Introduction
Synovial sarcoma is a type of mesenchymal tissue
cell tumor that exhibits epithelial differentiation, which most
frequently arises in the extremities, while a primary occurrence in
the mediastinum is quite rare (1,2).
Primary mediastinal synovial sarcomas are malignant tumors with a
low incidence, no specific clinical manifestations and a lack of
unified and effective treatments, highlighting the challenges
preventing its diagnosis and treatment in clinics. The present
study reports a case of primary giant mediastinal synovial sarcoma
of the neck in a patient admitted to The Second Xiangya Hospital of
Central South University (Hunan, China).
Case report
An 11-year-old male presented with a mass in the
left side of the neck that had been present for ~1 month, together
with mild dysphagia, without coughing, chest pain or shortness of
breath. A physical examination revealed a large lump with an area
of ~4×3 cm2 in the left thyroid gland, however nothing
of significance was revealed in the chest or abdominal regions.
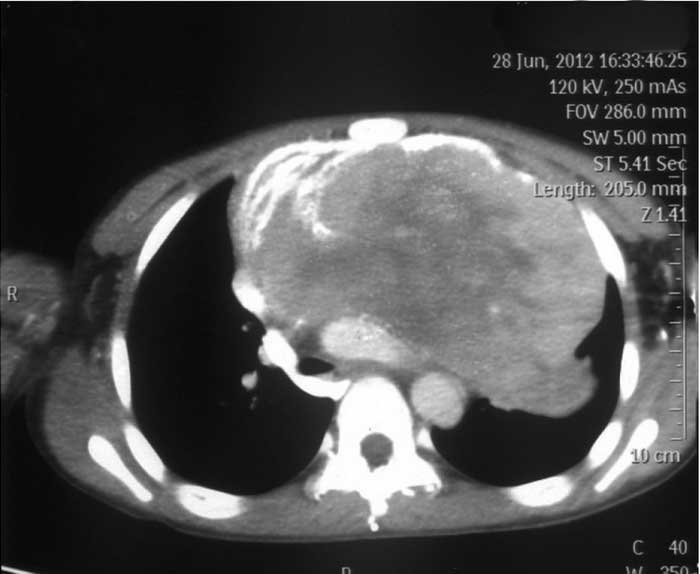
Computed tomography (CT) scans (Figs.
1 and 2) showed a large,
patchily enhanced mass in the anterior mediastinum, which extended
into the left thyroid gland. Patchy areas of necrosis, low-density
liquidity and calcification were observed in the mass. A
thoracotomy was performed, revealing a huge mass measuring 20×15×15
