Introduction
Dual specificity phosphatase 6 (DUSP6) is a
mitogen-activated protein kinase (MAPK) phosphatase that plays a
critical role as a negative regulator of the MAPK pathway (1,2). The
MAPK pathway controls a vast array of physiological processes,
including cell proliferation, cell cycle arrest, cell survival and
cell motility (3–6). Specifically, DUSP6 dephosphorylates
the threonine and tyrosine residues of extracellular
signal-regulated kinase (ERK) 1/2, and inactivates ERK1/2 in a
feedback loop (7–9). Disruption of this feedback loop may
give rise to increased exposure to growth factors, resulting in
neoplastic or even malignant transformation (2,10,11).
In different types of tumors, DUSP6 have various
roles depending on the tumor type and the stage of carcinogenesis.
Previous studies have indicated that DUSP6 acts as a tumor
suppressor gene in several types of tumors (12–23).
In pancreatic cancer, the expression of DUSP6 was downregulated,
which activated ERK excessively, and eventually led to improvement
of the carcinoma development and progression (12–14).
The downregulation of DUSP6 was caused by the hypermethylation of
CpG sequences in intron 1 of the DUSP6 gene in the progression of
pancreatic cancer (15). The DUSP6
expression correlated inversely with the growth activity and
histological grade of the tumor in lung cancer (16). In ovarian cancer, DUSP6 expression
is lost, particularly at the protein level, leading to the
hyperactivation of ERK1/2 and eventually resulting in
tumorigenicity and chemoresistance of human ovarian cancer cells
(17). Notably, DUSP6 has a
contrary effect in certain other tumor types. DUSP6 is upregulated
in myeloma (18), melanoma
(19), glioma (20), glioblastoma (21), keratinocytes (22) and breast cancer (23). In glioblastoma, the overexpression
of DUSP6 lessens tumor cell sensitivity to the anticancer
DNA-damaging drug cisplatin (21).
Overexpression of DUSP6 causes estrogen receptor-positive breast
cancer cells to become resistant to the growth inhibitory effects
of tamoxifen (23). Furthermore,
methylation of DUSP6 is infrequent in endometrial cancer (24). Therefore, silencing of DUSP6 may not
be involved in the constitutive activation of the ERK kinase
cascade in endometrial cancer (24).
However, few studies have reported the role of DUSP6
in esophageal cancer. Esophageal squamous cell carcinoma (ESCC) is
a potentially fatal disease with high incidence worldwide,
particularly in China (25).
Despite recent progress in ESCC diagnosis and treatment, the
survival rates for ESCC patients remain poor. Thus, there is a
requirement for studying new genes involved in ESCC tumorigenesis
and progression, in order to develop safer and faster diagnosis and
improved disease outcome predication following treatment of this
dangerous disease.
A previous investigation showed downregulation of
DUSP6 in ESCC in Hong Kong (26).
Furthermore, our previous study demonstrated that the exogenous
overexpression of DUSP6 confirmed the growth suppression in ESCC
cells (27). In the present study,
we focus on the correlation between DUSP6 expression and
clinicopathological features, the mechanisms by which DUSP6 affects
the ESCC cells and the possible epigenetic mechanisms involved in
the abrogation of DUSP6 in ESCC cells.
Materials and methods
Cell lines and primary tissues
The 19 paired ESCC and normal esophageal tissues
were obtained from patients who underwent surgery at Henan Cancer
Hospital (Zhengzhou, China). The tissue microarrays were purchased
from Biomax (Rockville, MD, USA; ES1202 and ES8010). The arrays
included a total of 89 benign esophageal tissue samples and 95
localized esophageal cancer samples. Two human esophageal squamous
cell carcinoma cell lines, EC9706 and KYSE150, were used in this
study. The EC9706 cell line was established and studied by Han
et al(28), while the
KYSE150 cell line was kindly provided by Dr Shimada (First
Department of Surgery, Faculty of Medicine, Kyoto University,
Japan). The two cell lines were cultured in accordance with their
original methods (28,29). The present study was approved by the
institutional review boards of the Cancer Institute and Hospital,
Chinese Academy of Medical Sciences and Peking Union Medical
College (Beijing, China). Written informed consent was obtained
from the patients.
Cellular transfection
The empty vector (pCMV-AC) and the plasmid
containing the complementary DNA sequence of human DUSP6
(pCMV-DUSP6) were purchased from OriGene (Beijing, China). The ESCC
cells, EC9706 and KYSE150, were transiently transfected with
pCMV-AC or pCMV-DUSP6, using Lipofectamine™ 2000 according to the
manufacturer’s instructions (Invitrogen Life Technologies,
Carlsbad, CA, USA). Whole cell lysates for immunoblotting were
collected at 24 h after transfection in order to confirm the
appropriate plasmid DUSP6 expression.
qPCR
The total RNA collected from each sample was
extracted using TRIzol reagent according to manufacturer’s
instructions (Invitrogen Life Technologies). Total RNA was then
used to synthesize cDNA using PrimeScript Rtase (Takara, Shiga,
Japan). The RT product was used as the template to amplify DUSP6.
