MPNSTs). Hum Mutat.
29:74–82. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gutmann DH, Donahoe J, Brown T, James CD
and Perry A: Loss of neurofibromatosis 1 (NF1) gene expression in
NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol.
26:361–367. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bausch B, Borozdin W, Mautner VF, et al;
European-American Phaeochromocytoma Registry Study Group. Germline
NF1 mutational spectra and loss-of-heterozygosity analyses in
patients with pheochromocytoma and neurofibromatosis type 1. J Clin
Endocrinol Metab. 92:2784–2792. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Güran S and Safali M: A case of
neurofibromatosis and breast cancer: loss of heterozygosity of NF1
in breast cancer. Cancer Genet Cytogenet. 156:86–88.
2005.PubMed/NCBI
|
|
14
|
Ceccaroni M, Genuardi M, Legge F, et al:
BRCA1-related malignancies in a family presenting with von
Recklinghausen’s disease. Gynecol Oncol. 86:375–378.
2002.PubMed/NCBI
|