cm3, arising from the anterior mediastinum, violating
the lower pole of the thyroid and engulfing the left phrenic nerve
and innominate vein. The left innominate vein and phrenic and vagus
nerves were resected and bluntly separated, and then the mass was
removed intact whilst the left diaphragmatic muscle was
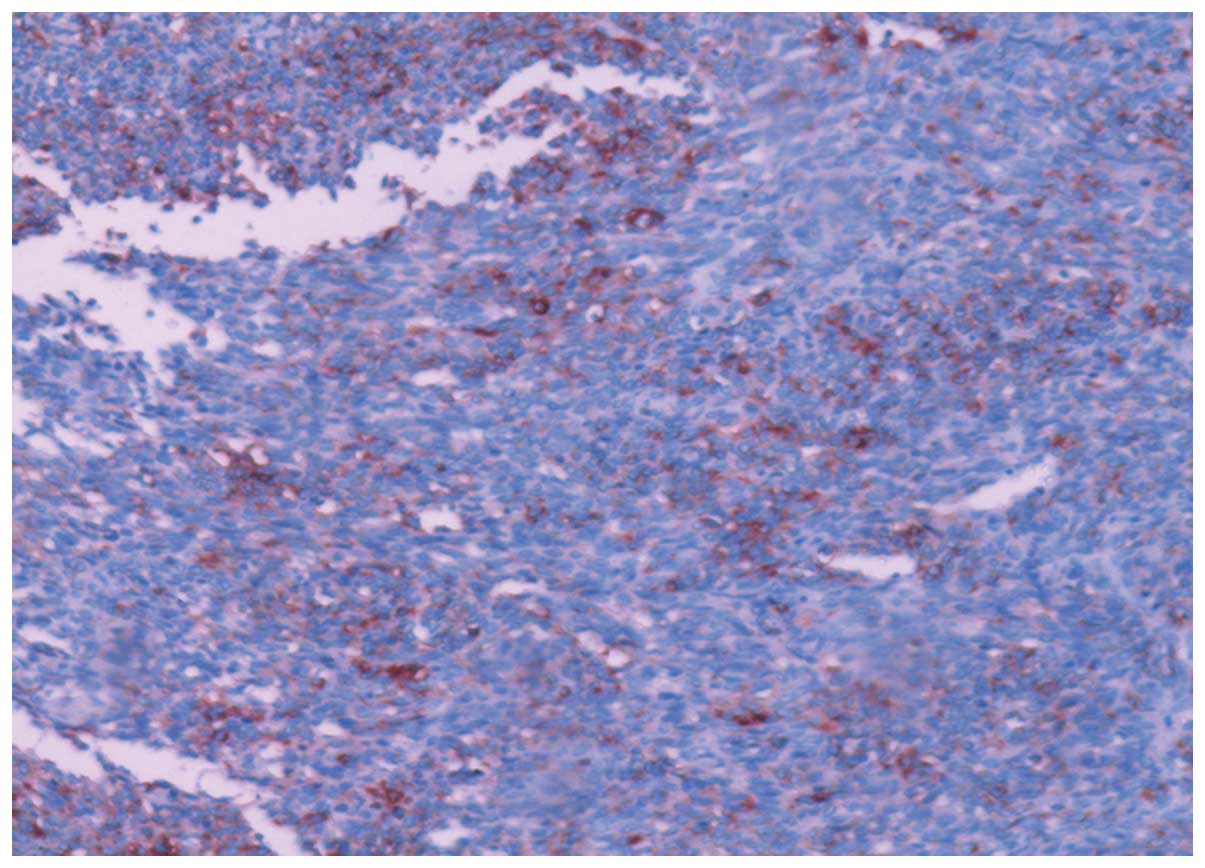
simultaneously suspended. A histological examination (Fig. 3) showed malignant mesenchymal tumor
tissue involving the left innominate vein. An immunochemistry
examination revealed the following results: EMA(+), Vim(+),
S100(+), CD99(+), Ki-67(+), CD117(−), CD34(−), SAM(−), HMB(−),
CK(−) and p53(−) (Figs. 4–7), confirming the lesion as a monophasic
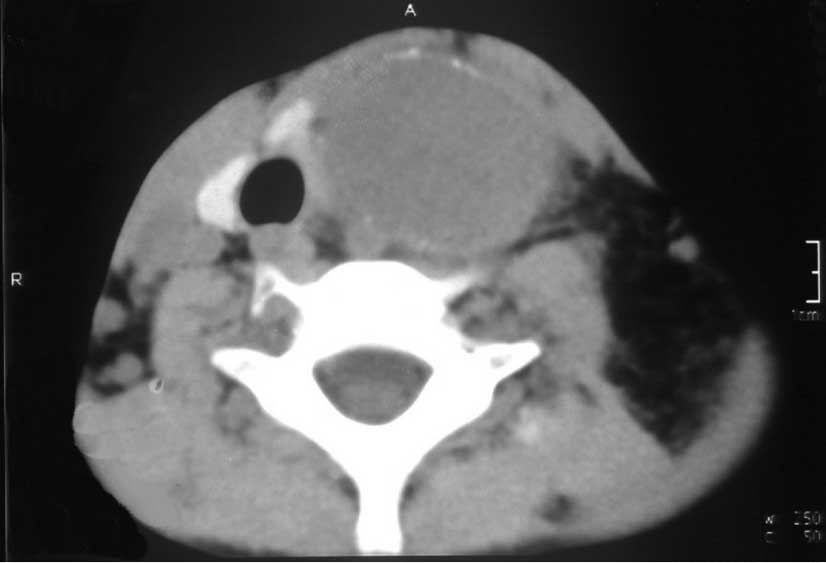
synovial sarcoma. One month after surgery, a follow-up CT scan
(Fig. 8) showed a patchily enhanced
soft tissue mass, 5 cm in diameter, in the anterior mediastinum,
while the neck structure was clear. Adjuvant chemotherapy
containing ifosfamide, 1.8 g days 1–4 and doxorubicin, 30 mg days
1–2, administered every 3 weeks for four cycles, was proposed for
the patient due to the residual mass. A thoracic CT (Fig. 9) was performed again following
chemotherapy and revealed no mass in the mediastinum or neck. A
total of 64 Gy adjuvant radiotherapy was applied to the primary
tumor with hope of an increased disease-free survival. The patient
recently completed treatment and is currently undergoing
follow-up.