The forward and reverse primers were 5′-AAC AGG GTT CCA GCA CAG
CAG-3′ and 5′-GGC CAG ACA CAT TCC AGC AA-3′, respectively. GAPDH
was used as an internal control. The products were resolved by
electrophoresis in 3% agar and stained with ethidium bromide. DUSP6
expression levels were evaluated by qPCR using StepOne RealTime PCR
system (Applied Biosystems, Beijing, China). The data were
normalized by the intensity of GAPDH.
Western blotting
Western blotting was performed as described
previously (30). The cells were
lysed in 1% NP-40 lysis buffer and cleared by centrifugation at
12,000 × g for 20 min. Supernatants were recovered as protein
extracts. The extracts containing equal amount of proteins were
separated using 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA). The membranes were
incubated with non-fat dry milk in 0.01 M/l Tris-buffered saline
containing 0.1% Tween-20 to block non-immunospecific protein
binding, and then with primary antibody against DUSP6 (OriGene) and
phosphorylated ERK (p-ERK; Santa Cruz Biotechnology, Santa Cruz,
CA, USA). Following incubation with a peroxidase-conjugated
affinipure goat anti-rabbit IgG secondary antibody (Jackson Immuno
Research, West Grove, PA, USA), protein signals were visualized by
enhanced chemiluminescence (Pierce, Rockford, IL USA).
Immunohistochemistry
Formalin-fixed tissue sections (4 μm) were
deparaffinized and rehydrated, and incubated with 3% hydrogen
peroxide, followed by antigen retrieval treatment, boiling in
citrate buffer (0.01 M/l, pH 6.0), to restore the masked epitope.
Goat serum (10%) was used to block endogenous peroxidase activities
and non-immunospecific protein binding. The sections were then
incubated with primary antibody against DUSP6 (OriGene) overnight
at 4°C. After washing with phosphate-buffered saline (PBS) with 1%
Tween-20, the sections were incubated with secondary horseradish
peroxidase-conjugated antibody for 30 min at room temperature.
Immunoreactivity was visualized with freshly prepared
diaminobenzidine substrate and the nuclei were counterstained with
hematoxylin.
DUSP6 expression levels in esophageal cancer cells
were subdivided into four categories, negative (0), faint (1), moderate (2) and strong (3). Negative (0) was defined as tissues
with no staining. Faint (1)
expression was defined as tissues with faint staining or moderate
to strong staining in <25% of cells. Moderate (2) was defined as a moderate or strong
staining in 25–50% of cells. Strong (3) was defined as a strong staining in
>50% of cells. The cut-off point to define high and low DUSP6
expression was 25% staining.
Treatment with
5-aza-2′-deoxycytidine
The ESCC cell lines were first cultured in 10-cm
plates and the cells were maintained until they reached 30%
confluence. Subsequently, the cells were treated with 10 μmol/l of
5-aza-2′-deoxycytidine (Sigma, St. Louis, MO, USA) for five days,
as described previously (16).
These cells were harvested for further investigation.
Methylation-specific PCR assays
As it was reported that the region in intron 1 of
the DUSP6 gene is highly methylated in pancreatic cancer (15), we designed primer sets for both
unmethylated (forward: 5′-GTA GGG GTT GTG AAT TGT GT-3′ and
reverse: 5′-AC CAC CAA TAC CCA CAA CCA-3′) and methylated (forward:
5′-GTA GGG GTC GCG AAT CGC GC-3′ and reverse 5′-ACC GCC GAT ACC CGC
AAC CG-3′) sequences at the highest methylated region in intron 1
of DUSP6. PCR conditions were as previously described (15).
Annexin/propidium iodide assays
After the EC9706 and KYSE150 cells were transiently
transfected with plasmid constructs, the two cell lines and their
transfectants were collected by trypsinization and washed with PBS,
and subsequently stained with annexin/PI. Samples were analyzed by
FACScan flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA),
according to the manufacturer’s instructions.
Statistical analysis
All statistical analyses was performed with the SPSS
software (version 17.0; SPSS Inc., Shanghai, China). Differences in
DUSP6 protein expression between normal and ESCC specimens, and
associations between DUSP6 expression and clinicopathological
characteristics of ESCC patients, were analyzed by the
χ2 test. The association between DUSP6 expression and
pathological grade of ESCC patients in the tissue microarray assay
was analyzed by Spearman’s rank correlation analysis. Student’s
two-sided t-test was used to compare values of test and control
samples. P<0.05 was considered to indicate a statistically
significant difference. Results of the experiments are depicted as
the means ± SD.
Results
DUSP6 is downregulated in ESCC
In this study, DUSP6 expression was examined at the
mRNA and protein levels in ESCC. The DUSP6 protein expression level
was measured by ESCC tissue microarray immunohistochemical staining
(Fig. 1C). DUSP6 protein was
predominantly observed in the cytoplasm. It was found that normal
esophageal epithelia showed moderate or strong positive staining,
yet cancer tissues demonstrated negative or weak immunoreactions.
In total, 81 of 89 (91%) normal esophageal specimens studied showed
DUSP6 protein expression and 61 of 95 (64.2%) ESCC specimens
studied exhibited reduced DUSP6 protein expression. Statistical
analysis revealed significant differences in DUSP6 protein
expression between normal and ESCC specimens (P<0.000). qPCR was
performed to evaluate the mRNA level of DUSP6 in 19 paired normal
and tumor tissues from the same patients (Fig. 1A). In total, 68.4% (13/19) of the
tumor biopsies expressed lower levels of DUSP6 than their
corresponding normal counterparts; and 36.8% (7/19) of the tumor
biopsies displayed at least two-fold downregulation of DUSP6
compared with their paired normal counterparts. The DUSP6 protein
level in ESCC cell lines and their transfectants was further
evaluated by western blotting (Fig.