Discussion
Synovial sarcoma was defined by the World Health
Organization (WHO) in 2002 as a type of mesenchymal tissue cell
tumor that exhibits epithelial differentiation (1), which most frequently arises in the
extremities and has been prevalent in adolescent and young adults
between the ages of 15–40 years (2). Although 85% of synovial sarcomas arise
in joint cavities, they may also occur in locations unassociated
with joint cavities, including the head and neck, thoracic wall,
abdominal region, genitourinary tract and other rarer sites
(3). In a retrospective study
conducted by Burt et al(4),
of the total 3,149 soft tissue sarcomas examined, primary
mediastinal sarcoma represented 1.4%. The most common primary
mediastinal sarcoma was the malignant peripheral nerve sheath
tumor, which accounted for 26%, while the synovial sarcoma only
accounted for 2%. To date, only 24 previous studies analyzing
mediastinal synovial sarcoma have been found through searching
PubMed, in which patients’ ages ranged between 3 and 83 years, with
a male-to-female ratio of ~3:1 and tumors measuring between 5 and
20 cm in their greatest diameter. Histologically, 36 cases
exhibited a biphasic growth pattern and monophasic type at a ratio
of 1:1 and 1 case exhibited a poorly-differentiated form. The
overall survival time ranged between 3 months and >5 years, with
a median overall survival time of 19.8 months.
Primary mediastinal synovial sarcoma is a type of
malignant tumor with no specific differences from other mediastinal
tumors with regard to clinical manifestation, imaging or histology,
therefore, it is difficult to diagnose. With regard to clinical
manifestations, mediastinal synovial sarcomas reveal various
initial symptoms due to their different scopes of infringement. The
common symptoms include chest pain (5–7),
shortness of breath and dyspnea (8,9), in
contrast to the present case whose initial symptom was the
accidental identification of a neck lump, which is a rare clinical
manifestation and is likely to be easily ignored and misdiagnosed.
Therefore, it is important to combine imaging and pathology to
assist in establishing a diagnosis. With regard to imaging
performances, the majority of mediastinal synovial sarcoma patients
visit their doctors following the onset of dyspnea or other
manifestations. Chest X-rays or CT scans may reveal space-occupying
lesions in the mediastinum, which exhibit no specific radiological
characteristics to other mediastinal stromal tumors, including
necrotic, hemorrhagic or cystic components on section, and
calcification may be found in the mass. In addition, magnetic
resonance imaging or positron emission tomography/CT may
demonstrate the adhesion and invasion scope of lesions to the
surrounding tissue, thus offering guidance for the selection of
appropriate treatment. The pathological diagnosis is important
since the multiformity of synovial sarcoma in clinical
manifestations and the absence of specificity relative to imaging
performances have limitations for the diagnosis of synovial
sarcoma. Usually, patients are confirmed to have synovial sarcoma
by B ultrasound or CT-guided fine-needle aspiration cytology from
the mass, or by post-operative pathological examination.