1B). The results revealed that the two ESCC cell lines (EC9706
and KYSE150) and their pCMV transfectants exhibited extremely low
DUSP6 protein expression, while their pCMV-DUSP6 transfectants
showed markedly high DUSP6 protein expression. These results
indicate that downregulated DUSP6 expression may be important in
the tumorigenesis of ESCC.
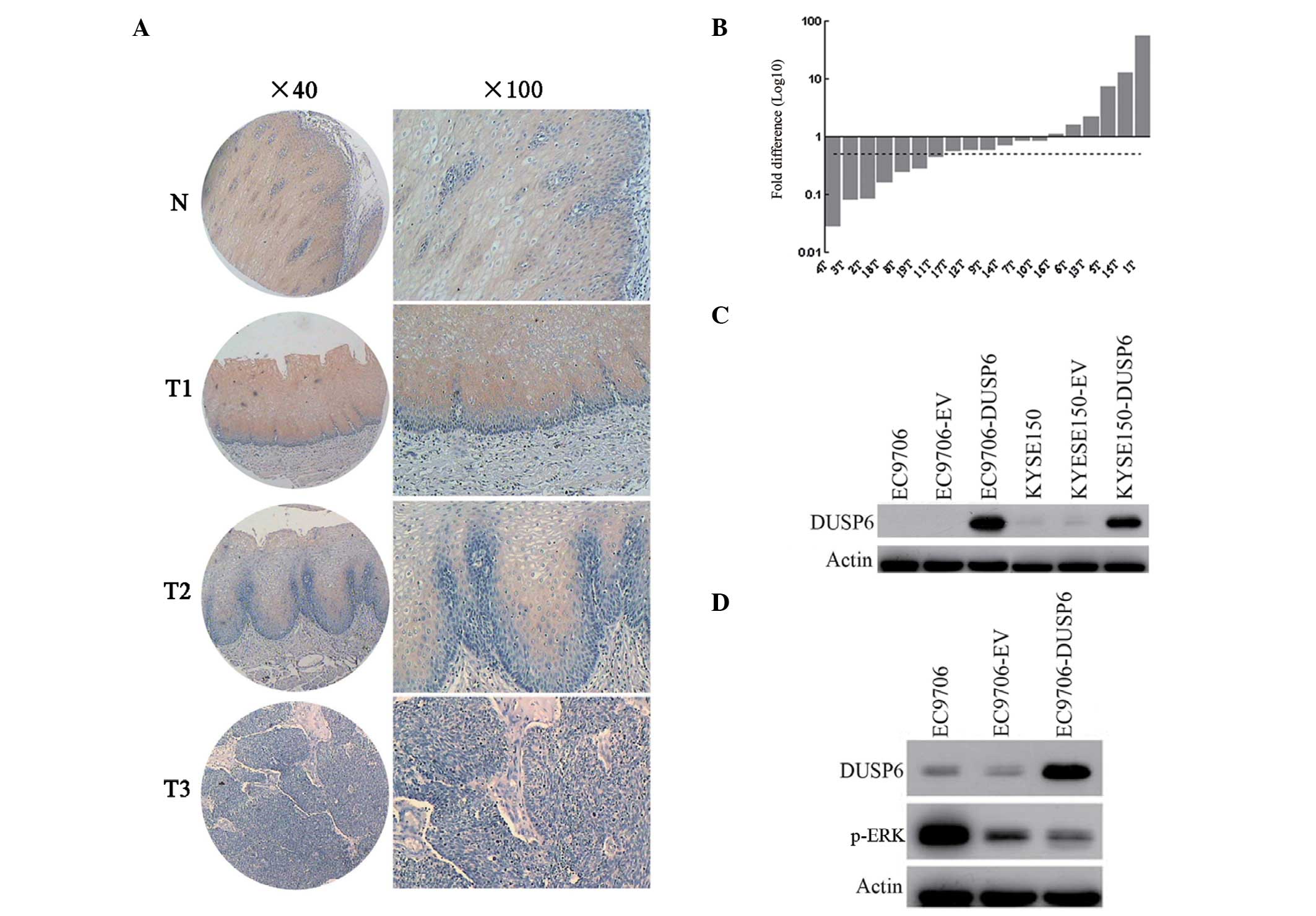 | Figure 1DUSP6 expression in esophageal
cancer. (A) Expression of DUSP6 in normal esophageal epithelia and
primary ESCC tumors were examined by immunohistochemistry. Normal
esophageal epithelia are shown in panel N and the primary
esophageal cancers are shown in panels T1, T2, and T3
(magnification: Left, ×40; right, ×100). (B) Expression of DUSP6 in
paired normal and tumor tissues from the same patients by qPCR. In
total, 36.8% (7/19) of the biopsies displayed at least two-fold
downregulation of DUSP6 compared with their corresponding normal
counterparts. The dotted line is shown to indicate the two-fold
threshold of downregulation. (C) Western blot analysis was used to
determine the DUSP6 expression in EC9706, empty vector-transfected
EC9706, pCMV-DUSP6 (DUSP6)-transfected EC9706, KYSE150, empty
vector-transfected KYSE150 and pCMV-DUSP6-transfected KYSE150
cells. (D) Western blot analysis was utilized to examine the DUSP6
and p-ERK expression in EC9706, empty vector-transfected EC9706 and
pCMV-DUSP6 (DUSP6)-transfected EC9706 cells. DUSP6,
dual-specificity phosphatase 6; ESCC, esophageal squamous cell
carcinoma; p-ERK, phosphorylated extracellular signal-regulated
kinase. |
It is widely accepted that DUSP6 is a negative
feedback regulator in the MAPK pathway that acts by
dephosphorylating the activated ERK (1). In the present study, DUSP6 and
phosphorylated ERK protein expression were detected in the EC9706
ESCC cell line and its transfectants using western blotting. As
shown in Fig. 1D, p-ERK expression
decreased, while DUSP6 expression increased in parental, pCMV-AC
transfected and pCMV-DUSP6 transfected EC9706 cells. Forced
expression of DUSP6 in EC9706 significantly suppressed the p-ERK
expression, indicating that increased DUSP6 expression is
associated with downregulation of p-ERK in vitro in
ESCC.
Correlation between DUSP6 expression and
clinicopathological features
We further analyzed the association between DUSP6
protein expression and clinicopathological features in ESCC
(Table I). The age, gender and
primary tumor size showed no significant correlations with the
expression of DUSP6. Notably, significant correlations were
observed between DUSP6 expression and pathological grade in ESCC
(r=−0.257, P=0.015). It was also found that upregulated expression
of DUSP6 was significantly correlated with regional lymph node
metastasis. We speculate that this observation may be caused by the
negative feedback loop of p-ERK to the tumorigenic signaling during
lymph node metastasis.
 | Table IAssociation between DUSP6 expression
and clinicopathological features in ESCC. |
Table I
Association between DUSP6 expression
and clinicopathological features in ESCC.
| DUSP6 staining, n
(%) | |
|---|
|
| |
|---|
| Variables | − | + | ++ | Correlation
(P-value) |
|---|
| Age, years |
| <60 | 21 (37.5) | 30 (53.6) | 5 (8.9) | 0.114 (0.275) |
| ≥60 | 13 (34.2) | 16 (42.1) | 9 (23.7) | |
| Gender |
| Male | 22 (34.9) | 33 (52.4) | 8 (12.7) | 0.009 (0.930) |
| Female | 12 (38.7) | 13 (41.9) | 6 (19.4) | |
| TNM
classification |
| pT |
|
pT1 | 3 (60) | 2 (40) | 0 (0) | 0.170 (0.104) |
|
pT2 | 10 (40) | 13 (52) | 2 (8) | |
|
pT3 | 20 (31.7) | 31 (49.2) | 12 (19.0) | |
| N |
|
N0 | 33 (39.8) | 40 (48.2) | 10 (12.0) | 0.253 (0.014) |
|
N1 | 1 (10.0) | 5 (50.0) | 4 (40.0) | |
|
N2 | 0 (0.0) | 1 (100.0) | 0 (0.0) | |
| Grade |
| G1 | 2 (15.4) | 8 (61.5) | 3 (23.1) | −0.257 (0.015) |
| G2 | 12 (27.9) | 24 (55.8) | 7 (16.3) | |
| G3 | 17 (51.5) | 12 (36.4) | 4 (15.7) | |
Promoter hypermethylation suppresses
DUSP6 expression
In the present study, we evaluated the role of
hypermethylation in the transcriptional suppression of DUSP6. The
two ESCC cell lines (EC9706 and KYSE150) were treated with DNA
methyl-transferase inhibitor 5-aza-2′-deoxycytidine at two
different concentrations (20 and 50 μM). The restored DUSP6
expression was upregulated when the drug concentration was higher,
as shown in Fig. 2A. The results
indicated that the hypermethylation of the promoter was important
in pathological suppression of DUSP6 transcription in ESCCs.
In order to determine whether hypermethylation
occurs in the expressional regulatory regions of DUSP6, we
performed methylation-specific PCR analysis. The high methylation
of the region between +544 and +627 in intron 1 of DUSP6 was
reported, accounting for the expressional suppression of DUSP6 in
pancreatic cancer that was shown previously (15). Therefore, the present study focused
on this region. The same methylation-specific PCR analysis was
employed as described previously (15). Methylation-specific products in both
ESCC cell lines (EC9706 and KYSE150) were observed, as expected
(Fig. 2B).
In this case, it was demonstrated that the
hypermethylation of CpG islands in intron 1 may be one main
mechanism leading to the silencing of DUSP6 in esophageal squamous
cancers.
DUSP6 expression promotes apoptosis in
ESCC
The functional effect of overexpressed DUSP6 on
cellular apoptosis in EC9706 and KYSE150 cells transfected with the
DUSP6 gene was examined.