Pathological diagnosis remains the gold standard, and synovial
sarcoma is divided into four types according to the various
histological observations of epithelial and spindle cells in the
mass (10), including monophasic
spindle and epithelial cell types, a poorly-differentiated form and
a biphasic pattern. In addition, characteristic histopathological
observations and immunochemistry examinations are important for the
differential diagnosis of a synovial sarcoma from other stromal
tumors (11). It has been indicated
that vimentin, cytokeratin and EMA positivity, in combination with
CD34 negativity, are the most useful protein biomarkers for the
diagnosis of monophasic synovial sarcoma (10). In the current case report, the tumor
was confirmed as a malignant mesenchymal tumor under the
microscope, but its subtype was difficult to determine. The tumor
cells were found to be positive for vimentin, EMA and S100, and
negative for CD34, which, in combination with the morphological
profile, confirmed the diagnosis of a monophasic synovial sarcoma.
With regard to a genetic diagnosis, cellular and molecular genetic
studies (10–13) have shown that the translocation
t(X;18)(p11.2; q11.2) exists in >90% of synovial sarcomas. The
translocation involves the SYT gene on chromosome 18 and the SSX1
or SSX2 gene on the X chromosome. In addition, this translocation
is not associated with other sarcomas and thus, may represent a
specific biomarker for diagnosing synovial sarcoma. However, this
fusion gene was not detected in the present case.
Synovial sarcomas, particularly mediastinal synovial
sarcomas, are highly aggressive tumors, which are more likely to
invade adjacent significant organs, including the heart, lung and
blood vessels, the majority of which exhibit no evident clinical
features, thus highlighting challenges for diagnosis and prompt
treatment. The following treatments have been applied for synovial
sarcomas: i) Although mediastinal synovial sarcomas have limited
clinical data and no standardized treatments, complete surgical
excision remains the cornerstone of therapy (5,6,8,9).
A broad surgical excision must be promoted for inchoate patients,
as presented in the current case report where the patient’s tumor
and left innominate vein and phrenic nerve were resected in hope of
increased disease-free survival. For patients with a neoplasm that
has invaded into the adjoining organs, the partial excision method
may provide appropriate clinical management. Ferrari et
al(14) retrospectively studied
271 patients with synovial sarcomas and found that the 5-year
disease-free survival rate was 42.5% for patients treated with
complete surgical excision and 31.6% for patients treated with
partial surgical excision. The remaining 31.9% of patients were not
treated with surgery. A partial surgical excision may not prolong
disease-free survival, as indicated by these results, but it may
solve the issues of obstruction or filling for terminal patients
who may not be treated by surgical excision due to a wide range of
violations, thus, there remains specific implications. ii)
Radiotherapy is an effective treatment method to kill cancer cells
and control local recurrence rates. In the retrospective study
conducted by Ferrari et al(14), adjuvant radiotherapy did not prolong
the 5-year local recurrence-free survival (LRFS) rate in patients
with completely resected disease and free histological margins; the
5-year LRFS rate for patients who received post-operative
radiotherapy and for those who had not was 77.8 and 66.9%,
respectively. However, adjuvant radiotherapy showed a marked impact
on patients with marginally resected disease, as in the 71 patients
who received partial surgical excision, the 5-year LRFS rate was
57.4% for patients who received radiotherapy and only 7.1% for
patients who had not. Similarly, Harb et al(15) also found that patients with synovial
sarcoma of the head and neck who were treated with surgery and
post-operative radiotherapy exhibited higher survival and lower
recurrence rates than those treated with surgery only or a
combination of surgery and chemotherapy. There has been a lack of
reliable data analysis to determine the significance of
post-operative radiotherapy in mediastinal synovial sarcoma, but
according to the aforementioned data, mediastinal synovial sarcoma
patients are commonly treated with adjuvant radiotherapy following
surgery, particularly those patients with positive histological
margins. In the current case report, a large tumor was found in the
mediastinum prior to surgery, and a small mass shadow remained
visible in the follow-up CT scan following surgery. After referring
to specific mediastinal synovial sarcoma treatments available
overseas (6,9), the patient was treated with adjuvant
post-operative radiotherapy with a total of 64 Gy, in the hope of
controlling local recurrence. iii) Chemotherapy is an additional,
predominant tool used for tumor treatments and is particularly
important for preventing tumors from distant metastasis. Synovial
sarcomas have moderate chemosensitivity with a response rate of 50%
to regimens containing ifosfamide and doxorubicin (16). In the study by Ferrari et
al(14), the 5-year
metastasis-free survival rates were 60.2% [standard error of the
mean (SEM), 6.8%] and 47.8% (SEM, 3.3%) for patients who had
accepted adjuvant chemotherapy and for those who had not,
respectively. Further analysis has shown that the greatest benefits
associated with chemotherapy usually appear in patients ≥17 years
old, with tumors measuring >5 cm. In addition, neoadjuvant
chemotherapy is likely to reduce tumor size and thus, may offer
surgical conditions for patients who cannot undergo surgery
immediately, and therefore improve radical surgical resection
rates. Balieiro et al(17)
recently reported a case of a giant primary mediastinal synovial
sarcoma treated with neoadjuvant chemotherapy. A large mass of 20
cm in its largest diameter was found in the upper section of the
mediastinum and had invaded the main anterior vessels and chest
wall, and compressed the heart and critical left mainstem bronchus,
thus, the patient exhibited a lack of surgical indications. The
mass was significantly reduced following 6 cycles of neoadjuvant
chemotherapy (ifosfamide and doxorubicin) followed by radical en
bloc resection, and the patient exhibited no signs of disease
recurrence following five years of routine follow-up examinations.