Following transfection with plasmids, the cells were
stained with annexin V-FITC and PI to analyze the proportion of
early and total apoptotic cells in both ESCC cell lines and their
transfectants (Fig. 3A). It was
found that both pCMV-DUSP6 transfectants displayed a marked
increased in early and total apoptosis by annexin/PI assay. The
mean early apoptotic cell proportion was 3.40±0.28, 2.40±0.46 and
8.05±0.56% in parental, pCMV-AC-transfected and
pCMV-DUSP6-transfected EC9706 cells, respectively, and was
5.85±0.79, 6.08±0.41 and 14.45±0.05% in parental,
pCMV-AC-transfected, and pCMV-DUSP6-transfected KYSE150 cells,
respectively (Fig. 3B).
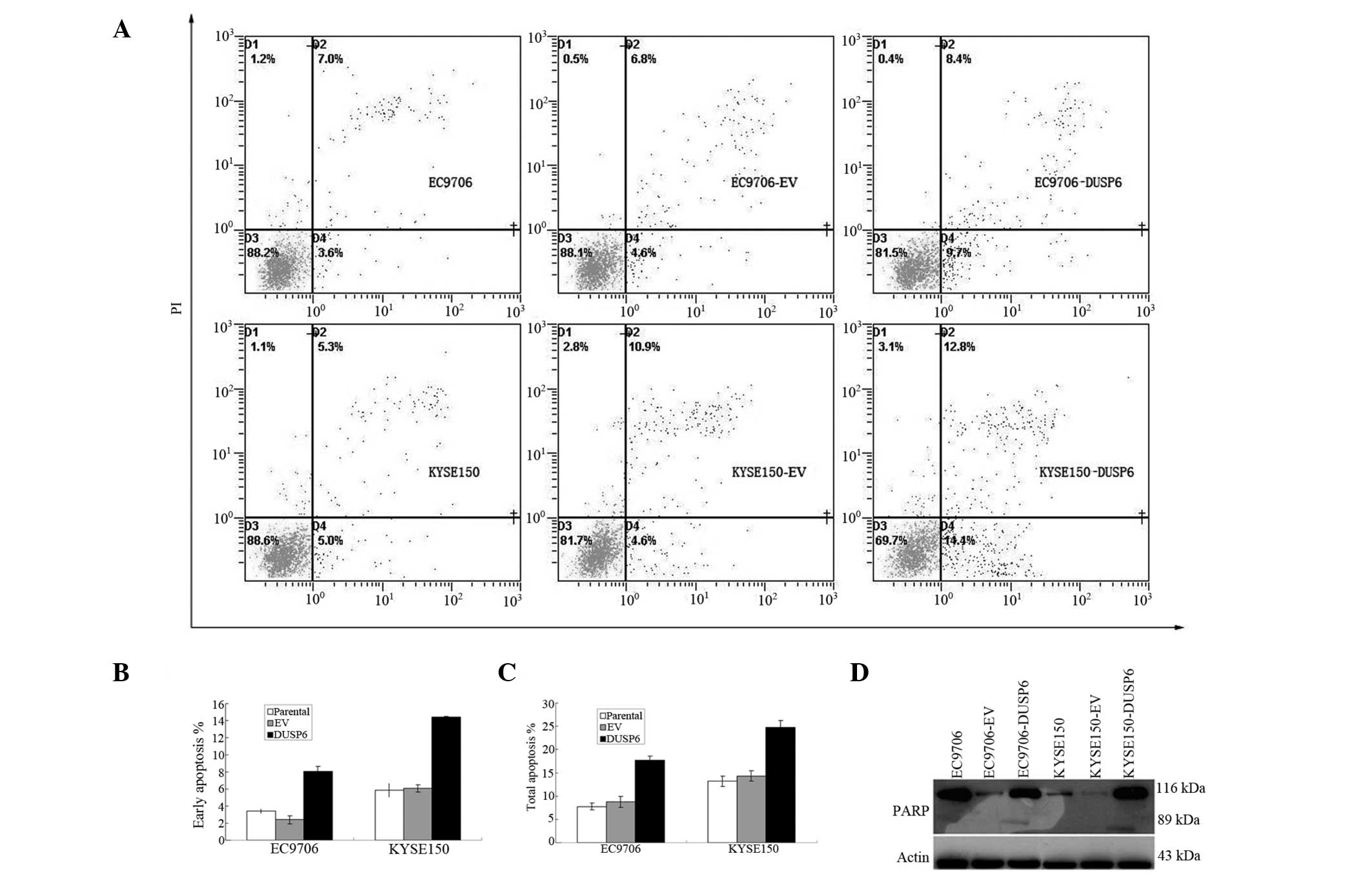 | Figure 3DUSP6 overexpression increased ESCC
cell apoptosis via the PARP pathway. (A–C) Annexin/PI assay: EC9706
and KYSE150 cells were transiently transfected with either EV or
pCMV-DUSP6 plasmids (DUSP6), stained with Annexin V-FITC and PI,
then analyzed by flow cytometry. Results with percentage of cells
listed for each quadrant: Lower left, viable cells; lower right,
cells in early apoptosis; upper right, cells in late apoptosis. (A)
Representative flow histograms. (B) Bar chart showing percentage of
cells in early apoptosis by two cell lines and their transfectants.
(C) Bar chart showing percentage of cells in total apoptosis by two
cell lines and their transfectants. (D) PARP assay: Apoptosis in
DUSP6-overexpressing EC9706 and KYSE150 transfectants confirmed by
immunoblot detection of the 89-kDa PARP cleavage product. DUSP6,
dual-specificity phosphatase 6; ESCC, esophageal squamous cell
carcinoma; PI, propidium iodide; EV, empty vector; FITC,
fluorescein isothiocyanate. |
The mean total apoptotic cell proportion was
7.78±0.76, 8.8±1.21 and 17.68±0.0.99% in parental,
pCMV-AC-transfected and pCMV-DUSP6-transfected EC9706 cells,
respectively, and was 13.2±1.08, 14.3±1.10 and 24.73±1.45% in
parental, pCMV-AC-transfected and pCMV-DUSP6-transfected KYSE150
cells, respectively (Fig. 3C).
Statistical analysis showed significant differences in early and
total apoptotic proportion between the control and experimental
groups of the two ESCC cell lines and their transfectants
(P<0.001).
Subsequently, cellular expression of PARP and its
cleaved product was assayed by immunoblotting in the two ESCC cell
lines and their transfectants. The presence of cleaved PARP
product, a marker of caspase-mediated apoptosis, was found to be
expressed in both pCMV-DUSP6 transfectants, in marked contrast to
the parental and pCMV transfected EC9706 and KYSE150 cells, further
confirming the induction of apoptosis by DUSP6 expression in these
cells (Fig. 3D).
Discussion
DUSP6 is an exclusive negative feedback regulator of
activated ERK during normal development (1,31). In
the present study, it was revealed that DUSP6 is a candidate tumor
suppressor gene in ESCC. Initially, the ESCC cell lines and primary
tumor specimens were screened, demonstrating that the DUSP6
expression level was downregulated at the mRNA and protein levels
in ESCC. Using Spearman’s rank correlation analysis, DUSP6
expression was observed to be negatively correlated to pathological
grade, indicating that DUSP6 is important in human ESCC
carcinogenesis, particularly in tumor progression and
differentiation. Subsequently, we demonstrated that the promoter
hypermethylation accounted for the frequent low expression of
DUSP6. Conversely, demethylation treatment restored DUSP6
expression in ESCC cells. Finally, we revealed that exogenous DUSP6
expression in both ESCC cell lines significantly induced apoptosis.
Overall, these results implied that DUSP6 may serve as a tumor
suppressor gene in ESCC, and loss of DUSP6 may be important in ESCC
tumorigenesis.
DUSP6, one of the DUSPs family, is a highly
selective phosphatase for ERK that appears to play a crucial role
in development and the pathology of various diseases. Accumulating
studies have demonstrated that DUSP6 is involved in tumor
progression and resistance. In various types of cancer, DUSP6 acts
in a contradictory manner. In pancreatic cancer, the DUSP6 gene was
not identified to be expressed in the vast majority of pancreatic
cancer cell lines and invasive primary pancreatic cancer tissues
(12,13). DUSP6 exerts apparent tumor
suppressive effects and is a strong candidate for a tumor
suppressor gene (13,32). Consistent with our findings, a
frequent loss of DUSP6 expression was observed as the histological
grade of the tumor increased in lung cancer. DUSP6 is a potential
tumor suppressor gene of lung cancer (16). However, DUSP6 is overexpressed and
acts as an oncogene in certain other types of cancer (21,23).
In the present study, we evaluated the expression at the mRNA and
protein levels in ESCC cell lines and primary tumor specimens. It
was demonstrated that DUSP6 was downregulated in ESCC. Tissue
microarray assays indicated that DUSP6 expression inversely
correlated with the histological grade, which suggested that lower
DUSP6 expression was involved in tumor progression and
differentiation. We hypothesized that DUSP6 may be a promising
prognostic biomarker in ESCC. Further studies are required to
validate the clinical utility of DUSP6 protein as a biomarker for
ESCC prognosis.
The crucial mechanisms inactivating tumor suppressor
genes are gene promoter hypermethylation, coding exon mutation and
loss of heterozygosity (LOH). Previous studies have demonstrated
that DUSP6 was downregulated by hypermethylation of intron 1
(12,15), LOH (16) or ubiquitination/proteasome
degradation (17). In ESCC, tumor
suppressor genes, such as FHIT, ECRG4 and DIRAS1, are downregulated
by gene promoter hypermethylation (30,33,34).
In the current study, our preliminary investigation of the DUSP6
downregulation focused on the epigenetic gene silencing, which is
important in the initiation and progression of cancer (35,36).
It was observed that methylation-specific products in ESCC cell
lines were produced by methylation-specific PCR. Further
pharmocological demethylation treatment restored the DUSP6
expression. These results indicated that the promoter
hypermethylation may be one important factor resulting in the loss
of DUSP6 expression in ESCC. Consistent with our results, a
previous study showed that hypermethylation was also pivotal in
downregulation of DUSP6 in ESCC in Hong Kong (37).