The therapeutic regimen of ifosfamide and doxorubicin has been
regularly used for the treatment of synovial sarcomas to prolong
overall survival rates (6,7,9,17). The
patient in the present case report also accepted 4 cycles of this
combination regimen and the follow-up CT scans (Fig. 8 and 9) showed that the mass in the mediastinum
had shrunk, confirming that the chemotherapy had worked. iv) The
final treatment for synovial sarcoma is targeted therapy. In total,
>90% of synovial sarcoma patients have the aforementioned fusion
genes, SYT-SSX1 or SYT-SSX2. In a study conducted by Sarver et
al(18), it was revealed that
the SYT-SSX gene may target EGR1 receptors to inhibit the
expression of EGR1, a type of cancer suppressor gene, and thus, is
involved in cell migration. Studies with regard to the role of this
highly-expressed fusion gene in synovial sarcoma (19) must be investigated in hope of
identifying novel targeted therapy drugs.
A previous study (3)
showed that the 5-, 10- and 15-year survival rates of synovial
sarcoma patients who received complete surgical excision and
adjuvant radiotherapy were 76, 63 and 57%, respectively. However,
synovial sarcomas in the mediastinum have a poorer prognosis
compared with synovial sarcomas presenting in the extremities,
since synovial sarcoma may be more likely to invade the surrounding
organs, including the heart, lungs and blood vessels. The existing
clinical data have shown that the overall survival time for
mediastinal synovial sarcoma is between 3 months and >5 years
(7–9,17).
Poor prognostic factors may include being of the male gender, being
>20 years old, tumor diameters of ≥9 cm, the existence of
extensive tumor necrosis, neurovascular invasion and the presence
of the SYT-SSX1 or SYT-SSX2 fusion genes (9).
Mediastinal synovial sarcomas are malignant tumors
with a low incidence, no specific clinical manifestations and a
lack of unified and effective treatments. These factors highlight
the challenges preventing its diagnosis and treatment in clinics.
The existing data supports surgery as the preferred treatment for
mediastinal synovial sarcoma, but appropriate auxiliary treatments,
including radiotherapy and chemotherapy, must be taken into
consideration according to the factors affecting prognosis. In
addition, the SYT-SSX fusion gene in synovial sarcoma must be
further investigated in hope of identifying novel targeted
therapies.
References
|
1
|
Fletcher CDM, Unni KK and Mertens F: World
Health Organization Classification of Tumours. Pathology and
Genetics of Tumours of Soft Tissue and Bone. IARC Press; Lyon:
2002
|
|
2
|
Baptista AM, Camargo OP, Croci AT, et al:
Synovial sarcoma of the extremities: prognostic factors for 20
nonmetastatic cases and a new histologic grading system with
prognostic significance. Clinics (Sao Paulo). 61:381–386. 2006.