Functionally, DUSP6 has demonstrated suppressive
effects in tumor formation and cancer cell mobility in ESCC in
previous studies (27,37). DUSP6 played an important role in
inducing cellular apoptosis in a previous study; the downregulation
of DUSP6 mRNA by nitric oxide exerted antiapoptotic effects in
endothelial cells (38). The
exogenous expression of DUSP6/MKP-3 has been shown to induce
apoptosis by the attenuation of ERK activation in pancreatic and
lung cancer (13,39). In the present study, we investigated
the effect of DUSP6 on cellular apoptosis in ESCC. Consistent with
a previous study (39), our data
showed that exogenous DUSP6 expression markedly increased early and
total apoptosis in vitro, further supporting the fact that
DUSP6 may be an important candidate tumor suppressor gene in
ESCC.
It has been reported that the activation of the
MEK/Erk pathway may inhibit cellular apoptosis (40–42)
and that the inhibition of ERK is crucial for the induction of
apoptosis (43). Given the
established role of ERK1/2 in the antiapoptotic defense network and
the fact that DUSP6 induced apoptosis and ERK was downregulated
in vitro in ESCC, we speculated that the apoptotic effect of
DUSP6 in ESCC may be induced by attenuation of ERK activation. More
comprehensive investigations are required to determine the
associations between DUSP6, ERK and apoptosis.
In conclusion, the current study has suggested that
DUSP6 expression decreases with the depth of invasion in ESCC,
predicting tumor progression independent of tumor grade. Enforced
DUSP6 expression induced cellular apoptosis in vitro in
ESCC. We hope that our data may provide new evidence to improve the
understanding of the carcinogenesis of ESCC and that it will help
to provide new therapeutic strategies for the treatment of
ESCC.
Acknowledgements
This study was supported by the State Key Basic
Research Programs 973 of China (2009CB521803). The authors would
like thank Dr Cao Yan for the histological evaluation of
tumors.
References
|
1
|
Arkell RS, Dickinson RJ, Squires M, Hayat
S, Keyse SM and Cook SJ: DUSP6/MKP-3 inactivates ERK1/2 but fails
to bind and inactivate ERK5. Cell Signal. 20:836–843. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eblaghie MC, Lunn JS, Dickinson RJ, et al:
Negative feedback regulation of FGF signaling levels by Pyst1/MKP3
in chick embryos. Curr Biol. 13:1009–1018. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lewis TS, Shapiro PS and Ahn NG: Signal
transduction through MAP kinase cascades. Adv Cancer Res.
74:49–139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ekerot M, Stavridis MP, Delavaine L, et
al: Negative-feedback regulation of FGF signalling by DUSP6/MKP-3
is driven by ERK1/2 and mediated by Ets factor binding to a
conserved site within the DUSP6/MKP-3 gene promoter. Biochem J.
412:287–298. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nichols A, Camps M, Gillieron C, et al:
Substrate recognition domains within extracellular signal-regulated
kinase mediate binding and catalytic activation of
mitogen-activated protein kinase phosphatase-3. J Biol Chem.
275:24613–24621. 2000. View Article : Google Scholar
|
|
8
|
Muda M, Theodosiou A, Gillieron C, et al:
The mitogen-activated protein kinase phosphatase-3 N-terminal
noncatalytic region is responsible for tight substrate binding and
enzymatic specificity. J Biol Chem. 273:9323–9329. 1998. View Article : Google Scholar
|
|
9
|
Zhou B, Wu L, Shen K, Zhang J, Lawrence DS
and Zhang ZY: Multiple regions of MAP kinase phosphatase 3 are
involved in its recognition and activation by ERK2. J Biol Chem.
276:6506–6515. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sridhar SS, Hedley D and Siu LL: Raf
kinase as a target for anticancer therapeutics. Mol Cancer Ther.
4:677–685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kohno M and Pouyssegur J: Targeting the
ERK signaling pathway in cancer therapy. Ann Med. 38:200–211. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Furukawa T, Yatsuoka T, Youssef EM, et al:
Genomic analysis of DUSP6, a dual specificity MAP kinase
phosphatase, in pancreatic cancer. Cytogenet Cell Genet.
82:156–159. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Furukawa T, Sunamura M, Motoi F, Matsuno S
and Horii A: Potential tumor suppressive pathway involving
DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 162:1807–1815. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Furukawa T, Fujisaki R, Yoshida Y, et al:
Distinct progression pathways involving the dysfunction of
DUSP6/MKP-3 in pancreatic intraepithelial neoplasia and intraductal
papillary-mucinous neoplasms of the pancreas. Mod Pathol.
18:1034–1042. 2005. View Article : Google Scholar
|
|
15
|
Xu S, Furukawa T, Kanai N, Sunamura M and
Horii A: Abrogation of DUSP6 by hypermethylation in human
pancreatic cancer. J Hum Genet. 50:159–167. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okudela K, Yazawa T, Woo T, et al:
Down-regulation of DUSP6 expression in lung cancer: its mechanism
and potential role in carcinogenesis. Am J Pathol. 175:867–881.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan DW, Liu VW, Tsao GS, et al: Loss of
MKP3 mediated by oxidative stress enhances tumorigenicity and
chemoresistance of ovarian cancer cells. Carcinogenesis.
29:1742–1750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Croonquist PA, Linden MA, Zhao F and Van
Ness BG: Gene profiling of a myeloma cell line reveals similarities
and unique signatures among IL-6 response, N-ras-activating
mutations, and coculture with bone marrow stromal cells. Blood.
102:2581–2592. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bloethner S, Chen B, Hemminki K, et al:
Effect of common B-RAF and N-RAS mutations on global gene
expression in melanoma cell lines. Carcinogenesis. 26:1224–1232.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramnarain DB, Park S, Lee DY, et al:
Differential gene expression analysis reveals generation of an
autocrine loop by a mutant epidermal growth factor receptor in
glioma cells. Cancer Res. 66:867–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Messina S, Frati L, Leonetti C, et al:
Dual-specificity phosphatase DUSP6 has tumor-promoting properties
in human glioblastomas. Oncogene. 30:3813–3820. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Warmka JK, Mauro LJ and Wattenberg EV:
Mitogen-activated protein kinase phosphatase-3 is a tumor promoter
target in initiated cells that express oncogenic Ras. J Biol Chem.
279:33085–33092. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cui Y, Parra I, Zhang M, et al: Elevated
expression of mitogen-activated protein kinase phosphatase 3 in
breast tumors: a mechanism of tamoxifen resistance. Cancer Res.
66:5950–5959. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chiappinelli KB, Rimel BJ, Massad LS and
Goodfellow PJ: Infrequent methylation of the DUSP6 phosphatase in
endometrial cancer. Gynecol Oncol. 119:146–150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
No authors listed. Esophageal cancer:
epidemiology, pathogenesis and prevention. Nat Clin Pract
Gastroenterol Hepatol. 5:517–526. 2008. View Article : Google Scholar
|
|
26
|
Leung AC, Wong VC, Yang LC, et al:
Frequent decreased expression of candidate tumor suppressor gene,
DEC1, and its anchorage-independent growth properties and impact on
global gene expression in esophageal carcinoma. Int J Cancer.
122:587–594. 2008. View Article : Google Scholar
|
|
27
|
Wang Y, Fan H, Zhou B, et al: Fusion of
human umbilical cord mesenchymal stem cells with esophageal
carcinoma cells inhibits the tumorigenicity of esophageal carcinoma
cells. Int J Oncol. 40:370–377. 2012.PubMed/NCBI
|
|
28
|
Han Y, Wei F, Xu X, et al: Establishment
and comparative genomic hybridization analysis of human esophageal
carcinomas cell line EC9706. Zhonghua Yi Xue Yi Chuan Xue Za Zhi.
19:455–457. 2002.(In Chinese).
|
|
29
|
Shimada Y, Imamura M, Wagata T, Yamaguchi
N and Tobe T: Characterization of 21 newly established esophageal
cancer cell lines. Cancer. 69:277–284. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li LW, Yu XY, Yang Y, Zhang CP, Guo LP and
Lu SH: Expression of esophageal cancer related gene 4 (ECRG4), a
novel tumor suppressor gene, in esophageal cancer and its
inhibitory effect on the tumor growth in vitro and in vivo. Int J
Cancer. 125:1505–1513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li C, Scott DA, Hatch E, Tian X and
Mansour SL: Dusp6 (Mkp3) is a negative feedback regulator of
FGF-stimulated ERK signaling during mouse development. Development.
134:167–176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Furukawa T and Horii A: Molecular
pathology of pancreatic cancer: in quest of tumor suppressor genes.
Pancreas. 28:253–256. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shimada Y, Sato F, Watanabe G, et al: Loss
of fragile histidine triad gene expression is associated with
progression of esophageal squamous cell carcinoma, but not with the
patient’s prognosis and smoking history. Cancer. 89:5–11. 2000.
|
|
34
|
Zhu YH, Fu L, Chen L, et al:
Downregulation of the novel tumor suppressor DIRAS1 predicts poor
prognosis in esophageal squamous cell carcinoma. Cancer Res.
73:2298–2309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baylin SB and Ohm JE: Epigenetic gene
silencing in cancer - a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feinberg AP, Ohlsson R and Henikoff S: The
epigenetic progenitor origin of human cancer. Nat Rev Genet.
7:21–33. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wong VC, Chen H, Ko JM, et al: Tumor
suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell
invasion and epithelial-mesenchymal transition (EMT)-associated
phenotype. Int J Cancer. 130:83–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rössig L, Haendeler J, Hermann C, et al:
Nitric oxide down-regulates MKP-3 mRNA levels: involvement in
endothelial cell protection from apoptosis. J Biol Chem.
275:25502–25507. 2000.PubMed/NCBI
|
|
39
|
Zhang Z, Kobayashi S, Borczuk AC, et al:
Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative
feedback mediator of oncogenic ERK signaling in lung cancer cells.
Carcinogenesis. 31:577–586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Le Gall M, Chambard JC, Breittmayer JP,
Grall D, Pouyssegur J and Van Obberghen-Schilling E: The p42/p44
MAP kinase pathway prevents apoptosis induced by anchorage and
serum removal. Mol Biol Cell. 11:1103–1112. 2000.PubMed/NCBI
|
|
41
|
Bonni A, Brunet A, West AE, Datta SR,
Takasu MA and Greenberg ME: Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent
mechanisms. Science. 286:1358–1362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Erhardt P, Schremser EJ and Cooper GM:
B-Raf inhibits programmed cell death downstream of cytochrome c
release from mitochondria by activating the MEK/Erk pathway. Mol
Cell Biol. 19:5308–5315. 1999.PubMed/NCBI
|
|
43
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|