View Article : Google Scholar
|
|
3
|
Korula A, Shah A, Philip MA, Kuruvila K,
Pradhip J, Pai MC and Chacko RT: Primary mediastinal synovial
sarcoma with transdiaphragmatic extension presenting as a
pericardial effusion. Singapore Med J. 50:e26–e28. 2009.PubMed/NCBI
|
|
4
|
Burt M, Ihde JK, Hajdu SI, Smith JW, Bains
MS, Downey R, et al: Primary sarcomas of the mediastinum: results
of therapy. J Thorac Cardiovasc Surg. 115:671–680. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeganathan R, Davis R, Wilson L, McGuigan
J and Sidhu P: Primary mediastinal synovial sarcoma. Ulster Med J.
76:109–111. 2007.
|
|
6
|
Pal M, Ghosh BN, Roy C and Manna AK:
Posterior mediastinal biphasic synovial sarcoma in a 12 year-old
boy: a case report and review of literature. J Cancer Res Ther.
6:564–566. 2010.PubMed/NCBI
|
|
7
|
Ravikumar G, Mullick S, Ananthamurthy A
and Correa M: Primary synovial sarcoma of the mediastinum: a case
report. Case Rep Surg. 2011:6028532011.PubMed/NCBI
|
|
8
|
Loggos S, Kondrafouris K, Oikonomopoulos G
and Mitropoulos F: Large monophasic synovial sarcoma of the
mediastinum in a 15-year old boy. Interact Cardiovasc Thorac Surg.
15:909–911. 2012.PubMed/NCBI
|
|
9
|
Tezcan Y, Koc M, Kocak H and Kaya Y:
Primary intrathoracic biphasic synovial sarcoma. Arch Iran Med.
15:331–333. 2012.PubMed/NCBI
|
|
10
|
Arafah M and Zaidi SN: Poorly
differentiated monophasic synovial sarcoma of the mediastinum.
Indian J Pathol Microbiol. 54:384–387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jang JW, Lee JK, Seo BR and Kim SH:
Synovial sarcoma of the posterior neck: a case report and review of
literature. J Korean Neurosurg Soc. 47:306–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inagaki H, Nagasaka T, Otsuka T, Sugiura
E, Nakashima N and Eimoto T: Association of SYT-SSX fusion types
with proliferative activity and prognosis in synovial sarcoma. Mod
Pathol. 13:482–488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Antonescu CR, Kawai A, Leung DH, Lonardo
F, Woodruff JM, Healey JH and Ladanyi M: Strong association of
SYT-SSX fusion type and morphologic epithelial differentiation in
synovial sarcoma. Diagn Mol Pathol. 9:1–8. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ferrari A, Gronchi A, Casanova M, et al:
Synovial sarcoma: a retrospective analysis of 271 patients of all
ages treated at a single institution. Cancer. 101:627–634. 2004.
View Article : Google Scholar
|
|
15
|
Harb WJ, Luna MA, Patel SR, Ballo MT,
Roberts DB and Sturgis EM: Survival in patients with synovial
sarcoma of the head and neck: association with tumor location,
size, and extension. Head Neck. 29:731–740. 2007. View Article : Google Scholar
|
|
16
|
Dennison S, Weppler E and Giacoppe G:
Primary pulmonary synovial sarcoma: a case report and review of
current diagnostic and therapeutic standards. Oncologist.
9:339–342. 2004. View Article : Google Scholar
|
|
17
|
Balieiro MA, Lopes AJ, Costa BP, Veras GP,
Perelson PS, Acatauassú Nunes R and Saito EH: The surprising
outcome of a giant primary mediastinal synovial sarcoma treated
with neoadjuvant chemotherapy. J Thorac Dis. 5:94–96. 2013.
|
|
18
|
Sarver AL, Li L and Subramanian S:
MicroRNA miR-183 functions as an oncogene by targeting the
transcription factor EGR1 and promoting tumor cell migration.
Cancer Res. 70:9570–9580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Randall RL, Schabel KL, Hitchcock Y,
Joyner DE and Albritton KH: Diagnosis and management of synovial
sarcoma. Curr Treat Options Oncol. 6:449–459. 2005. View Article : Google Scholar
|